

Abstract
Rett syndrome (Rett) is the leading genetic cause of mental retardation in females. Most cases of Rett are caused by loss-of-function mutations in the gene coding for the transcriptional regulator methyl-CpG binding protein 2 (MeCP2), but despite much effort, it remains unclear how a loss of MeCP2 function generates the neurological deficits of Rett. Here we show that MeCP2 plays an essential and cell-autonomous role in homeostatic synaptic scaling up in response to reduced firing or reduced sensory drive in rat visual cortical pyramidal neurons. We found that acute RNAi knockdown of MeCP2 blocked synaptic scaling within targeted neocortical pyramidal neurons. Furthermore, MeCP2 knockdown decreased excitatory synapse number without affecting basal mEPSC amplitude or AMPAR accumulation at spared synapses, demonstrating that MeCP2 acts cell-autonomously to maintain both excitatory synapse number and synaptic scaling in individual neocortical neurons. Finally, we used a mouse model of Rett to show that MeCP2 loss prevents homeostatic synaptic scaling up in response to visual deprivation in vivo, demonstrating for the first time that MeCP2 loss disrupts homeostatic plasticity within the intact developing neocortex. Our results establish MeCP2 as a critical mediator of synaptic scaling and raise the possibility that some of the neurological defects of Rett arise from a disruption of homeostatic plasticity.
Introduction
The neurodevelopmental disorder Rett syndrome (Rett) is the leading genetic cause of mental retardation in females. Most cases of Rett are caused by loss-of-function mutations in the X-linked gene coding for methyl-CpG binding protein 2 (MeCP2) and are neurologically characterized by epileptic seizures and mental retardation. Mutations in MeCP2 have also been identified in other neurodevelopmental disorders, including autism and Angelman syndrome (Chahrour and Zoghbi, 2007; Guy et al., 2011; Banerjee et al., 2012). The severe neurological symptoms of Rett suggest that transcriptional regulation by MeCP2 is required for proper neuronal function, but despite much effort, it remains unclear how a loss of MeCP2 generates the neurological deficits of Rett. Here we show that MeCP2 plays an essential role in homeostatic synaptic scaling up in neocortical neurons in response to reduced firing or reduced sensory drive.
Homeostatic plasticity mechanisms are believed to be necessary for proper development and function of neuronal networks (Turrigiano and Nelson, 2004). The best-characterized form of homeostatic plasticity is synaptic scaling expressed at glutamatergic synapses (Turrigiano, 2008; Pozo and Goda, 2010). Synaptic scaling bidirectionally regulates synaptic strengths in response to chronic changes in network activity, in the correct direction to stabilize firing rates. Although these homeostatic changes are transcription dependent, the relevant transcriptional regulators and their targets are not known (Turrigiano, 2011). Previously, we found an imbalance of synaptic excitation and inhibition within neocortical circuits in a mouse model of Rett, without significant defects in Hebbian plasticity (Dani et al., 2005; Dani and Nelson, 2009). This excitation/inhibition balance is thought to be maintained by homeostatic mechanisms, raising the interesting possibility that homeostatic plasticity might be disturbed in Rett. Consistent with this, a recent study found that MeCP2 is required for synaptic scaling down induced by elevated network activity in dissociated hippocampal cultures (Qiu et al., 2012). The molecular pathways mediating scaling up and down are, in many cases, distinct (Rutherford et al., 1998; Shepherd et al., 2006; Peng et al., 2009; Anggono et al., 2011; Sun and Turrigiano, 2011), so here we investigated whether MeCP2 is also necessary for synaptic scaling up, both in vitro and in the intact neocortex.
Here we show that sparse and acute knockdown (KD) of MeCP2 blocks synaptic scaling up of miniature EPSCs (mEPSCs), and the associated increase in synaptic AMPAR accumulation, within targeted neurons. Furthermore, MeCP2 knockdown decreased excitatory synapse number without affecting basal mEPSC amplitude or AMPAR accumulation at spared synapses. These data demonstrate that MeCP2 acts in a cell-autonomous way to maintain both excitatory synapse number and synaptic scaling in individual neocortical neurons. Finally, we used a mouse model of Rett to show that MeCP2 loss prevents homeostatic synaptic scaling up in response to visual deprivation in vivo, demonstrating for the first time that MeCP2 loss disrupts homeostatic plasticity within the intact developing neocortex. Our results establish MeCP2 as a critical mediator of synaptic scaling and raise the possibility that some of the neurological defects of Rett arise from a disruption of homeostatic plasticity.
Materials and Methods
All experimental procedures were approved by the Animal Care and Use Committee of Brandeis University and followed the guidelines of the National Institutes of Health.
Neuronal cultures and transfection.
Neuronal dissociated cultures were prepared from primary visual cortex of postnatal day 0 (P0) to P2 Long–Evans rats of either sex and plated on a bed of confluent astrocytes, as described previously (Pratt et al., 2003). Cultures were used for imaging or electrophysiology after 7–9 d in vitro. Electrophysiology and immunocytochemistry were performed on morphologically identified pyramidal neurons, as described previously (Pratt et al., 2003). All experimental conditions were compared with age-matched sister cultures. All transfections were done using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions; cultures were transfected 3 d before the experimental procedure. Two short hairpin RNAs (shRNAs) directed against two different sequences of MeCP2 were used. The targeted sequences were as follows: hp1, 5′CTAAAGTAG-3′ [described by Jin et al. (2008)]; hp2, 5′GTCAGAAGACCAGGATCTC-3′ [described by Zhou et al. (2006) and Wood et al. (2009)]. Empty vectors (ev) were identical to the hp vectors except for absence of the shRNA insert. Chronic activity blockade was accomplished by adding 20 μm DNQX or 5 μm TTX, as indicated in the text, to the culture dishes 1 d before the experiment (and 2 d after transfection).
Immunostaining.
To quantify nuclear MeCP2 levels, immunostaining was performed using the following antibodies: NeuN (1:500, MAB377; Millipore) and MeCP2 (1:800, 07-013; Millipore). To visualize surface AMPAR, immunostaining was performed under nonpermeant conditions using antibodies against GluR1 (1:10, PC246; Calbiochem) or GluR2 (1: 200, MAB397; Millipore) as described previously (Wierenga et al., 2005); neurons were counterstained with the presynaptic marker VGLUT-1 (1:200, 135 304; Synaptic Systems). Alexa Fluor 568 and 647, Texas Red, and Cascade Blue (1:500; Invitrogen) were used as secondary antibodies. All images were acquired on an IX-81 microscope (Olympus) using a 60× oil-immersion objective and an Orca ER camera (Hamamatsu). Images were obtained with either Openlab or Volocity (Improvision) and analyzed with MetaMorph software (Molecular Devices) as described previously (Sun and Turrigiano, 2011).
