

Abstract
Activation of the extracellular signal-regulated kinase 1 (ERK1) and ERK2 by neurotrophins, neuronal activity, or cAMP has been strongly implicated in differentiation, survival, and adaptive responses of neurons during development and in the adult brain. Recently, a new member of the mitogen-activated protein (MAP) kinase family, ERK5, was discovered. Like ERK1 and ERK2, ERK5 is expressed in neurons, and ERK5 stimulation by epidermal growth factor is blocked by the MAP kinase/ERK kinase 1 (MEK1) inhibitors PD98059 and U0126. This suggests the interesting possibility that some of the functions attributed to ERK1/2 may be mediated by ERK5. However, the regulatory properties of ERK5 in primary cultured neurons have not been reported. Here we examined the regulation of ERK5 signaling in primary cultured cortical neurons. Our data demonstrate that, similar to ERK1/2, ERK5 is activated by neurotrophins including brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and NT-4. BDNF stimulation of ERK5 required the activity of MEK5. Surprisingly, ERK5 was not stimulated by cAMP or neuronal activity induced by glutamate or membrane depolarization. In contrast to ERK1/2, ERK5 strongly activated the transcriptional activity of myocyte enhancer factor 2C (MEF2C) in pheochromocytoma 12 (PC12) cells and was required for neurotrophin stimulation of MEF2C transcription in both PC12 cells and cortical neurons. Furthermore, ERK1/2, but not ERK5, induced transcription from Elk1 and the cAMP/ Ca2+ response element in PC12 cells. Our data suggest that mechanisms for regulation of ERK5 and downstream transcriptional pathways regulated by ERK5 are distinct from those of ERK1/2 in neurons. Furthermore, ERK5 is the first MAP kinase identified whose activity is stimulated by neurotrophins but not by neuronal activity.
- signal transduction
- CNS
- cortical neurons
- neurons
- MAP kinase
- ERK1/2
- ERK5
- BMK1
- CREB
- CRE
- MEF2C
- BDNF
- glutamate
- membrane depolarization
- neuronal activity
- neurotrophin
- cAMP
Activation of extracellular signal-regulated kinase 1 (ERK1) and ERK2 is important for several neuronal functions that are regulated by neurotrophins and neuronal activity. This includes neuronal differentiation and survival during development, as well as survival and adaptive responses of mature neurons including long-term potentiation (LTP) and memory formation. For example, stimulation of the ERK1/2-signaling pathway promotes neuronal survival (Xia et al., 1995; Bonni et al., 1999;Hetman et al., 1999) and is important for LTP as well as memory formation in vertebrates (English and Sweatt, 1997; Atkins et al., 1998; Impey et al., 1998a, 1999). The Ca2+response element-binding protein (CREB)/CRE transcriptional pathway is a major regulatory target of ERK1/2 signaling and may be pivotal for plasticity and neuronal survival mediated by ERK1/2 (Montminy and Bilezikjian, 1987; Impey et al., 1998a,b; Bonni et al., 1999;Riccio et al., 1999). The transcription factor Elk1 may be another nuclear target of ERK1/2 important for neuronal plasticity (Berman et al., 1998). Elk1 is directly phosphorylated and activated by ERK1/2 and plays an important role in glutamate-induced gene expression in neurons (Gille et al., 1992; Xia et al., 1996; Sgambato et al., 1998).
ERK1/2 activity is regulated by the cAMP-signaling pathway. Although cAMP inhibits ERK1/2 in non-neuronal cells (Burgering et al., 1993;Graves et al., 1993), it activates ERK1/2 in pheochromocytoma 12 (PC12) cells and neurons (Erhardt et al., 1995; Martin et al., 1997; Vossler et al., 1997; Wei et al., 1998). cAMP is required for ERK1/2 activation of gene expression (Wei et al., 1998; Yao et al., 1998), and stimulation of ERK1/2 by cAMP and Ca2+ is critical for long-lasting LTP (English and Sweatt, 1996; Impey et al., 1998b).
ERK5 or big mitogen-activated protein (MAP) kinase 1 is the newest member of the MAP kinase family (Lee et al., 1995; Zhou et al., 1995). Upstream-signaling proteins of the ERK5 pathway include MEK5, MEKK3, and Cot (English et al., 1995; Zhou et al., 1995; Chao et al., 1999; Chiariello et al., 2000). Although ERK5 contains a TEY dual phosphorylation motif similar to that of ERK1/2, a large C terminal and a unique loop-12 sequence distinguish it from ERK1/2 and other MAP kinase family members. ERK5 is activated by serum, epidermal growth factor (EGF), nerve growth factor (NGF), and G-protein-coupled receptors and weakly by phorbol esters (Kato et al., 1997, 1998;English et al., 1998; Chao et al., 1999; Kamakura et al., 1999;Marinissen et al., 1999). ERK5 contributes to EGF-induced cell proliferation and cell cycle progression (Kato et al., 1998) as well as Ras-dependent cellular transformation (English et al., 1999).
The MEK1/2 inhibitors PD98059, SL327, and U0126 have been extensively used to implicate ERK1/2 in neuroplasticity (Impey et al., 1999) and neuronal survival (Villalba and Journot, 1997; MeyerFranke et al., 1998; Skaper et al., 1998; Anderson and Tolkovsky, 1999; Singer et al., 1999; Bi et al., 2000). Interestingly, ERK5 activation by EGF in COS7 cells is also blocked by these inhibitors (Kamakura et al., 1999), suggesting that the ERK5 pathway may also regulate cellular processes credited previously to ERK1/2. However, the regulatory properties of ERK5 and the downstream transcriptional events involved in the ERK5 signaling in neurons have not been reported. Consequently, it is crucial to define ERK5-signaling mechanisms in neurons.
In this study, we examined the regulation of ERK5 signaling in primary cultures of cortical neurons and in PC12 cells. We measured ERK5 activity by two well established methods: ERK5 autophosphorylation (Abe et al., 1997; Yan et al., 1999) and reduced electrophoretic mobility (phosphorylation shift) (Kato et al., 1997, 1998). We report that ERK5 is activated by neurotrophins but not by neuronal activity or cAMP in cortical neurons. Furthermore, ERK1/2 and ERK5 activate distinct transcription pathways in PC12 cells and cortical neurons. These data suggest that ERK5 and ERK1/2 are differentially regulated in cortical neurons.
