Article Text

Abstract
In MCF-7 breast cancer cells, the insulin-like growth factor 1 receptor (IGF-1R) and the oestrogen receptor (ER) are coexpressed and the two signalling systems are engaged in a crosstalk that results in synergistic growth. However, coupling between the signalling cascades is poorly understood. Oestradiol enhances IGF-1R signalling by inducing the expression of insulin receptor substrate 1 (IRS-1), a substrate of the IGF-1R. Oestradiol induced expression of IRS-1 results in enhanced tyrosine phosphorylation of IRS-1 after IGF-1 stimulation, followed by enhanced mitogen activated protein kinase, phosphoinositide 3′ kinase, and Akt activation. Oestradiol can also potentiate the effect of IGF-1 on the expression of cyclin D1 and cyclin E, and on the phosphorylation of the retinoblastoma protein (RB). These effects are greatly diminished in SX13 cells, which exhibit a 50% reduction in IGF-1R expression but few functional IGF-1Rs at the surface. Oestradiol and IGF-1 regulate the expression of two cyclin dependent kinase inhibitors, p21 and p27, differently. Whereas IGF-1 increases p21 expression and reduces p27 expression, oestradiol has no effect on p21. In summary, in MCF-7 cells, oestrogen potentiates the effect of IGF-1 on IGF-1R signalling and its effects on certain cell cycle components.
- breast cancer
- oestrogen receptor
- insulin-like growth factor 1 receptor
- cell cycle
Statistics from Altmetric.com
Insulin-like growth factors (IGFs) are both ubiquitous in the expression of their ligands, receptors, and binding proteins and varied in their cellular functions. During development they function as growth factors, influencing the size of the various tissues as well as the differentiation of specialised cells. In the adult organism their function is more specialised, as determined by the differentiation of the particular tissue.
IGFs have long been known to affect cellular proliferation by stimulating cell cycle progression and to protect cells from premature death by their antiapoptotic effects. IGFs working through their cognate receptor, the IGF-1 receptor (IGF-1R), are not necessarily powerful mitogens when compared with other growth factors such as fibroblast growth factor, vascular endothelial growth factor, and platelet derived growth factor, to name but a few examples. On the other hand, they often synergise with these growth factors to produce an enhanced mitogenic response in a particular cell.
Their role in the process of tumour growth has received much attention recently with the finding that raised circulating IGF-1 concentrations correlate very closely with the relative risk for the development of several common cancers including cancers of the breast,1 prostate,2 colon3, 4 and lung.5 This led investigators to re-examine the role of the IGFs, their receptors, and binding proteins in cancer development and growth.
In vitro systems have shown that activation of the IGF-1R signalling cascades counteracts the apoptotic effects of various cancer treatments.6 Interruption of the IGF-1/IGF-1R pathway results in reduced growth of breast cancer cell lines.7, 8 Finally, overexpression of the IGF-1R in oestrogen receptor (ER) positive breast cancer cell lines may result in enhanced cellular proliferation in an oestrogen independent manner.9
In this review, we will examine the effect of IGFs on breast cancer and in particular the potentiation of two different signalling pathways, the IGF-1R and the ER pathways. This is particularly relevant to breast tissue and breast cancer. Although there is no evidence to suggest that either hormonal system initiates cancer, there is growing evidence of an effect of these hormonal pathways on cancer progression.
IGF-1R signalling
The widely expressed IGF-1R is a tyrosine kinase receptor that is activated after ligand binding. After autophosphorylation, it binds several adapter molecules, such as the insulin receptor substrates (IRS1–4) and Shc, which are in turn phosphorylated on tyrosine molecules.10 These docking molecules are then capable of binding proteins that contain SH2 domains through an interaction with the phosphotyrosine motifs. IRS molecules bind the p85 regulatory subunit of phosphoinositide 3′ kinase (PI3′-K), which leads to the activation of the p110 catalytic subunit. Activation of PI3′-'K leads to the activation of protein kinase B (Akt), a pathway known to prevent apoptosis and to stimulate cellular proliferation.11 Tyrosine phosphorylated Shc binds Grb2/mSos, activating the Ras/Raf/mitogen activated protein kinase (MAPK) pathway, which is also active in mitogenesis.12 Although these two well established pathways have different downstream effectors, they do not act independently. For example, IRS proteins also bind Grb2, thereby activating the MAPK pathway. Furthermore, each pathway can interact with the other in certain cell types.