Culture electrophysiology.
Whole-cell AMPA-mediated mEPSCs were recorded and analyzed as described previously (Turrigiano et al., 1998; Wierenga et al., 2005). mEPSCs were recorded from pyramidal neurons voltage clamped at −70 mV using an Axopatch 200B amplifier (Molecular Devices) and perfused at 25°C in the following artificial CSF (ACSF): 126 mm NaCl, 5.5 mm KCl, 2 mm MgSO4, 1 mm NaH2PO4, 25 mm NaHCO3, 2 mm CaCl2, 14 mm dextrose, 0.1 μm TTX, 50 μm APV, and 25 μm picrotoxin. The internal recording solution contained the following (in mm): 120 KMeSO4, 10 KCl, 2 MgSO4, 10 K-HEPES, 0.5 EGTA, 3 K2ATP, 0.3 Na-GTP, and 10 Na2 phosphocreatine. Recordings with Vm > −50 mV, Rs > 20 MΩ, and Rin < 125 MΩ and < 25 mEPSCs were excluded. In-house Igor Pro (Wavemetrics) software was used to detect and measure mEPSCs.
Visual deprivation and slice electrophysiology.
Mecp2 knock-out (KO) mice (Mecp2tm1.1Jae) were bred as described previously (Chen et al., 2001; Dani et al., 2005). Male wild-type (WT) and Mecp2 KO mice were raised in a normal 12 h light/dark cycle. Dark exposure (DE) was started on P20–P21 for a period of 4 d. At P24–P25, DE or control (Ctl) animals were killed, and coronal brain slices containing primary visual cortex were obtained as described previously (Dani et al., 2005). Whole-cell recordings were obtained from layer 2/3 pyramidal neurons of monocular visual cortex using a Multiclamp 700B amplifier (Molecular Devices), and AMPA-mediated mEPSCs were recorded as described previously (Desai et al., 2002; Maffei and Turrigiano, 2008). Pyramidal neurons were identified using differential interference contrast/IR optics and morphology verified by post hoc reconstitution of biocytin fills as described previously(Desai et al., 2002). The internal recording solution contained the following: 100 mm K-gluconate, 20 mm KCl, 10 mm K-HEPES, 0.3 mm Na-GTP, 4 mm Mg-ATP, 10 mm Na-phosphocreatine, and 0.1% biocytin. Neurons were voltage clamped to −70 mV in ACSF containing the following: 126 mm NaCl, 3 mm KCl, 2 mm MgSO4, 1 mm NaH2PO4, 26 mm NaHCO3, 2 mm CaCl2, 20 mm dextrose, 1 μm TTX, 50 μm APV, and 20 μm picrotoxin. Recordings were excluded if the Vm > −60 mV, Rs > 25 MΩ, and Rin < 50 MΩ, or if any of these properties changed by >10% during the course of the recording.
Scaled cumulative distributions.
Cumulative mEPSC amplitude distributions were constructed by including the first 25 mEPSCs (culture) and 100 mEPSCs (slice) recorded from each neuron for each condition. Scaling was analyzed as described previously (Turrigiano et al., 1998); briefly, the rank-ordered control and experimental distributions were plotted against each other, the best linear fit obtained, and the experimental distribution transformed by this equation. A Kolmogorov–Smirnov (KS) test was used to determine if the control and scaled experimental distributions were different. For DNQX versus control, slope was 1.64 and intercept (−2.6) was close to that needed to compensate for the 5 pA cutoff (−3.1).
Statistics.
All data are expressed as the mean ± SEM for the number of neurons indicated; n is the number of neurons unless otherwise noted in the text. Statistical comparisons were made using the unpaired Student's t test; for multiple comparisons, single-factor ANOVAs followed by corrected t tests were performed. For comparisons of cumulative distributions, a KS test was used. A p value of <0.05 was considered statistically significant.
Results
It remains unclear to what extent the synaptic defects that contribute to Rett are attributable to cell-autonomous loss within particular cell types, or global and prolonged loss throughout development. To determine whether MeCP2 is necessary for cell-autonomous synaptic scaling up in neocortical pyramidal neurons, we began by taking an RNAi approach that allowed us to acutely and sparsely decrease MeCP2 levels within individual pyramidal neurons in vitro and measure the impact on basal synaptic properties and synaptic scaling.
MeCP2 knockdown does not affect basal quantal amplitude
To investigate whether MeCP2 plays a cell-autonomous role in synaptic scaling up, cultured neocortical neurons were transiently transfected with an shRNA construct targeted against MeCP2 (Jin et al., 2008); vectors also expressed soluble EGFP to allow visualization of transfected neurons. This allowed us to reduce MeCP2 protein levels in a small number of neurons (5–15 per culture dish) in an otherwise unaffected network. To characterize the efficiency of KD, cultures transfected with either the hairpin (hp1) or the ev were immunolabeled with antibodies against MeCP2 and the pan neuronal marker NeuN (Fig. 1A). Quantification of the fluorescence intensity of the MeCP2 signal in the nucleus showed that expression of hp1 for 3 d reduced endogenous MeCP2 protein levels to ∼40% of untransfected control neurons. Expression of the empty vector had no significant effect on MeCP2 levels (Fig. 1B). Here and below, all experiments were performed on neurons with a pyramidal morphology (Fig. 1A) as described previously (Rutherford et al., 1998; Watt et al., 2000; Pratt et al., 2003).