MATERIALS AND METHODS
Materials. The following plasmids have been described: pON260 (Cherrington and Mocarski, 1989), the dominant-negative and constitutively active MEK1 (Mansour et al., 1994), the pGEX–GST–ERK5 [C-terminal 100 amino acids (aa)] (Yan et al., 1999), and CRE(α168)-luciferase (Matthews et al., 1994). The following materials were obtained from Dr. J. D. Lee at Scripps Institute, La Jolla, CA (Kato et al., 1997): the Flag-tagged wild-type ERK5 expression vector, the hemagglutinin (HA)-tagged dominant-negative and constitutively active MEK5 expression vectors, the pGEX–GST–ERK5 (M; short form), and the polyclonal anti-peptide body against the C-terminal sequence of ERK5 (EGHGMNPADIESLQREIQMDSPML). The polyclonal anti-phospho-ERK1/2 antibody (anti-ACTIVE mitogen-activated protein kinase) was purchased from Promega (Madison, WI).
Cell cultures. Primary cortical neurons were prepared from newborn Sprague Dawley rats as described (Hetman et al., 1999, 2000). Briefly, dissociated cortical neurons were plated in 60 mm culture dishes for biochemistry experiments or in 35 mm dishes for transfection experiments at a density of 4 × 106cells/60 mm dish or 2 × 106 cells/35 mm dish, respectively; cultured in basal medium Eagle (BME) supplemented with 10% heat-inactivated bovine calf serum (BCS), 35 mm glucose, 1 mml-glutamine, 100 U/ml penicillin, and 0.1 mg/ml streptomycin; and maintained in a humidified incubator with 5% CO2 at 37°C. Plates and glass coverslips were coated with poly-d-lysine and laminin. Cytosine-β-d-arabinofuranoside (Ara-C; 2.5 μm; Sigma, St. Louis, MO) was added to cultures on the second day after seeding (DIV2) to inhibit the proliferation of non-neuronal cells. Previous studies demonstrated that >90% of the cells in this culture preparation are neurons (Hetman et al., 1999). Cortical neurons were cultured for 6 d (DIV6) before drug treatment. PC12 cells were maintained in DMEM (Life Technologies, Gaithersburg, MD) supplemented with 10% BCS, 5% fetal bovine serum (Life Technologies), 100 U/ml penicillin, and 0.1 mg/ml streptomycin.
Transient transfection of primary cortical neurons for kinase assays. Cortical neurons (2 × 106 cells/35 mm dish) were transiently transfected at DIV3 using a calcium phosphate coprecipitation protocol as described (Xia et al., 1996; Hetman et al., 1999). Briefly, the DNA–calcium phosphate precipitates were prepared by mixing 1 vol of DNA in 250 mm CaCl2 with an equal volume of 2× HEPES-buffered saline (HBS; 274 mmNaCl, 10 mm KCl, 1.4 mmNa2HPO4, 15 mmd-glucose, and 42 mm HEPES, pH 7.07). The precipitates were allowed to form for 25–30 min at room temperature before addition to the cultures. The conditioned culture media were removed and saved. Cells were washed three times with BME, and 1.5 ml of transfection media was added to each 35 mm dish. The transfection media consist of BME supplemented with 1 mmsodium kynurenate, 10 mm MgCl2, and 5 mm HEPES. The pH of the transfection media was kept high by incubating BME in a dish at 37°C and 0% CO2for 30 min to “degas.” Sixty microliters of the DNA–calcium phosphate precipitates were added dropwise to each 35 mm dish and mixed gently. Plates were incubated at room temperature and ambient air for 5 min and then in a humidified incubator with 5% C02 at 37°C for 35–45 min. The incubation was stopped 20–25 min after the layer of precipitate formed on the plates by “shocking” the cells for 2 min with 1× HBS, 1 mmsodium kynurenate, and 10 mm MgCl2 in 5 mm HEPES, pH 7.5, and 5% glycerol. Cells were then washed three times with 2 ml of BME. The saved conditioned media were added back to each plate, and cells were returned to the 5% CO2 incubator at 37°C for 48 hr before treatment or harvesting.
Drug treatment. Drug treatment was performed at DIV6 for cortical neurons. Forskolin was dissolved in ethanol. Ethanol was used as vehicle control for forskolin treatment. K252a was dissolved in water and added 30 min before BDNF stimulation. Doses and times of drug treatment are described in detail in the figure legends.
Generation of a polyclonal anti-ERK5 antibody. We made a polyclonal antibody against ERK5 by immunizing rabbits (Cocalico Biologicals, Reamstown, PA) with the GST–ERK5 (C-terminal 100 aa) fusion protein. This anti-ERK5 antibody was used in the ERK5 autophosphorylation kinase assay. The specificity of this antibody was confirmed by showing that ERK5 autophosphorylation was not seen when the anti-ERK5 antibody was preincubated with the GST–ERK5 fusion protein used to generate the antibody or when preimmune sera were used for immune precipitation of ERK5 (data not shown).
Kinase assays. Cell lysates were prepared as described previously (Dérijard et al., 1995; Xia et al., 1995), and protein concentrations were assayed by the Bradford method. Equal amounts of protein extracts (300 μg) were used for each kinase assay. To measure endogenous ERK5 activity, cell lysates were incubated at 4°C for 2.5 hr with 6 μl of the antisera against the GST–ERK5 C-terminal 100 aa fusion protein that we generated. Protein A–Sepharose beads (60 μl) were then added, and the mixture was incubated at 4°C for an additional hour. The endogenous ERK5 activity in the immune precipitates was then quantitated by an autophosphorylation assay as described (Abe et al., 1997; Yan et al., 1999).
To measure the activity of transfected ERK5 or ERK2, cell lysates (200–300 μg) were incubated with an anti-Flag antibody prebound to a slurry of 80% protein G–Sepharose and 20% protein A–Sepharose. The activity of transfected ERK5 or ERK2 in the immune precipitates was quantitated by a kinase assay using recombinant GST–MEF2C (10 μg) or MBP (2.5 μg) as the substrate, respectively (Xia et al., 1995; Kato et al., 1997, 1998; Hetman et al., 1999).