The process of tumorigenesis is considered to be the result of dysregulation of the cell cycle machinery. Cell division is a process used by all eukaryotic cells to control growth.13 The cell cycle is described in four distinct stages: G1, S, and G2, where RNA and protein synthesis occurs, and an M phase where the cell undergoes mitosis and divides into two daughter cells (fig 1). In the G1 phase, cells monitor their environment to determine whether to divide or to remain in a state of quiescence (known as G0). Considerable progress has been made in understanding the molecules and mechanisms that control and coordinate cell cycle progression. As cells emerge from quiescence in response to mitogenic stimuli, the synthesis of D-type cyclins is induced. Once synthesised, the D-type cyclins associate with the cyclin dependent kinase (CDKs), CDK4 and CDK6. The cyclin D–CDK4/6 complexes phosphorylate the retinoblastoma (RB) protein, which is normally found in a hypophosphorylated form bound to the transcription factor, E2F. Phosphorylation of RB (shown as P) leads to its dissociation from E2F and the induction of genes necessary for S phase progression (for example the genes encoding cyclins A and E). The activity of the CDKs is regulated by changes in cyclin concentrations, interaction with CDK inhibitors (CDKIs), and by regulatory phosphorylation.14 The CDKI molecules include two families, the CDK2 interacting protein (CIP)/CDK inhibitory protein (KIP) family (p21WAF-1/CIP, p27KIP1, and p57KIP2), members of which inhibit the formation of cyclin D–CDK4/6 and cyclin E–CDK2 complexes, and the inhibitors of CDK4 (INK4) family (p15INK4b, p16INK4a, p18INK4c, and p19INK4d), which inhibit the cyclin D–CDK4/6 complexes only.15, 16 Because of their inhibitory activity on cell cycle progression, CDKIs are considered to be potential tumour suppressor genes. The importance of the various cyclins and CDKI molecules in controlling normal cellular growth and differentiation has been demonstrated in vitro and in vivo in a series of overexpression and knockout studies. In vitro, in MCF-7 cells, microinjection of anti-cyclin D1 antibody resulted in a pronounced reduction of entry into the S phase of the cell cycle in response to oestrogen or mitogens.17 In vivo, cyclin D1 knockout mice display a pronounced defect in breast epithelial development during pregnancy,18 and the overexpression of cyclin D1 specifically in the mammary gland in