MeCP2 KD does not affect basal quantal amplitude. A, Pyramidal neurons transfected for 3 d with ev or MeCP2 shRNA1 (hp1) and untransfected control neurons (unt), fixed, and stained with antibodies against endogenous MeCP2 (red) and NeuN (orange). The ev and hp1 constructs also express soluble EGFP (green). White arrowheads show the position of the same neuron's cell body. Yellow arrowheads indicate a nearby untransfected neuron. Scale bar, 20 μm. B, Quantification of the nuclear MeCP2 fluorescence intensity for unt and ev- and hp1-expressing neurons (n = 164, 48, and 39, respectively). A.U., Arbitrary units. C, Left, Example raw traces of mEPSCs recorded from ev- and hp1-expressing neurons. Right, Average mEPSC waveforms for the same conditions. D, Cumulative histograms of mEPSC amplitudes for ev- and hp1-expressing neurons. Inset, Average mEPSC amplitude for ev-expressing (n = 22) and hp1-expressing (n = 20) neurons. E, Peak scaled average mEPSC waveform for the same conditions. **p < 0.001, different from control.
Previous studies have shown that MeCP2 KO reduces excitatory synaptic transmission in L5 neocortical pyramidal neurons, both through a small reduction in mEPSC amplitude and through a reduction in connectivity (Dani et al., 2005; Dani and Nelson, 2009). To determine whether an acute and cell-autonomous loss of MeCP2 is able to reduce baseline mEPSC amplitude, we recorded AMPA-mediated mEPSCs from neurons expressing either hp1 or the ev for 3 d. MeCP2 knockdown had no significant effect on mEPSC amplitude (Fig. 1C,D). mEPSC frequency was variable, and although there was a trend toward reduction, this was not significant (ev, 0.63 ± 0.10 Hz; hp1, 0.58 ± 0.08 Hz; p = 0.67). In addition, there were no significant changes in passive neuronal properties, and mEPSC kinetics were also unaffected, as can be seen from a comparison of the peak-scaled average mEPSC waveforms (Fig. 1E). These data indicate that an acute and cell-autonomous reduction of MeCP2, unlike a chronic and global loss, does not reduce baseline mEPSC amplitude.
MeCP2 knockdown reduces excitatory synapse number without affecting receptor accumulation at spared synapses
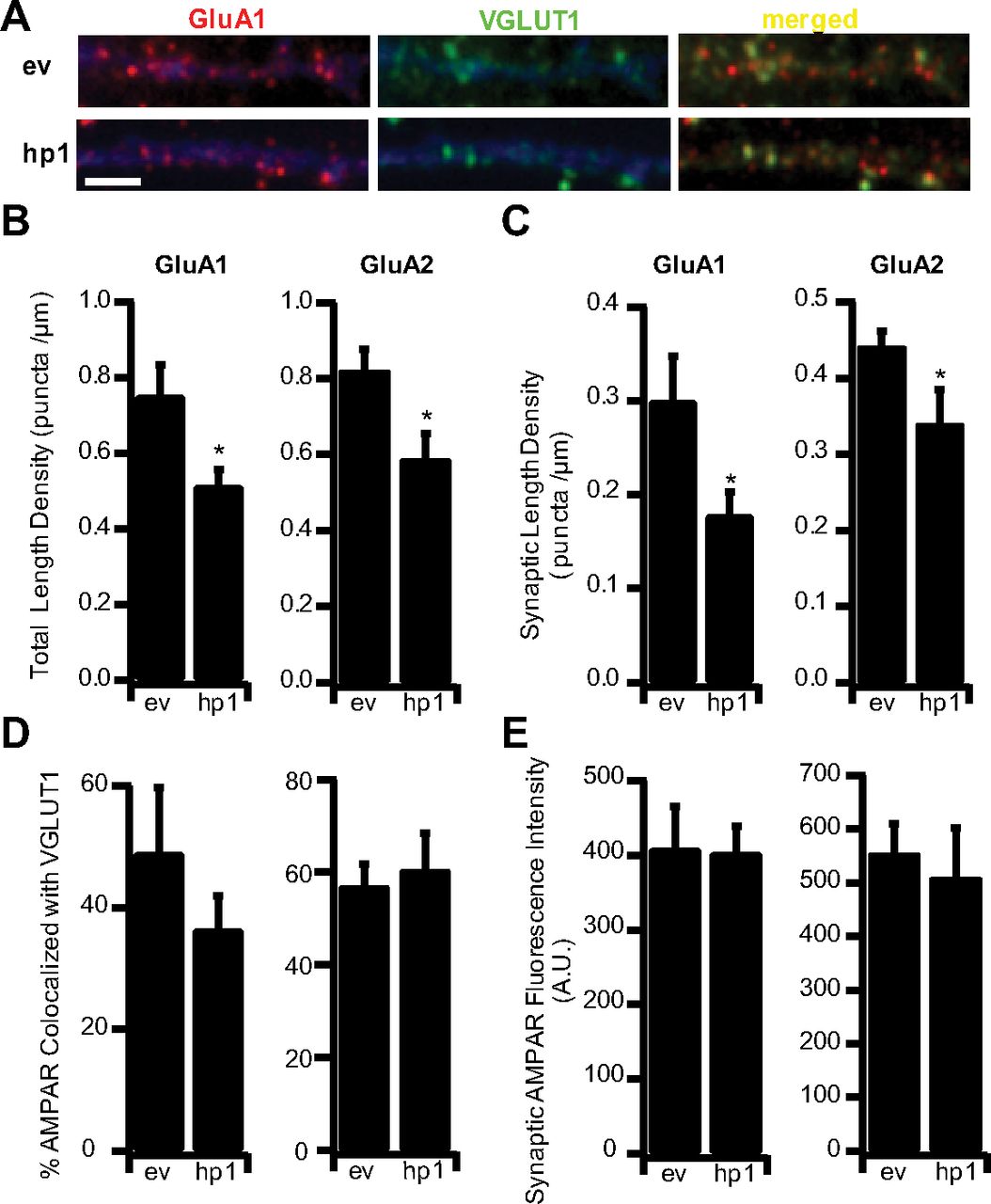
Previous studies have shown that MeCP2 KO reduces the number of glutamatergic synapses (Nelson et al., 2006; Chao et al., 2007; Tropea et al., 2009). However, it was not clear whether this reduction in synapse number was a direct result of postsynaptic MeCP2 loss, presynaptic and postsynaptic loss together (Chao et al., 2007), or changes in network function caused by global MeCP2 KO (Tropea et al., 2009). To investigate this, we examined the effects of acute and sparse MeCP2 KD on excitatory synapse number. Three days after transfection, cultures were surface labeled with an antibody against the GluA1 subunit of the AMPA receptor and against VGLUT1 (to label presynaptic excitatory terminals), as described previously (Wierenga et al., 2005); sites where postsynaptic GluA1 puncta were apposed to or colocalized with VGLUT1 puncta were considered putative synapses (Fig. 2A). MeCP2 KD significantly reduced both total GluA1 puncta and the number of colocalized GluA1/VGLUT1 puncta by ∼30% (Fig. 2B,C), without significantly affecting the colocalization rate between GluA1 and VGLUT1 (Fig. 2D). In keeping with the observation that MeCP2 KD did not affect mEPSC amplitude, there was no change in the fluorescence intensity of the GluA1 signal at synaptic sites, indicating that GluA1 accumulation at spared synapses is unaffected by MeCP2 loss (Fig. 2E). Similar results were obtained when GluA2 was used as the postsynaptic marker (Fig. 2B–E). Interestingly, although MeCP2 has been implicated in the transcriptional regulation of GluA2 (Qiu et al., 2012), MeCP2 KD had no impact on baseline synaptic GluA2 levels. Together, these data indicate that MeCP2 plays a critical and cell-autonomous role in the maintenance of excitatory synapse density, but not baseline quantal amplitude, at spared synapses.