To measure endogenous MEK5 activity, cell lysates (300 μg) were incubated at 4°C for 3 hr with an anti-MEK5 antibody (N-19; Santa Cruz Biotechnology, Santa Cruz, CA) prebound to protein G–Sepharose beads. The MEK5 activity in the immune precipitates was quantitated by a kinase assay using a recombinant GST–ERK5(M) as the substrate (Kato et al., 1997, 1998; Kamakura et al., 1999; Marinissen et al., 1999). Quantitation of kinase activity was achieved by PhosphorImager analysis (Molecular Dynamics, Sunnyvale, CA) or by using the ImageQuant program after scanning the autoradiographic images.
Western analysis. Western blot analysis of ERK5 and anti-phospho-ERK1/2 was performed as described (Kato et al., 1997; Chao et al., 1999; Hetman et al., 1999). The polyclonal anti-ERK5 peptide antibody used for Western analysis was kindly provided by Dr. J. D. Lee and was used at a dilution of 1:5000.
Reporter gene assays. PC12 cells were transfected using Transfast (Promega) as described by the manufacturer. Briefly, 1 × 105 cells were plated onto each well of a 24-well plate coated with poly-d-lysine (Sigma). One day later, cells were transfected with a CRE–luciferase reporter gene (1.2 μg/3 wells) or a Gal4–luciferase reporter gene (0.2 μg/3 wells) together with various expression vectors for Gal4 fusion proteins (0.4 μg/3 wells). The EF1a.LacZ DNA (Invitrogen, San Diego, CA) was added at 0.125 μg/3 wells for normalization of transfection efficiency. Cortical neurons were transfected using LipofectAMINE 2000 (Life Technologies) (Impey et al., 1996; Poser et al., 2000). Briefly, 0.5 × 106 cells were plated onto each well of a 24-well plate coated with poly-d-lysine (Research Collaborative). At DIV4–DIV5, neurons were cotransfected with the Gal4–luciferase reporter gene (1.4 μg/4 wells), Gal4–MEF2C fusion protein (0.9 μg/well), and EF1a.LacZ DNA (0.55 μg/4 wells). Where indicated, PC12 cells and cortical neurons were also cotransfected with various expression vectors for the ERK5- and ERK1/2-signaling pathways. For PC12 cells, cells were serum starved at 1 d after transfection for 24 hr and then treated with NGF (50 ng/ml) for 6 hr when indicated. For cortical neurons, cells were treated at 2 d after transfection with 10 ng/ml BDNF or 55 mm KCl for 6 hr when indicated. Cell lysates were prepared, and the activities of luciferase or β-galactosidase were measured as described (Impey et al., 1996). The reporter gene luciferase activity was normalized to β-galactosidase activity and expressed as the fold induction relative to control.
RESULTS
ERK5 is activated by neurotrophins in primary cortical neurons
ERK1/2 is activated by growth factors and neurotrophins in several cell types including neurons (Ahn et al., 1992; Castellino and Chao, 1996; Segal and Greenberg, 1996). Similarly, ERK5 is activated by EGF and NGF in PC12 and non-neuronal cells (Kato et al., 1998; Kamakura et al., 1999). However, the regulation of ERK5 by neurotrophins in primary cultured neurons derived from CNS has not been reported. Therefore, we examined whether ERK5 is stimulated by various physiological stimuli in cortical neurons and compared the regulation of ERK5 with that of ERK1/2. Cortical neurons were treated with various concentrations (0–50 ng/ml) of BDNF for 1 hr, and the cell lysates were analyzed by Western analysis using antibodies against ERK5 or phospho-ERK1/2 (Fig.1A). BDNF treatment, at concentrations as low as 5 ng/ml, caused a reduced electrophoretic mobility (phosphorylation shift) of ERK5, indicative of ERK5 activation (Kato et al., 1997, 1998). The phosphorylation shift of ERK5 was maximal at 10 ng/ml BDNF, a concentration of BDNF used in subsequent studies. ERK1/2 activation was measured by Western analysis using the anti-phospho-ERK1/2 antibody that recognizes phosphorylated and activated ERK1/2. As reported previously (Marsh et al., 1993; Hetman et al., 1999), BDNF activated ERK1/2 in cortical neurons, and the dose–response curves for activation of ERK5 and ERK1/2 were similar.
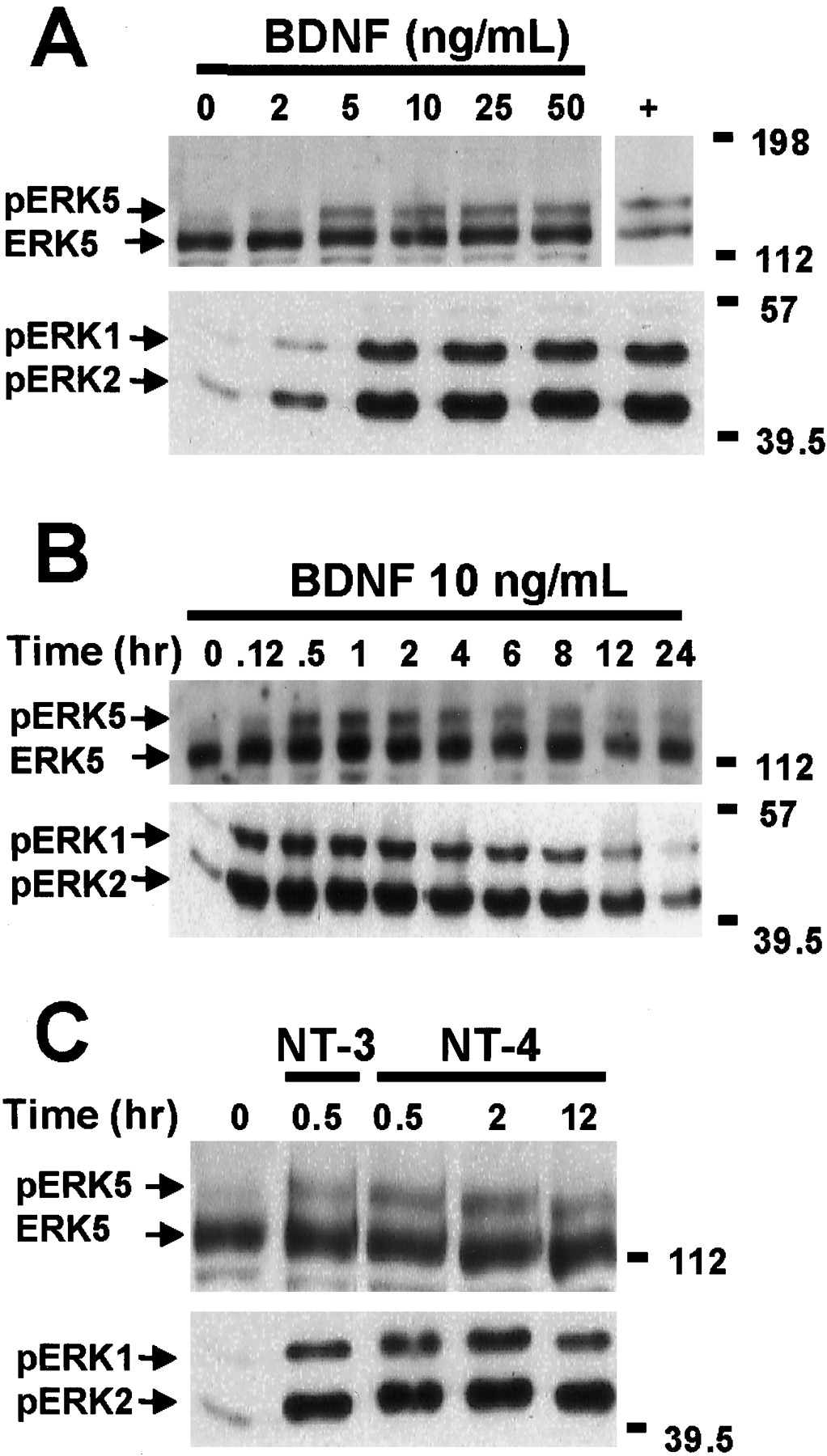