mice results in mammary hyperplasia and adenocarcinoma,19 suggesting that cyclin D1 plays an important role in regulating the proliferation of breast cells.20 In addition, mice carrying a targeted deletion of the Ink4a (p16) gene are viable but develop spontaneous tumours at an early stage,21 and mice that carry a deficiency in the Kip 1 (p27) gene are larger than their wild-type littermates and have a proportional enlargement of many organs of the body.22 Not surprisingly, IGFs, like other growth factors, have been shown to regulate both the expression and activity of many proteins involved in cell cycle progression. In Rat L6E9 skeletal muscle cells, IGF-1 treatment upregulates the expression of the CDK4 and cyclin D1 genes and increases RB protein phosphorylation.23 In MCF-7 cells, IGF-1 induces cyclin D1 expression and hyperphosphorylation of RB through the PI3′-K pathway and not the MAPK/MAPK kinase (MEK-1) route.24 IGF-1 has also been shown to regulate inhibitory pathways of cell cycle progression. For example, in cardiomyocytes overexpressing E2F, IGF-1 induces a specific downregulation of total p21 and p27 protein concentrations and their dissociation from CDKs and, consequently, the activation of CDK2/4 and CDK6 and release from cell cycle arrest.25 In melanoma cells, Kanter-Lewensohn et al have shown that IGF-1R is involved in the redistribution of p27, which could be a mechanism for growth arrest.26 It has also been shown that p21 expression is increased by growth factors such as IGF-1.24, 27 This suggests that p21 might provide a positive rather than a negative stimulus to passage through the cell cycle. This is consistent with the earlier report of Zhang et al that p21 containing cyclin–CDK complexes exist in both catalytically active and inactive forms in untransformed cells.28
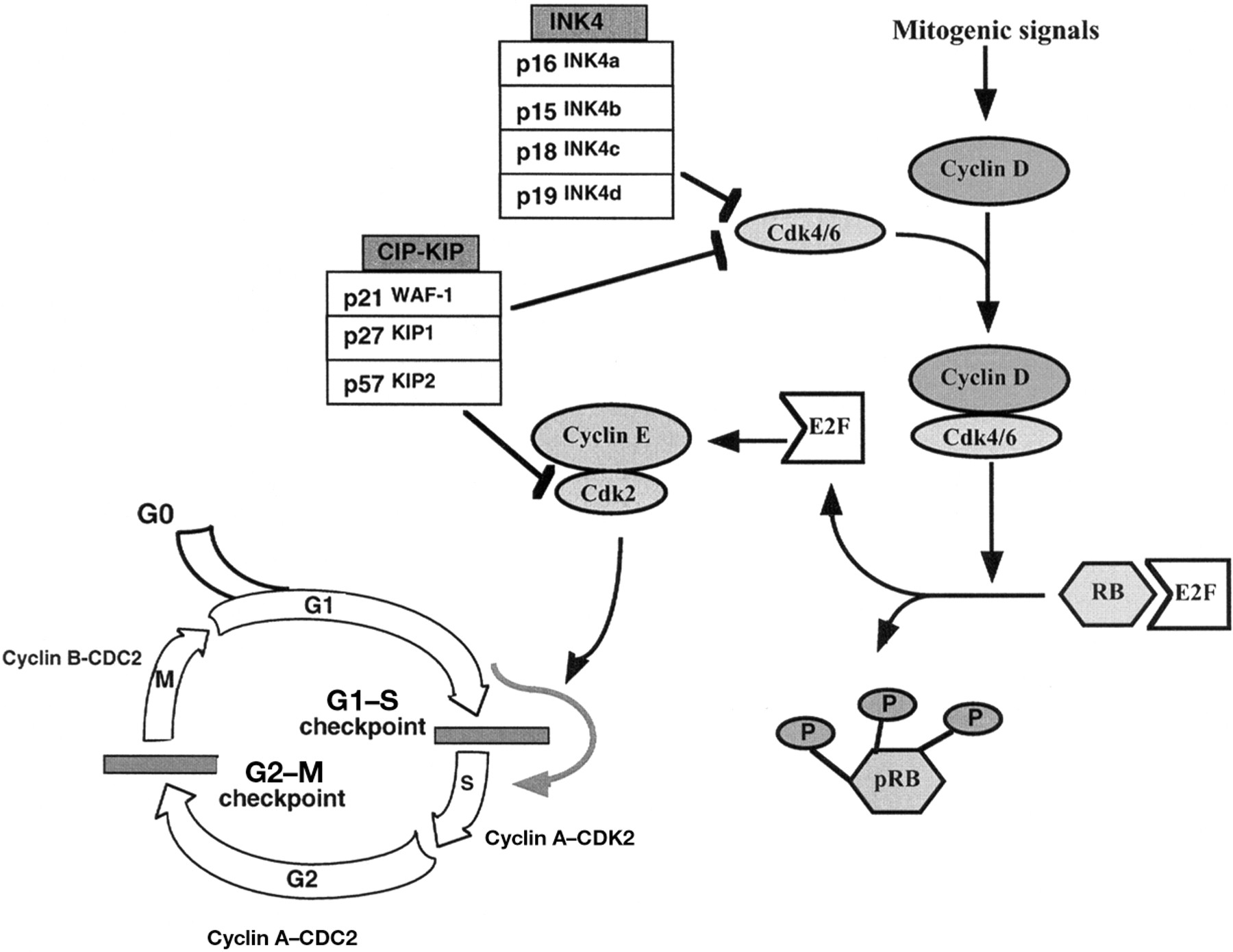
Schematic representation of the cell cycle. G0, M, G1, S, and G2 refer to the quiescent, mitosis, first gap, DNA synthesis, and second gap phases of the cell cycle, respectively. The two checkpoints (G1–S and G2–M) are shown. The decision to replicate is made at a point during G1 referred to as the G1–S checkpoint or restriction point. Progression up to and through this point in the cell cycle is usually driven by mitogenic growth factors that push the cell into a proliferative mode. RB and pRB represent the unphosphorylated and hyperphosphorylated forms of the retinoblastoma protein.