MeCP2 KD reduces excitatory synapse number. A, Example dendrites from ev- or hp1-expressing neurons, stained against endogenous surface GluA1 (red) and the excitatory synapse marker VGLUT-1 (green); EGFP is shown in pseudo color blue. Scale bar, 2 μm. B, Length density for total GluA1 and GluA2 puncta in ev- and hp1-expressing cells: ev (n = 5) and hp1 (n = 7) for GluA1; ev (n = 12) and hp1 (n = 6) for GluA2. C, Synaptic length density for GluA1 and GluA2 puncta in ev- and hp1-expressing cells. D, Colocalization of GluA1 and GluA2 with VGLUT-1 in ev- and hp1-expressing cells. E, Synaptic GluA1 and GluA2 surface receptor fluorescent intensity in ev- and hp1-expressing cells. *p < 0.05, different from ev.
MeCP2 knockdown blocks or attenuates synaptic scaling up
Neocortical neurons compensate for changes in synapse number or sensory drive by homeostatically regulating synaptic strength (Desai et al., 2002; Maffei et al., 2006; Goel and Lee, 2007; Maffei and Turrigiano, 2008), yet the reduction in synapse number induced by MeCP2 KD did not result in a compensatory increase in mEPSC amplitude, either in vitro (Fig. 1C,D) or in vivo (Dani et al., 2005). This raised the possibility that synaptic scaling up is impaired after MeCP2 KD. To determine whether acute loss of MeCP2 impairs scaling up, transfected cultures were treated with the AMPAR antagonist DNQX for 24 h to block activity, and AMPA mEPSCs were recorded from ev- and hp1-expressing neurons. As described previously (Turrigiano et al., 1998; Thiagarajan et al., 2005; Sun and Turrigiano, 2011), DNQX treatment significantly increased mEPS amplitude in ev-expressing neurons and scaled up the entire distribution of mEPSC amplitudes (Fig. 3A,B; DNQX scaled not different from control; KS test, 0.89). In marked contrast, DNQX had no impact on mEPSC amplitude in neurons expressing hp1 (Fig. 3A,C). TTX treatment similarly increased mEPSC amplitude in untransfected neurons (TTX, 119% of ctl; p = 0.008) but not hp1-expressing neurons (TTX, 105% of ctl; p = 0.51).
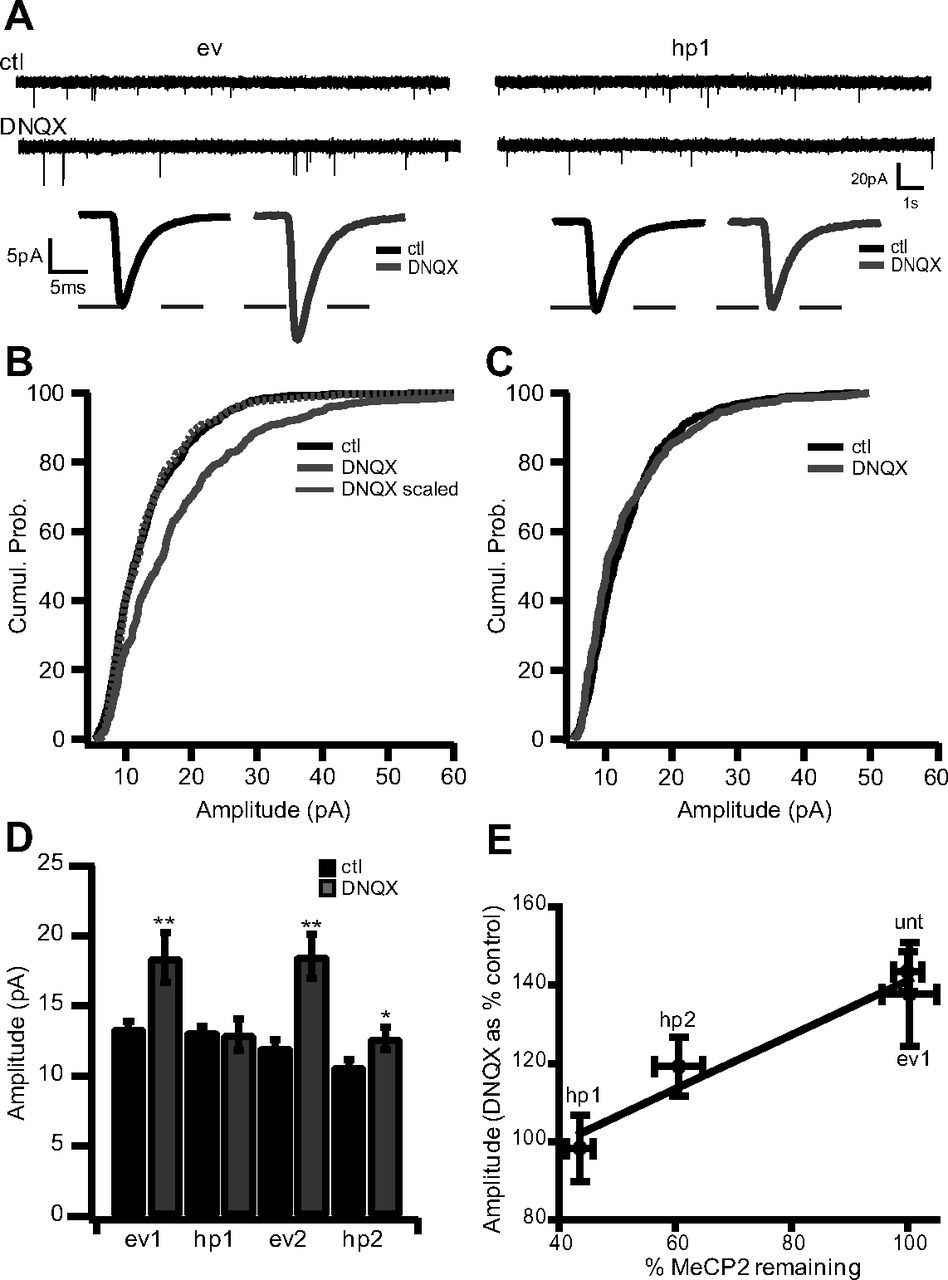
MeCP2 KD blocks or attenuates synaptic scaling up. A, Top, Example raw traces of mEPSCs recorded from ev-expressing (left) and hp1-expressing (right) neurons in untreated control (ctl; top) and DNQX-treated (bottom) conditions. Bottom, Average mEPSC waveform for each condition. B, Cumulative histograms of mEPSC amplitudes from ev-expressing neurons in ctl and DNQX-treated conditions. C, Cumulative histograms of mEPSC amplitudes from hp1-expressing neurons in ctl and DNQX-treated conditions. D, Average amplitude for ctl and DNQX-treated cultures for neurons expressing ev1 (n = 22 and 10, respectively), hp1 (n = 20 and 10, respectively), ev2 (n = 13 and 6, respectively), and hp2 (n = 15 and 7, respectively). E, Plot comparing the percentage change in mEPSC amplitude during scaling (DNQX as a percentage of control) versus the percentage of MeCP2 remaining after KD for unt, ev1, hp1, and hp2 neurons. *p < 0.05, different from ctl; **p < 0.005, different from ctl.