Neurotrophins induce ERK5 and ERK1/2 phosphorylation in cortical neurons. A, Dose–response relationship of BDNF stimulation of ERK5 and ERK1/2 phosphorylation is shown. At DIV5, cortical neurons were treated with 0, 2, 5, 10, 25, or 50 ng/ml BDNF for 1 hr. Cell lysates from human embryonic kidney 293 cells transiently transfected with a constitutively active MEK5 and a wild-type ERK5 were used as a positive control (+) for the ERK5 phosphorylation shift. B, Kinetics of BDNF stimulation of ERK5 and ERK1/2 phosphorylation is shown. At DIV5, cortical neurons were treated with 10 ng/ml BDNF for the indicated times.C, NT-3 and NT-4 also induce ERK5 and ERK1/2 phosphorylation. At DIV5, cortical neurons were treated with 10 ng/ml NT-3 or NT-4 for 0.5, 2, or 12 hr. Cell lysates were prepared, and 20 μg of total protein was submitted to Western analysis using antibodies recognizing ERK5 (Abe et al., 1996) or phosphorylated (p) ERK1/2. Phosphorylation of ERK5 was observed as a shift in ERK5 mobility, indicative of ERK5 activation. The anti-phospho-ERK1/2 antibody recognizes the phosphorylated and activated ERK1/2, indicative of ERK1/2 activation. Data are representative of four (A, B) or three (C) independent experiments. Thepositions of molecular weight markers are indicated on the right of the figures.
To determine the kinetics of ERK5 activation, cortical neurons were treated with 10 ng/ml BDNF for various times. Like ERK1/2 activation, ERK5 activation was prolonged and sustained for up to 24 hr after BDNF treatment (Fig. 1B). However, the peak activation of ERK5 was slower than that of ERK1/2. Although ERK5 activation was detectable at 5 min, it did not reach a maximum until 1–2 hr after BDNF treatment. The slow kinetics of ERK5 activation was also confirmed by the autophosphorylation assay (e.g., see Fig. 3). In contrast, ERK1/2 was maximally activated by BDNF at 30 min under the same conditions using the same cultured neuron preparations. In addition to BDNF, other neurotrophins, including neurotrophin-3 (NT-3), NT-4, and NGF, also activate ERK1/2 (Castellino and Chao, 1996; Segal and Greenberg, 1996). Similarly, ERK5 was activated by NT-3 or NT-4 treatment of cortical neurons (Fig. 1C). In agreement with other reports (Kamakura et al., 1999), NGF treatment of PC12 cells activated ERK5 (data not shown).
To determine whether BDNF activation of ERK5 requires TrkB tyrosine kinase activity, cortical neurons were treated with K252a, an inhibitor of receptor tyrosine kinases. Like ERK1/2, ERK5 activation was inhibited by K252a (Fig. 2), suggesting that inhibition of receptor tyrosine kinase prevents BDNF stimulation of both ERK5 and ERK1/2.

BDNF stimulation of ERK5 phosphorylation requires receptor tyrosine kinase activity. Cortical neurons (DIV5–DIV6) were pretreated with 0, 2, or 5 μm K252a for 30 min and then stimulated with 10 ng/ml BDNF for 0.5, 1, or 2 hr as indicated. Phosphorylation (p) of ERK5 and ERK1/2 was measured by Western analysis as described in Figure 1. Similar results were obtained in two independent experiments.
To confirm ERK5 activation by BDNF, ERK5 activity was quantitated using an ERK5 autophosphorylation assay after immune precipitation of ERK5. This alternative assay was used because activation of ERK5 leads to increased ERK5 autophosphorylation (Abe et al., 1997; Yan et al., 1999). BDNF stimulation of cortical neurons increased ERK5 autophosphorylation fourfold, and the kinetics of activation was comparable with that measured using the phosphorylation shift assay (Fig. 3). Together, the phosphorylation shift and autophosphorylation data indicate that neurotrophins activate both ERK5 and ERK1/2 in cortical neurons.

BDNF activates ERK5 in cortical neurons. Cortical neurons (DIV5–DIV6) were treated with 10 ng/ml BDNF for various times. Three hundred micrograms of total protein were used to measure ERK5 activity by the autophosphorylation assay. A, A representative autoradiograph of the ERK5 autophosphorylation kinase assay. B, Quantitation of ERK5 autophosphorylation.Inset, A more detailed profile of ERK5 activation at early time points. Data are the average of five to seven experiments. Error bars represent SEM.