Oestrogen receptor signalling
Oestrogens mediate their activity through binding to a specific intranuclear receptor protein, the ER, encoded by two genes: ERα and Erβ.29, 30 The ERs are members of the steroid, thyroid, retinoic family of receptors, and have traditionally been thought to act primarily by binding to DNA, via cis elements, to regulate gene transcription. The genes that respond to the ER are numerous and participate in the growth and development of oestrogen sensitive tissues, such as breast tissue and the uterus.31 Although this mechanism is well established, there is now convincing evidence that these receptors may also have non-nuclear mechanisms of action, either at the plasma membrane, like tyrosine kinase receptors or G protein coupled receptors, or within the cytoplasm.32 Thus, in human endothelial cells, E2 induces rapid (within minutes) release of nitric oxide (NO) and the activation of guanylate cyclase and MAPK, which might be mediated by ERs at the cell surface.33 In the same cells, E2 induces the activation of endothelial NO synthase (eNOS) through an Akt dependent mechanism, which is mediated by ERα via a non-genomic effect.34 Moreover, Simoncini et al showed that E2 acting through ERα activates the PI3′-K pathway and eNOS activity independently of gene transcription.35 Several studies have also suggested that oestrogens in varying doses might potentiate the acetylcholine response in large vessels within a non-genomic time frame.36, 37 Finally, in breast cancer cells, E2 modulates apoptosis through a non-genomic action involving phosphotyrosine activation.38 The mechanism(s) involved are as yet undefined but could involve interaction with and activation of plasma membrane receptors.
Potentiation of the effects of IGF-1 and E2 on MCF-7 cell proliferation
To study the potentiation of cellular proliferation by the IGF-1R and ER signalling pathways, we used ER positive MCF-7 breast cancer derived cells as a model. In addition, we performed similar studies on an MCF-7 derived cell line that was engineered to express an antisense RNA directed towards to IGF-1R, thereby reducing the expression of the IGF-1R by approximately 50%.39 These cells (named SX-13) enabled us to determine the relative strength of the IGF-1R signalling pathways.
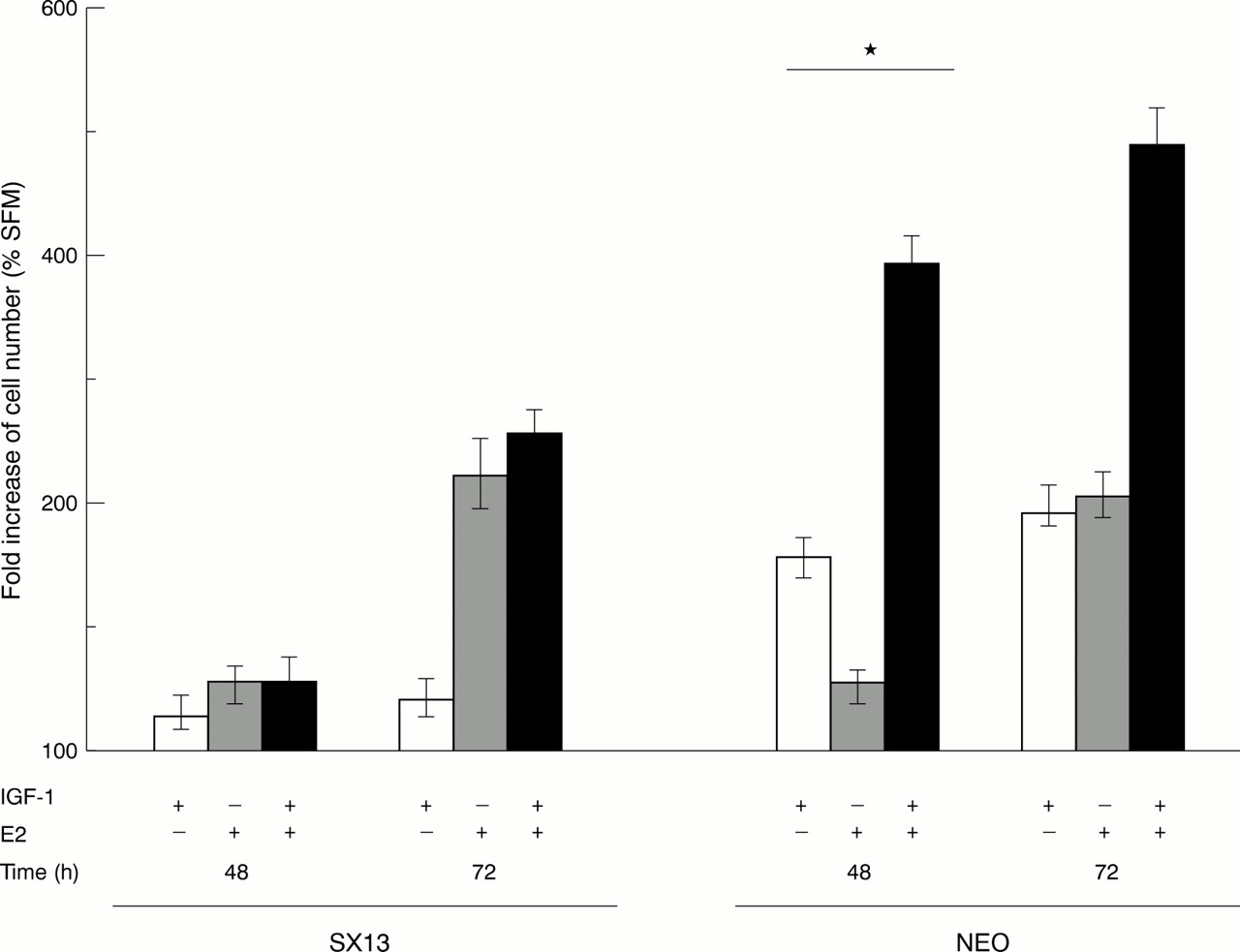
Cells were synchronised in G0 phase by culturing in the presence of the anti-oestrogen ICI 182,780 using serum free, phenol free, oestrogen stripped medium. After stimulation with IGF-1 (1 nM), or oestradiol (10 nM), or a combination of both, cell proliferation was measured indirectly at 48 and 72 hours using the MTT assay. In MCF-7 cells, after 48 hours of stimulation, IGF-1 caused an approximately 1.7 fold increase in cellular proliferation, whereas E2 alone induced only an approximately 1.3 times greater response. In contrast, the combination of IGF-1 and E2 induced a four to fivefold increase. In cells with a reduced number of IGF-1 receptors (SX-13 cells) this effect was absent. Thus, we interpret these results as suggesting that E2 potentiates the effect of IGF-1 on cellular proliferation (fig 2).