To control for potential off-target effects of hp1, we repeated these experiments with a second hairpin (hp2) targeted against a different unique sequence of MeCP2 (Zhou et al., 2006; Wood et al., 2009). Hp2 greatly attenuated (but did not completely block) the normal increase in mEPSC amplitude induced by DNQX (Fig. 3D). To determine whether the impact on scaling was related to the efficacy of the two hps at knocking down MeCP2, we compared the increase in mEPSC amplitude induced by DNQX (DNQX as a percentage of control) with the fraction of MeCP2 protein remaining after KD. The hp2 reduced MeCP2 levels significantly less (to 60% of control) than hp1 (to 40% of control), and there was an approximately linear relationship between the degree of synaptic scaling and the fraction of MeCP2 remaining (Fig. 3E). These data establish an important role for MeCP2 in synaptic scaling up and suggest that there is a graded relationship between the amount of scaling and the levels of MeCP2 in the nucleus. On the other hand, our data also suggest there is a threshold level of MeCP2 below which synaptic scaling is completely blocked. These data support the emerging view that MeCP2 levels must be tightly controlled to allow for normal neural circuit function (Chahrour and Zoghbi, 2007).
Previous studies have established that synaptic scaling is associated with an increase in AMPAR accumulation at synapses (Lissin et al., 1998; O'Brien et al., 1998; Turrigiano et al., 1998; Wierenga et al., 2005, 2006; Ibata et al., 2008). Therefore, as a second means of assessing synaptic scaling, we compared the fluorescence intensity of the synaptic GluA1 signal in control and DNQX-treated transfected cultures (Fig. 4A). Surface synaptic GluA1 was quantified as described previously, using an antibody directed against an extracellular epitope of GluA1 under nonpermeant conditions (Wierenga et al., 2005). DNQX treatment causes a significant increase in the fluorescence intensity of synaptic GluA1 in ev-expressing neurons, but not in neurons expressing hp1 (Fig. 4A,B). DNQX treatment had no significant effect on the percentage of GluA1 puncta colocalized with VGLUT1, or on the synaptic length density in either ev- or hp1-expressing neurons (Fig. 4C,D). These data indicate that although MeCP2 knockdown does not affect baseline AMPAR accumulation (Fig. 2E), it prevents the regulated enhancement of synaptic AMPAR that underlies scaling up.
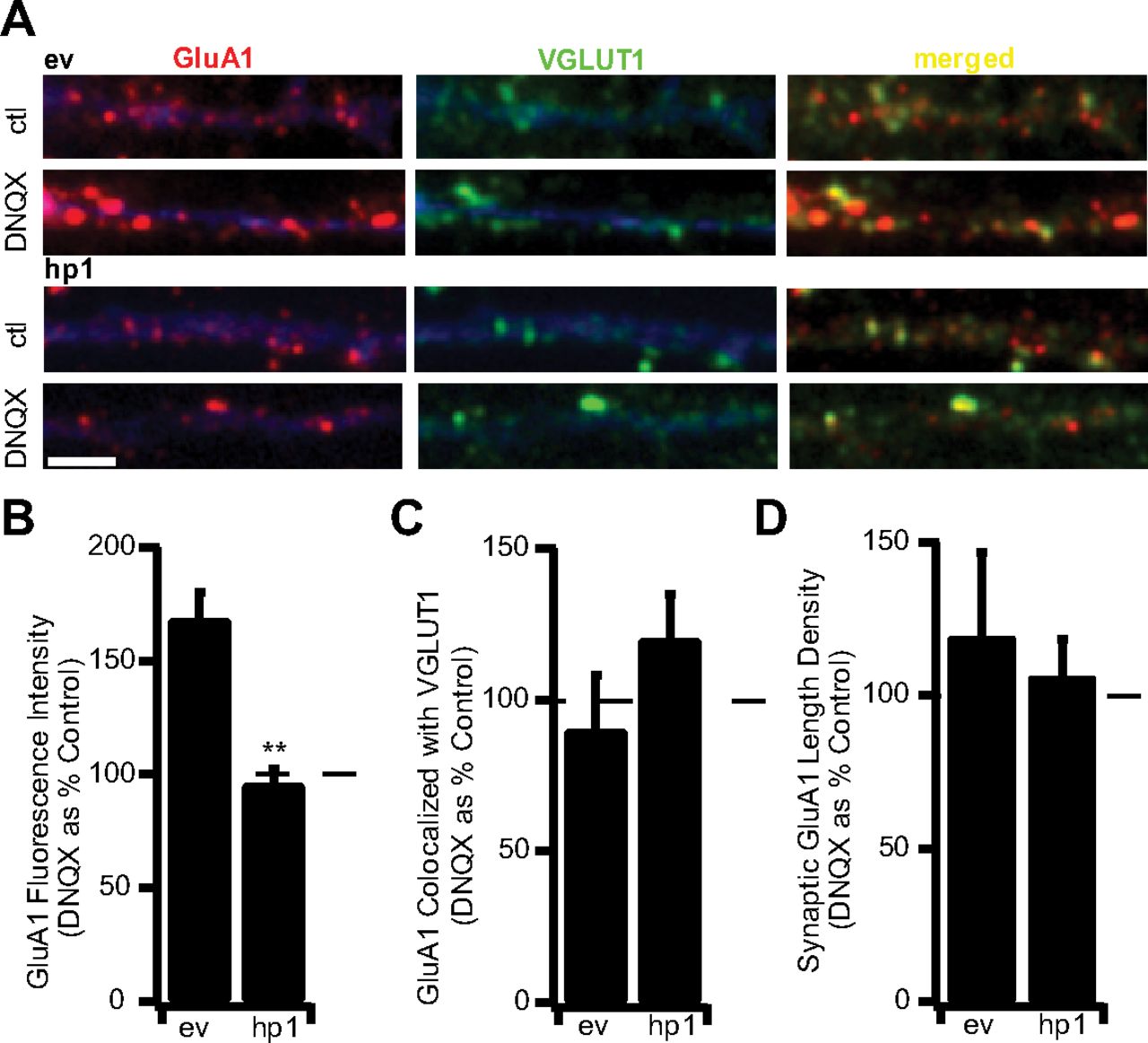
MeCP2 KD prevents the inactivity-induced accumulation of AMPAR at synapses. A, Examples of staining for endogenous GluA1 (red) and VGLUT-1 (green) for ev-expressing (top) and hp1-expressing (bottom) neurons treated with DNQX for 24 h. Scale bar, 2 μm. B, Fluorescence intensity of synaptic GluA1 puncta for ev- and hp1-expressing neurons treated with DNQX. Here and below, values are from DNQX-treated neurons expressed as a percentage of untreated control neurons. ev, n = 5 and 8, respectively; hp1, n = 7 and 9, respectively. C, Colocalization of GluA1 with VGLUT-1 for ev- and hp1-expressing neurons. D, Length density for synaptic GluA1 for ev- and hp1-expressing neurons. **p < 0.005, different from ev.