MEK5 is activated by BDNF and is required for BDNF stimulation of ERK5
MEK5 is an upstream kinase that phosphorylates and activates ERK5 in several non-neuronal cells (Zhou et al., 1995; Kato et al., 1997,1998; English et al., 1999; Kamakura et al., 1999). To determine whether MEK5 mediates BDNF stimulation of ERK5 in cortical neurons, MEK5 kinase activity was monitored by an immune complex kinase assay using GST–ERK5(M) as the substrate (Kato et al., 1997). BDNF activated MEK5 in cortical neurons, and like ERK5, MEK5 activation was maximal at 1 hr and persisted for up to 24 hr after BDNF stimulation (Fig.4A).

MEK5 is activated by BDNF and is required for BDNF stimulation of ERK5 in cortical neurons. A, Endogenous MEK5 is activated by BDNF. At DIV5, cortical neurons were treated with 10 ng/ml BDNF for various times. Three hundred micrograms of total protein were used for an MEK5 immune complex kinase assay with truncated GST–ERK5(M) as the substrate. Inset, A more detailed profile of MEK5 activation at early time points is shown. Data shown are averages of two independent experiments. Error bars represent SEM. B, C, Expression of a dominant-negative MEK5 blocks BDNF stimulation of ERK5 (B) but not ERK2 (C). Cortical neurons (DIV3; 2 × 106 cells/35 mm dish) were cotransfected with 2 μg each of plasmid DNA encoding a wild-type Flag-tagged ERK5 (ERK5wt) or ERK2 (ERK2wt), a dominant-negative HA-tagged MEK5 (MEK5DN), or a vector control (pCMV5) as indicated. Two days later, cells were treated with 10 ng/ml BDNF for 1 hr. Three hundred micrograms of total protein were used for immunoprecipitation with anti-Flag antibody. The transfected ERK5 and ERK2 kinase activities in the precipitates were assayed using GST–MEF2C or MBP as the respective substrates. Data shown are averages of three independent experiments. Error bars represent SEM.
To determine whether MEK5 is required for BDNF stimulation of ERK5, cortical neurons were transiently cotransfected with plasmid DNA encoding a Flag-tagged, wild-type ERK5 and a dominant-negative MEK5 or the MEK5 vector control (Fig. 4B). To determine the specificity of MEK5 for the ERK5 pathway, dominant-negative MEK5 was cotransfected with a Flag-tagged, wild-type ERK2. Two days after transfection, neurons were stimulated with 10 ng/ml BDNF for 1 hr. The activities of transfected ERK5 or ERK2 were determined by anti-Flag immune precipitation followed by an immune complex kinase assay using MEF2C or MBP as the substrate, respectively (Xia et al., 1995; Kato et al., 1997; Hetman et al., 1999). Similar to endogenous ERK5 and ERK2, the transfected wild-type ERK5 and ERK2 were activated by BDNF (Fig. 4B,C). Cotransfection of a dominant-negative MEK5, but not the vector control, inhibited BDNF stimulation of ERK5 (Fig. 4B). However, BDNF stimulation of ERK2 was not inhibited by this dominant-negative MEK5 (Fig. 4C). These data suggest that MEK5 is an upstream kinase that mediates BDNF activation of ERK5.
Glutamate, membrane depolarization, and cAMP do not activate ERK5
Because ERK1/2 activation by neuronal activity is critical for many aspects of neuronal function including neuronal survival (Curtis and Finkbeiner, 1999; Grewal et al., 1999) and neuronal plasticity (Siegelbaum and Kandel, 1991; Impey et al., 1999; Vanhoutte et al., 1999), it was important to determine whether ERK5 is also regulated by neuronal activity. Because ERK5 and ERK1/2 are both activated by neurotrophins, one might expect that ERK5, like ERK1/2, would also be activated by neuronal activity. To mimic neuronal activity in vitro, cultured neurons were treated with membrane-depolarizing concentrations of KCl (55 mm) or with the excitatory neurotransmitter glutamate. Surprisingly, ERK5 was not stimulated by treatment with 55 mm KCl for up to 2 hr (Fig.5A). Similarly, 30 or 100 μm glutamate treatment for various times (5–120 min) did not activate ERK5 (Fig. 5B; data not shown). In contrast, both glutamate and membrane depolarization induced ERK1/2 phosphorylation (Fig. 5A,B), confirming that the cortical neurons used in the experiment showed normal responses to membrane depolarization and activation of glutamate receptors.

cAMP, KCl, and glutamate activate ERK1/2 but not ERK5 in cortical neurons. Cortical neurons (DIV5) were treated with vehicle control, 55 mm KCl (A), 100 μm glutamate (B), or 2, 5, 10, or 50 μm forskolin (C) that increases intracellular cAMP for the indicated times. For a positive control, neurons were treated with BDNF (10 ng/ml; 1 hr). Phosphorylation (p) of ERK5 and ERK1/2 was measured by Western analysis as described in Figure 1. Similar results were obtained in three independent experiments. Fsk, Forskolin.
In neurons and PC12 cells, cAMP activates the ERK1/2 regulatory pathway. To determine whether ERK5 is also regulated by cAMP, cortical neurons were treated with forskolin, a general activator of adenylyl cyclases. Treatment of cortical neurons with 2–50 μmforskolin for 0.5, 2, or 12 hr stimulated ERK1/2 (Fig. 5C), consistent with previous reports (Erhardt et al., 1995; Martin et al., 1997; Vossler et al., 1997; de Rooij et al., 1998; Kawasaki et al., 1998; Wei et al., 1998). However, under identical conditions, forskolin did not stimulate ERK5. We also quantitated ERK5 activity after glutamate, KCl, or forskolin treatment using the ERK5 autophosphorylation assay (Fig. 6). In agreement with the results obtained using the phosphorylation shift assay, ERK5 was not activated after treatment with 30 μm glutamate, 55 mm KCl, or 2 μm forskolin (Fig. 6). These data indicate that ERK5 is stimulated by neurotrophins but not neuronal activity or cAMP.

ERK5 autophosphorylation is not increased by KCl, glutamate, or forskolin treatment of cortical neurons. Cortical neurons (DIV5–DIV6) were treated with 10 ng/ml BDNF, 55 mm KCl, 30 μm glutamate, or 2 μm forskolin for various times. Three hundred micrograms of total protein were used to measure ERK5 activity by the autophosphorylation assay. Similar results were obtained with 50 μm forskolin. Data shown are the averages of four to seven experiments. Error bars represent SEM.