Abrogation of the synergistic effect of E2 and insulin-like growth factor 1 (IGF-1) on cell growth in SX13 cells. MCF-7 cells were maintained in SFM (serum free medium) in the absence or presence of IGF-1 (1 nM), E2 (10 nM), or a combination of these two mitogens for three days. Cell number was determined indirectly each day using the colorimetric MTT method. Results are expressed as the mean ± SD of percentage of cell number increase as compared with cells maintained in SFM. The results are obtained from three independent experiments using five determinations in each experiment for each condition. *p < 0.05 indicates a significant synergistic effect between IGF-1 and E2.27
Mechanisms of the potentiation of IGF-1 by E2
PROXIMAL SIGNALLING MECHANISMS
The potentiation of the effects of IGF-1 by E2 was studied by looking at the components of the PI3′-K pathway and the MAPK pathway. Cells were treated for 48 hours with E2 and with IGF-1 for five minutes. IGF-1R expression was unaffected by the E2 treatment. This result, which differs from the results obtained by other investigators,40, 41 could be explained by the use of the E2 antagonist, ICI 182,780 to synchronise the cells in G0 phase. However, IGF-1 induced tyrosine phosphorylation of IGF-1R was enhanced by E2 treatment of the MCF-7 cells. IRS-1 protein values were increased by E2, as was the IGF-1 induced tyrosine phosphorylation of IRS-1. PI3′-K activity and Akt phosphorylation after IGF-1R activation were also enhanced by E2 treatment of the cells; E2 alone had no effect on this process. Thus, the effect of E2 is seen as a potentiation of the response to IGF-1. Similar effects were seen with Erk1 and Erk2, where E2 potentiated the IGF-1 induced phosphorylation of these proteins (fig 3A and B).

Effects of E2 and insulin-like growth factor 1 (IGF-1) on the expression and phosphorylation of (A) the IGF-1 receptor β subunit (βIGF-1R), insulin receptor substrate 1 (IRS-1), and Erk1/2 and (B) the phosphoinositide 31 (PI31) kinase pathways. (A) NEO and SX13 cells were stimulated with IGF-1 (1 nM) for five minutes, E2 (10 nM) for 48 hours, or with both agents. Cells were lysed, proteins separated on sodium dodecyl sulphate polyacrylamide gel electrophoresis and immunoblotted with antibodies to phosphotyrosine (PY20, panels 1 and 3), IGF-1R (panel 2), IRS-1 (panel 4), phospho-Erk1/2 (panel 5), and Erk1 (panel 6, this antibody crossreacts to a lesser extent with Erk2). (B) Cell lysates were immunoprecipitated with αIRS-1 and the PI3′ kinase activity associated with IRS-1 was measured (upper panel). The lower panel represents the protein and serine 473 phosphorylation of Akt in stimulated NEO and SX13 cells (adapted from Dupont and colleagues27).