MeCP2 knock-out blocks homeostatic synaptic scaling in vivo
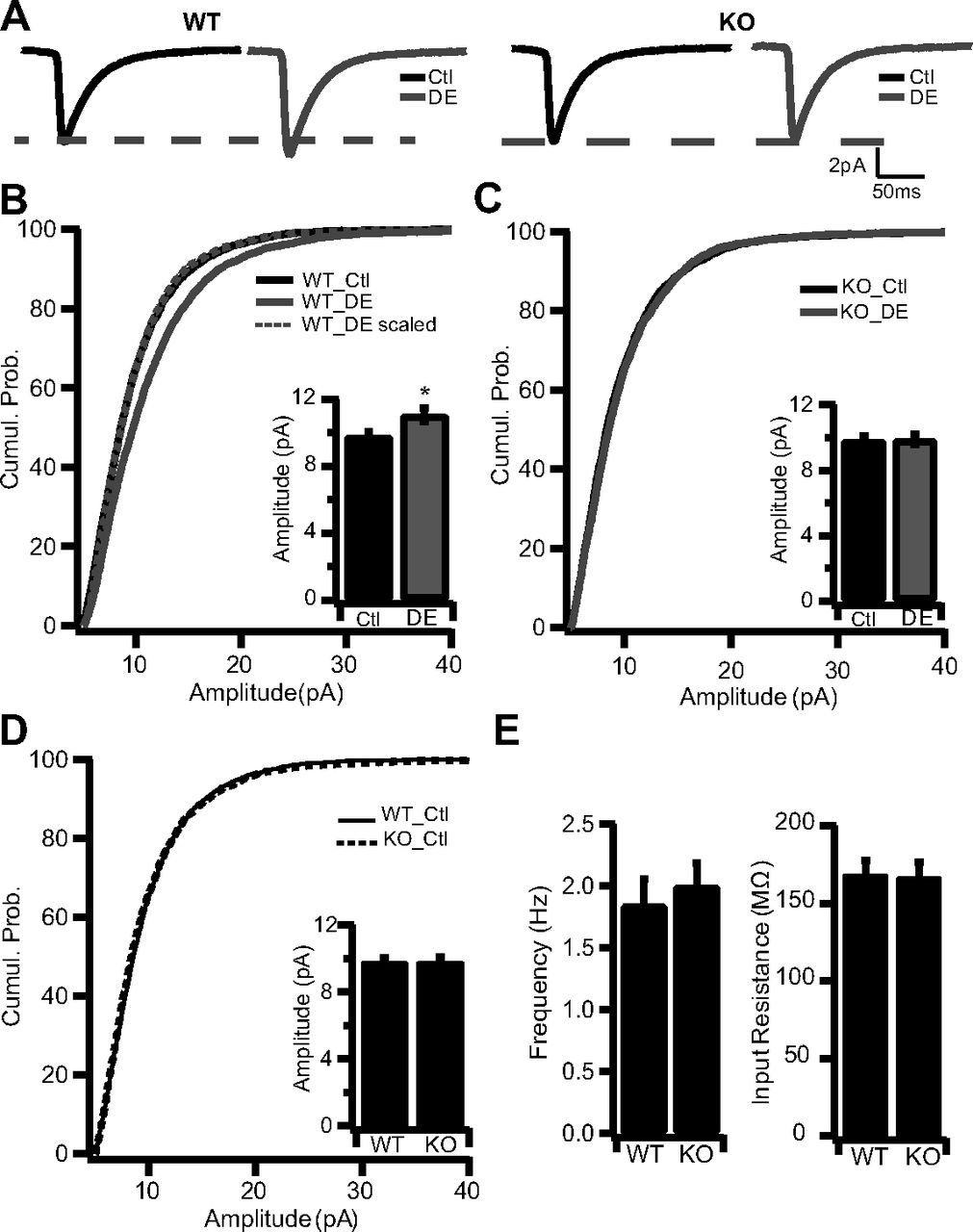
Synaptic scaling up can be induced in the intact visual cortex in response to visual deprivation, suggesting that synaptic scaling plays an important role in experience-dependent neocortical development (Desai et al., 2002; Maffei et al., 2006; Goel and Lee, 2007; Maffei and Turrigiano, 2008). In particular, placing animals in the dark for several days (DE) during the classical visual system critical period scales up mEPSC amplitude onto layer 2/3 pyramidal neurons (Goel and Lee, 2007). To determine whether MeCP2 is required for synaptic scaling in vivo, WT or Mecp2 KO mice were visually deprived using 4 d of DE starting at P20/P21. We chose to use this KO mouse model, rather than KD in vivo, to test whether synaptic scaling is absent in this widely used mouse model of Rett. Acute cortical slices from primary visual cortex were then obtained from DE and normally reared animals, and AMPA mEPSCs were recorded from layer 2/3 pyramidal neurons as described previously (Desai et al., 2002; Maffei and Turrigiano, 2008). Mecp2 KO mice were compared with age-matched, normally reared control mice. As described previously (Goel and Lee, 2007), 4 d of DE caused a significant increase in mEPSC amplitude in WT mice (Fig. 5A,B). In contrast, in KO mice DE had no impact on mEPSC amplitude (Fig. 5A,C). As for synaptic scaling in culture, DE scaled up the entire mEPSC amplitude distribution in WT mice (Fig. 5B; WT_DE scaled not different from WT_ctl; KS test, 0.52), as expected (Desai et al., 2002; Goel and Lee, 2007). This increase in mEPSC amplitude was not associated with significant changes in mEPSC frequency (WT_Ctl, 1.84 ± 0.22; WT_DE, 2.30 ± 0.23; KO_Ctl, 1.20 ± 0.19; KO_DE, 1.79 ± 0.21) or kinetics. Furthermore, there was no difference in baseline mEPSC amplitude, frequency, or passive neuronal properties between WT and KO mice (Fig. 5D,E). These data demonstrate that MeCP2 is essential for visual deprivation-induced synaptic scaling in vivo, suggesting that MeCP2 likely plays a role in experience-dependent visual cortical plasticity.

MeCP2 knock-out blocks homeostatic synaptic scaling in vivo. A, Average waveform for mEPSCs recorded from WT (left) and KO (right) neurons from DE and normal 12 h light/dark cycle Ctl mice: WT_Ctl (n = 5 animals), WT_DE (n = 4 animals), KO_Ctl (n = 2 animals), and KO_DE (n = 2 animals). B, Cumulative histogram of mEPSC amplitudes recorded from Ctl (n = 23) and DE (n = 32) WT mice. Inset, Average mEPSC amplitudes for the same conditions. C, Cumulative histogram of mEPSC amplitudes recorded from Ctl (n = 24) and DE (n = 35) KO mice. Inset, Average mEPSC amplitudes for the same conditions. D, Cumulative histogram of mEPSC amplitudes for WT and KO neurons from Ctl mice. Inset, Average mEPSC amplitude for the same conditions. E, Average frequency (left) and input resistance (right) for WT and KO neurons from ctl mice. *p < 0.05, different from WT_Ctl. Cumul. Prob., Cumulative probability.
Discussion