ERK5 and ERK1/2 regulate different downstream transcriptional pathways in PC12 cells and cortical neurons
Although ERK5 and ERK1/2 activate some of the same transcription pathways in non-neuronal cells, there are differences in their downstream transcriptional targets. For example, they both phosphorylate transcription factors c-Myc and Sap1a (Kato et al., 1997;English et al., 1998; Yang et al., 1998; Kamakura et al., 1999;Marinissen et al., 1999). However, MEF2A and MEF2C are phosphorylated and activated by ERK5 but not by ERK1/2, whereas Elk1 is phosphorylated and activated by ERK1/2 but not by ERK5 (Gille et al., 1992; Janknecht et al., 1993; Marais et al., 1993; Kato et al., 1997; English et al., 1998; Yang et al., 1998; Marinissen et al., 1999). To determine whether ERK5 and ERK1/2 activate distinct transcriptional pathways in neurons, we examined the effect of ERK5 and ERK1/2 activation on transcription mediated by transcription factors MEF2C and Elk1 and on transcription initiated from the CRE using PC12 cells and cortical neurons. These transcription events were analyzed because MEF2C and CREB have been implicated in neuronal survival (Bonni et al., 1999; Mao et al., 1999;Riccio et al., 1999). Furthermore, the Elk1 and the CREB/CRE transcription pathways are thought to contribute to memory formation (Berman et al., 1998; Impey et al., 1998b, 1999; Sgambato et al., 1998).
PC12 cells were transiently transfected with a constitutively active MEK5 and a wild-type ERK5 or a constitutively active MEK1 and a wild-type ERK2 to activate ERK5 or ERK1/2 signaling specifically. Activation of the ERK5- or ERK1/2-signaling pathway caused 22-fold and 5-fold increases in Gal4–MEF2C-mediated transcription, respectively, indicating that MEF2C transcription is preferentially activated by ERK5 (Fig. 7A). This is consistent with previous reports (Kato et al., 1997, 2000; English et al., 1998;Yang et al., 1998; Marinissen et al., 1999).

MEF2C-transactivating activity is stimulated by NGF and ERK5 in PC12 cells. PC12 cells were transfected with a Gal4–luciferase reporter gene (0.2 μg/3 wells) and an expression vector for Gal4–MEF2C fusion protein (0.4 μg/3 wells) to measure MEF2C transcriptional activity. An EF promoter-driven LacZ expression vector was cotransfected in all cases to normalize for transfection efficiency. A, MEF2C-mediated transactivation is preferentially stimulated by constitutive activation of the ERK5 pathway. To activate ERK5 or ERK1/2, cells were cotransfected with expression vectors (0.1 μg each/3 wells) encoding a constitutively active MEK5 (MEK5CA) with a wild-type ERK5 (ERK5wt) or a MEK1CA with anERK2wt, respectively. Data shown are the averages of 12 independent experiments ± SEM. B, MEF2C-mediated transcription is activated by NGF via an ERK5-dependent mechanism. To block ERK5 signaling, PC12 cells were transiently transfected with a dominant-negative MEK5 (0.9 μg/4 wells; MEK5DN) together with a dominant-negative ERK5 (0.9 μg/4 wells;ERK5DN) or the corresponding vector controls. Cells were treated with 50 ng/ml NGF (+NGF) or vehicle control (−NGF) for 6 hr. Data shown are the averages of three independent experiments ± SEM.Luc, Luciferase.
It has been reported that MEF2C transcription is stimulated by membrane depolarization in cerebellar neurons (Mao and Wiedmann, 1999;Mao et al., 1999). However, it is not known whether neurotrophins stimulate MEF2C transcription in neurons. To address this issue, PC12 cells and cortical neurons were transiently transfected with a Gal4–MEF2C expression vector and a Gal4–luciferase reporter gene and treated with NGF or BDNF, respectively (Figs. 7B,8A). NGF and BDNF both activated Gal4–MEF2C-induced transcription, suggesting that MEF2C transcription is also regulated by neurotrophins in neurons. Neurotrophins activate several kinase pathways in addition to ERK5 including the p38 MAP kinase pathway (Xing et al., 1998) that can also stimulate MEF2C transcription (Han et al., 1997). To determine the contribution of ERK5 signaling in neurotrophin stimulation of MEF2C transcription, we cotransfected PC12 cells and cortical neurons with a dominant-negative MEK5 together with a dominant-negative ERK5 to inhibit neurotrophin activation of the ERK5 pathway. Expression of the dominant-negative MEK5 plus dominant-negative ERK5 inhibited MEF2C transcription induced by NGF and BDNF (Figs. 7B,8A). To determine the specific involvement of the ERK5 pathway in neurotrophin stimulation of MEF2C transcription, we used a dominant-negative MEK1 as a negative control. Expression of this dominant-negative MEK1 construct blocked CRE-mediated transcription in cortical neurons after KCl stimulation (Fig. 8B), consistent with other reports (Impey et al., 1998a). However, it did not affect BDNF stimulation of MEF2C transcription (Fig.8A). Together, these data suggest that MEF2C transcription is activated by neurotrophins via a mechanism involving the ERK5 but not the ERK1/2 pathway.

BDNF activates MEF2C in cortical neurons that require ERK5 signaling. A, MEF2C-mediated transcription is activated by BDNF via an ERK5-dependent mechanism. Cortical neurons (DIV4–DIV5; 0.5 × 106 cells/well) were transiently transfected with a Gal4–luciferase reporter gene (1.4 μg/4 wells) and an expression vector for Gal4–MEF2C fusion protein (0.9 μg/4 wells) to measure MEF2C transcriptional activity. To block ERK5 or ERK1/2 signaling, neurons were cotransfected with a dominant-negative MEK5 (0.9 μg/4 wells; MEK5DN) together with a dominant-negative ERK5 (0.9 μg/4 wells;ERK5DN) or with a dominant-negative MEK1 (1.8 μg/4 wells; MEK1DN), respectively. The corresponding vectors were used as controls. Cells were treated with 10 ng/ml BDNF (+BDNF) or vehicle control (−BDNF) for 6 hr. Data are representative of quadruplicate determinations from three independent experiments.B, KCl-activated CRE transcription is inhibited by the dominant-negative MEK1 used in A. To confirm that theMEK1DN used in A functioned properly as a dominant negative, cortical neurons were cotransfected with a CRE–luciferase reporter (2.4 μg/4 wells), a MEK1DN, or its vector control (1.8 μg/4 wells). Cells were treated with 55 mm KCl for 6 hr. Data shown are the averages of quadruplicate determinations. For both A andB, an EF promoter-driven LacZ expression vector (0.55 μg/4 wells) was cotransfected to normalize for transfection efficiency, and error bars indicate SEM. Luc, Luciferase.