Although Rett arises from loss of function of a single gene, MeCP2, the nature of the synaptic defects underlying the neurological symptoms of Rett remains unclear. Here we investigated the role of MeCP2 in synaptic scaling, a form of homeostatic synaptic plasticity widely believed to contribute to the maintenance of the proper excitation–inhibition balance within central neural circuits. We find that acute and cell-autonomous loss of MeCP2 in neocortical pyramidal neurons is sufficient to prevent scaling up of synaptic strengths in response to activity blockade. Furthermore, we show that MeCP2 is required for synaptic scaling in the intact neocortex in response to sensory deprivation. These data demonstrate that the correct expression level of MeCP2 within individual neurons is essential for the expression of homeostatic synaptic scaling and suggest that circuit defects arising from loss of scaling contribute to the pathogenesis of Rett.
A hotly debated question in the field is when and where MeCP2 is needed for proper brain function. MeCP2 levels increase postnatally (Shahbazian et al., 2002; Balmer et al., 2003; Cohen et al., 2003; Kishi and Macklis, 2004; Mullaney et al., 2004; Skene et al., 2010), prenatal and early postnatal development is essentially normal in Rett and in mouse models of Rett (Chen et al., 2001; Guy et al., 2001; Schüle et al., 2008), and loss of MeCP2 in adult mice generates neurological symptoms (McGraw et al., 2011), suggesting that MeCP2 plays an important role in postnatal and mature brain function. Our data provide strong support for the idea that MeCP2 is essential for the ongoing maintenance of synaptic function and plasticity. First, we found that brief (3 d) postnatal knockdown of MeCP2 was sufficient to induce a 30% reduction in excitatory synapse density, an effect comparable in magnitude to that observed for knock-out throughout development (Nelson et al., 2006; Chao et al., 2007). Thus, in neocortical pyramidal neurons, normal MeCP2 expression is critical for the ongoing maintenance of excitatory synapses. This likely accounts for the consistent observation that excitatory connectivity within neocortical circuits is reduced after loss of MeCP2 function (Dani et al., 2005; Dani and Nelson, 2009; Wood et al., 2009; Stuss et al., 2012). Second, we found that an acute reduction of MeCP2 was sufficient to block or attenuate synaptic scaling up in response to activity blockade. A recent study showed that more prolonged (9 d) knockdown of MeCP2 in postnatal hippocampal neurons was able to block the reduction in mEPSC amplitude induced by elevated activity (Qiu et al., 2012); together with this previous study, our data demonstrate that MeCP2 plays a critical role in both directions of synaptic scaling. Postnatal expression of MeCP2 is thus critical for several important aspects of central excitatory synaptic function, including synapse maintenance and bidirectional homeostatic synaptic plasticity. Whether these two aspects of MeCP2 function are mechanistically linked or operate through separate regulatory pathways remains unclear.