In contrast to MEF2C, Gal4–Elk1-induced transcription was enhanced by activation of ERK1/2 but not by ERK5 (Fig.9A). Furthermore, unlike ERK1/2, ERK5 did not stimulate transcription from CRE (Fig.9B). These data suggest that ERK5 and ERK1/2 activate distinct transcription pathways in PC12 cells and cortical neurons.

ERK5 does not stimulate Gal4–Elk1 transactivation or CRE-mediated transcription in PC12 cells. A, Transactivation of Elk1 is increased after NGF treatment or stimulation of the ERK1/2 but not the ERK5 pathway. PC12 cells were transfected with a Gal4–luciferase reporter gene (0.2 μg/3 wells) and an expression vector for the Gal4–Elk1 fusion protein (0.4 μg/3 wells). To activate ERK5 or ERK1/2, cells were cotransfected with expression vectors encoding a constitutively active MEK5 (0.2 μg/3 wells;MEK5CA) with a wild-type ERK5 (0.6 μg/3 wells;ERK5wt) or a MEK1CA (0.6 μg/3 wells), respectively. B, NGF treatment or constitutive activation of ERK1/2, but not ERK5, stimulates CRE-mediated transcription. PC12 cells were transfected with a CRE–luciferase reporter (1.2 μg/3 wells) to measure transcription initiated from CRE. To activate ERK5 or ERK1/2, cells were cotransfected with expression vectors encoding MEK5CA (0.1 μg/3 wells) and ERK5wt (0.3 μg/3 wells) or MEK1CA(0.1 μg/3 wells) and ERK2wt (0.3 μg/3 wells), respectively. An EF promoter-driven LacZ expression vector was cotransfected in all cases to normalize for transfection efficiency. When indicated, cells were treated with NGF (50 ng/ml) for 6 hr. Data shown are the averages of triplicate determinations ± SEM. Similar results were obtained in three to four independent experiments.Luc, Luciferase.
DISCUSSION
The objective of this study was to define the regulatory properties of ERK5 in primary cultures of cortical neurons in response to neurotrophins, neuronal activity, or cAMP. Neuronal activity was mimicked in vitro by treating cultured neurons with membrane-depolarizing concentrations of KCl or glutamate. The activity of endogenous ERK5 activity in cortical neurons was measured by two well established methods: the ERK5 autophosphorylation assay (Abe et al., 1997; Yan et al., 1999) and the reduced electrophoretic mobility assay (phosphorylation shift) (Kato et al., 1997, 1998). Neurotrophins including BDNF, NGF, NT-3, and NT-4 caused a sustained activation of MEK5 as well as ERK5 in PC12 cells and cortical neurons. BDNF activation of ERK5 was blocked by K252a, indicating a requirement for the receptor tyrosine kinase of TrkB. Furthermore, expression of a dominant-negative MEK5 blocked BDNF stimulation of ERK5, suggesting that MEK5 mediates neurotrophin stimulation of ERK5 in neurons.
Surprisingly, membrane depolarization, glutamate, or cAMP did not activate ERK5 in cortical neurons although they stimulated ERK1/2 activity. Furthermore, MEF2C was activated by neurotrophins, and the ERK5 signaling was required for neurotrophin stimulation of MEF2C transcription. On the other hand, ERK1/2, but not ERK5, activated Elk-1 transcriptional activity and stimulated transcription initiated from CRE. These data suggest that the regulatory properties of ERK5 as well as the downstream transcriptional pathways regulated by ERK5 are distinct from those of ERK1/2 in neurons.
Although neurotrophins activated both ERK5 and ERK1/2 pathways in cortical neurons, the kinetics of ERK5/MEK5 activation was slower than that of ERK1/2. One possibility for this difference is that the ERK5 and ERK1/2 pathways are activated by distinct upstream components, such as small G-proteins. For example, NGF activation of the ERK1/2 pathway in PC12 cells is both rapid and sustained. However, distinct mechanisms account for the two phases; the sustained activation of ERK1/2 by NGF requires the small G-protein Rap1, whereas the initial rapid activation requires Ras (York et al., 1998). It is possible that BDNF stimulation of the MEK5/ERK5 pathway is mediated by Rap1-like small G-proteins.
Because neuronal activity is critical for neuronal survival and synapse formation (Oppenheim, 1991; Bear and Malenka, 1994; Goldberg and Barres, 2000), it is important to elucidate mechanisms that translate activity changes to long-lasting modifications of neurons, particularly transcriptional changes. Activation of NMDA receptors or voltage-sensitive calcium channels increases intracellular Ca2+ and stimulates ERK1/2 in neuronal cells (Ely et al., 1990; Bading and Greenberg, 1991; Baraban et al., 1993; Fiore et al., 1993; Rosen et al., 1994; Kurino et al., 1995). ERK1/2 is also activated during LTP in mice and long-term facilitation in Aplysia (English and Sweatt, 1996; Martin et al., 1997;Impey et al., 1998b). Activity-dependent activation of ERK1/2 has been implicated in several important aspects of CNS neuron function including LTP (Brambilla et al., 1997; English and Sweatt, 1997; Impey et al., 1998b, 1999) and memory formation (Atkins et al., 1998;Berman et al., 1998; Impey et al., 1999). Similar to ERK1/2, several other kinase-signaling pathways are stimulated by both neurotrophins and neuronal activity including those of the phosphatidylinositol-3 kinase, Akt, the c-Jun N-terminal protein kinase, and p38 MAP kinases (Farnsworth et al., 1995; Rusanescu et al., 1995; Tan et al., 1996;Kawasaki et al., 1997; Miller et al., 1997; Schwarzschild et al., 1997;Chen et al., 1998; Xing et al., 1998; Yano et al., 1998; Zhang et al., 1998; Grewal et al., 2000). Our data indicate that ERK5 is not activated by glutamate or membrane depolarization in cortical neurons, distinguishing this kinase from all of the other MAP kinases. ERK5 is the first MAP kinase identified that is activated by neurotrophins but not by neuronal activity.