Interestingly, we found that basal quantal amplitude and AMPA receptor abundance at spared synapses were normal after MeCP2 loss, both in dissociated cultures after MeCP2 knockdown and in L2/3 pyramidal neurons in acute slices of primary visual cortex of MeCP2 knock-out mice. These data are consistent with several studies demonstrating no effect of postnatal MeCP2 loss on mEPSC amplitude in neocortical and hippocampal neurons (Nelson et al., 2006; Chao et al., 2007; Qiu et al., 2012). However, our slice data are in contrast to a previous study using the same mouse model where acute slice recordings from L5 pyramidal neurons in somatosensory cortex demonstrated a small reduction in mEPSC amplitude (Dani et al., 2005). This could be attributable to cell-type-specific differences (L2/3 vs L5 pyramidal neurons), brain region differences (visual vs somatosensory cortex), or differences in age, since we recorded at P24–P25 rather than P28–P35, as described previously (Dani et al., 2005). Whereas prolonged loss of MeCP2 may eventually impair the maintenance of baseline postsynaptic properties, our data strongly suggest that acute loss of MeCP2 does not impair baseline AMPAR accumulation but specifically prevents activity-dependent homeostatic adjustments in quantal amplitude. This, in turn, suggests that other processes independent of MeCP2 function help to set baseline quantal amplitude, but what these processes might be are currently unknown.
MeCP2 is widely expressed throughout the CNS and is X-linked, so due to X chromosome inactivation, females with Rett are mosaic for MeCP2 expression. Furthermore, the severity of the disease is thought to correlate with the fraction of cells expressing the mutated gene (Hoffbuhr et al., 2002; Chahrour and Zoghbi, 2007; Guy et al., 2011). In contrast, most mouse models of Rett involve global loss of function in male mice, and as a consequence, the relative contribution of cell-autonomous and non-cell-autonomous effects of MeCP2 loss are poorly understood. It has recently become clear that MeCP2 expression in glia is significant and that loss of this expression can itself induce non-cell-autonomous defects in neuronal morphology and synaptogenesis (Ballas et al., 2009); furthermore, specific loss of MeCP2 in GABAergic interneurons recapitulates some aspects of global loss (Chao et al., 2010). These studies make it clear that understanding the cell-type-specific functions of MeCP2, and the degree to which loss of MeCP2 induces cell-autonomous defects, is critical for understanding the consequences of mosaic MeCP2 loss in Rett. Here we used sparse knockdown in cultured neocortical pyramidal neurons to show that acute MeCP2 loss regulates synaptic scaling up in a cell-autonomous manner, demonstrating for the first time that the normal expression level of MeCP2 within individual postnatal pyramidal neurons is essential for the expression of synaptic plasticity. To determine whether synaptic scaling is also blocked by global and prolonged MeCP2 KO, we used a common mouse model of Rett (Chen et al., 2001; Dani et al., 2005) and a standard in vivo model of synaptic scaling (Desai et al., 2002; Goel and Lee, 2007) to show that synaptic scaling up in response to sensory deprivation is also blocked in the intact MeCP2-lacking neocortex. These data indicate that neurons are unable to compensate for prolonged developmental loss of MeCP2 to regain the ability to scale synaptic strength and suggest that loss of synaptic scaling is a major synaptic defect contributing to Rett syndrome. Loss of MeCP2 has recently been reported to disrupt experience-dependent changes at the retinogeniculate synapse (Noutel et al., 2011), supporting the idea that MeCP2 is critical for several aspects of normal experience-dependent plasticity in the visual system.
It is currently unclear how loss of MeCP2 prevents neurons from expressing synaptic scaling. Synaptic scaling up and down are both transcription dependent (Ibata et al., 2008; Goold and Nicoll, 2010), suggesting that MeCP2 may disrupt scaling by disrupting the regulation of transcription. What the targets of MeCP2 might be, however, is unclear. Synaptic scaling up and down are both critically dependent on trafficking steps involving the GluA2 subunit of the AMPAR (Ibata et al., 2008; Goold and Nicoll, 2010), and it was recently shown that elevated activity enhanced MeCP2 expression and suppressed the expression of GluA2 (Qiu et al., 2012), but because directly reducing GluA2 through RNAi does not impact mEPSC amplitude (Gainey et al., 2009), it is unlikely that this activity-dependent reduction in GluA2 expression is sufficient to drive scaling down. Along the same lines, MeCP2 loss had no significant impact on baseline GluA2 expression (Qiu et al., 2012) or on GluA2 abundance at spared synapses (Fig. 2E), strongly suggesting that block of scaling up is not a simple result of loss of GluA2. Disruption of MeCP2 has recently been shown to disregulate the induction of the immediate-early gene Arc (Su et al., 2012), which by influencing AMPAR internalization can occlude synaptic scaling (Rial Verde et al., 2006; Shepherd et al., 2006), suggesting another possible route by which MeCP2 disruption could attenuate synaptic scaling. Alternatively, rather than regulating the transcription of specific scaling-dependent genes, MeCP2 could play a more general role in chromatin remodeling during activity-dependent changes in transcription (Chahrour and Zoghbi, 2007; Skene et al., 2010; Cohen et al., 2011; Guy et al., 2011; Banerjee et al., 2012).
In summary, we demonstrate that MeCP2 acts in a cell-autonomous and acute manner to maintain both excitatory synapse number and synaptic scaling in individual neocortical pyramidal neurons. Furthermore, we show that MeCP2 is critical for the expression of synaptic scaling during postnatal development in vivo. Our results establish MeCP2 as a critical mediator of bidirectional synaptic scaling and raise the possibility that a disruption of homeostatic plasticity contributes to the imbalance in excitation and inhibition within neocortical circuits induced by MeCP2 loss.
Footnotes
This work was supported by National Institutes of Health Grant NS36853 (G.G.T.) and the International Rett Syndrome Foundation (G.G.T.). M.P.B. was supported by a fellowship from the American Psychological Association Diversity Program in Neuroscience and by the American Physiological Society Porter Physiology Development Fellowship. We thank Lirong Wang and Bryan Baxter for technical assistance and Xinhua Bao and Michael E. Greenberg for kindly providing DNA constructs.
- Correspondence should be addressed to Gina G. Turrigiano, Department of Biology and Center for Behavioral Genomics, Brandeis University, Waltham, MA 02454. turrigiano{at}brandeis.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}