Our observation that ERK5 in cortical neurons is not stimulated by Ca2+ is in accord with previous studies with non-neuronal cells showing that the Ca2+ ionophore A23187 does not activate ERK5 in COS7 and bovine aortic endothelial cells (BAEC) (Kamakura et al., 1999; Yan et al., 1999). However, the intracellular Ca2+ chelator BAPTA AM prevented ERK5 activation by EGF in mouse embryo fibroblasts and by shear stress in BAEC (Yan et al., 1999; Ji and Carpenter, 2000). This suggests that Ca2+ may be required, but is not sufficient, for ERK5 activation in non-neuronal cells.
cAMP inhibits growth factor stimulation of ERK1/2 in non-neuronal cells (Burgering et al., 1993; Graves et al., 1993) but stimulates ERK1/2 in neurons via Rap1 and B-raf (Erhardt et al., 1995; Vossler et al., 1997; de Rooij et al., 1998; Kawasaki et al., 1998). This emphasizes the importance of defining regulatory mechanisms for the MAP kinases in neurons because they are often different from those in non-neuronal cells. Activators of adenylyl cyclases markedly increase ERK1/2 activity in hippocampal neurons (Martin et al., 1997; Wei et al., 1998). Furthermore, activation of ERK1/2 by cAMP is critical for long-lasting LTP (English and Sweatt, 1996; Impey et al., 1998b). In contrast, our results suggest that ERK5 is not activated by increases in intracellular cAMP, which further distinguishes ERK5 from ERK1/2.
The MEF2 proteins constitute a family of transcription factors: MEF2A, MEF2B, MEF2C, and MEF2D. They cooperate with members of the MyoD family in muscle differentiation (Kaushal et al., 1994; Molkentin et al., 1995). In addition to muscle, MEF2A and MEF2C are also expressed in developing and adult brain including cortex and cerebellum (Leifer et al., 1993, 1994; McDermott et al., 1993; Lyons et al., 1995; Lin et al., 1996; Mao et al., 1999; Marinissen et al., 1999). Cortex contains a high level of MEF2C protein (Lin et al., 1996). However, the function and regulation of MEF2 in the nervous system have not been extensively studied. Although MEF2 mediates T cell receptor-induced apoptosis in T cells (Youn et al., 1999), a recent study suggests that MEF2 mediates activity-dependent survival of cortical and cerebellar neurons (Mao et al., 1999). Although MEF2C transcription is stimulated by membrane depolarization in cerebellar neurons (Mao and Wiedmann, 1999; Mao et al., 1999), it has not been reported whether neurotrophins stimulate MEF2C transcription in neurons. Furthermore, the kinase-signaling mechanisms that mediate this transcription are unknown. Our data suggest that neurotrophins (NGF and BDNF) activate MEF2C transcription in both neuron-like PC12 cells and in primary-cultured cortical neurons. Furthermore, ERK5 but not ERK1/2 signaling contributes to neurotrophin stimulation of MEF2C transcription.
The CREB/CRE transcriptional pathway is a major downstream target of ERK1/2 signaling that contributes to neuroplasticity (Impey et al., 1998b, 1999; Sgambato et al., 1998) and neuronal survival (Bonni et al., 1999; Riccio et al., 1999). Although CREB is not directly phosphorylated by ERK1/2, it is phosphorylated and transactivated by the ERK1/2-activated Rsk family of protein kinases p90rsk (Xing et al., 1996, 1998; Impey et al., 1998a). In addition to CREB, the transcription factor Elk1 may also be a major nuclear target of ERK1/2 important for synaptic plasticity (Berman et al., 1998; Sgambato et al., 1998). Elk1 is directly phosphorylated and activated by ERK1/2 (Gille et al., 1992,1995; Janknecht et al., 1993; Marais et al., 1993) and plays an important role in NMDA-induced gene expression in neurons via serum response element (Xia et al., 1996). Our data suggest that ERK5 does not stimulate the transcriptional activity of Elk1 or transcription initiated from CRE. Together with the fact that ERK5 is not activated by neuronal activity, these results emphasize the importance of ERK1/2 signaling for activity-dependent neural plasticity. Although ERK5 may not be directly stimulated by neuronal activity, it may be subsequently activated as a result of neurotrophin synthesis. For example, the promoter for BDNF contains a CRE element, and neuronal activity induces BDNF synthesis via the CREB/CRE transcriptional pathway (Shieh et al., 1998; Tao et al., 1998). BDNF expression is increased in brain during training for associative fear learning (Hall et al., 2000), a process that stimulates CRE-mediated transcription (Impey et al., 1998a) and is inhibited by MEK inhibitors (Atkins et al., 1998). Consequently, BDNF stimulation of ERK5 may contribute to synaptic plasticity and memory formation by stimulating other transcriptional pathways, e.g., MEF2C-mediated transcription.
In summary, our data identify several key differences between ERK5 and ERK1/2 in their response to neuronal activity and cAMP, as well as in downstream transcriptional targets. This suggests that ERK5 may have unique functions in the nervous system. For example, ERK5 may be particularly important for promoting neurotrophin-mediated neuronal survival during development when activity is not crucial for neuronal survival (Shatz, 1990; Oppenheim, 1991; Johnson and Deckwerth, 1993;Linden, 1994; Ikonomidou et al., 1999, 2000; Goldberg and Barres, 2000). It may also play a key role in neurotrophin-promoted survival when ERK1/2 expression is low during early development (Boulton et al., 1991) or in regions of the brain where ERK1/2 activity is low (Thomas and Hunt, 1993). Furthermore, the ERK5 survival mechanism may differ from and complement the ERK1/2 survival mechanism by activating distinct downstream transcriptional pathways.
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke Grant NS37359 and the Burroughs Wellcome Fund for New Investigator Award in Toxicology Grant APP#3010 (Z.X.). J.E.C. was supported by the National Institutes of Health Postdoctoral Training Grant Genetic Approaches to Aging (2 T32 AG00057-21). We thank Dr. J. D. Lee for providing anti-ERK5 antibody and expression vectors for ERK5, MEK5, and GST–ERK5 (M). We thank Dr. J. Silvio Gutkind for GST-MEF2C. We thank Dr. J. Han for providing the Gal4-MEF2C and Flag-tagged wild-type ERK2 plasmids.
Correspondence should be addressed to Dr. Zhengui Xia, Department of Environmental Health, Box 357234, University of Washington, HSB, Room F561C, Seattle, WA 98195. E-mail: zxia{at}u.washington.edu.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}