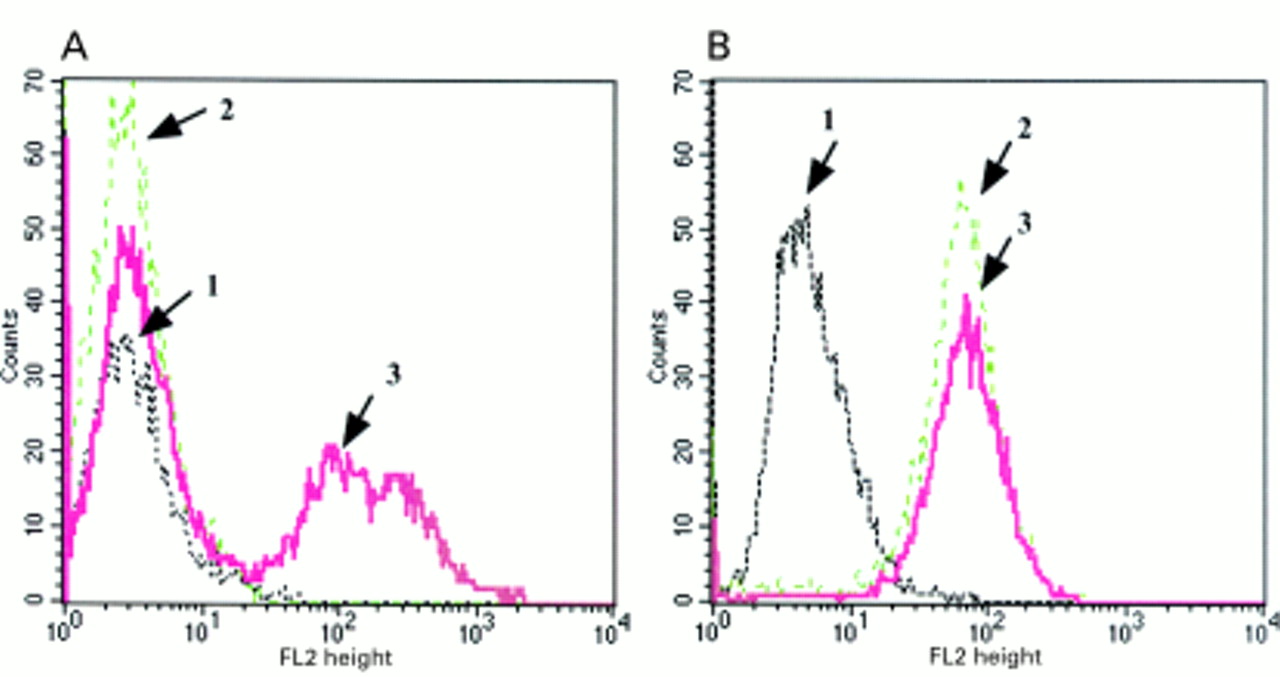
In SX13 cells, stably expressing the IGF-1R antisense construct, there was a pronounced reduction in all the IGF-1 induced signalling events described above. However, in the presence of E2 there was an increase in these responses (fig 3A and B). Fluorescence activated cell sorter analysis demonstrated that SX13 cells have a greatly reduced cell surface expression of the IGF-1R, despite only a 50% reduction in total IGF-1R values when compared with control cells (NEO, MCF-7 cells transfected with the empty vector; fig 4). Interestingly, with E2 treatment of the SX-13 cells, IGF-1R surface expression was significantly increased and could be the explanation for the induction of IGF-1 responsiveness in these cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Expression of surface insulin-like growth factor 1 receptors (IGF-1Rs) in SX13 and NEO cells in response to E2 treatment. Both cell lines were synchronised in G0 phase by serum deprivation and using the anti-oestrogen ICI 182,780 and then maintained in serum free medium in the absence or presence of E2 (10 nM) for 48 hours. Cells were trypsinised and their surface IGF-1R analysed by flow cytometry. In both panels, 1 represents the isotype control (non-specific), 2 represents unstimulated SX13 or NEO cells (starved for 48 hours), and 3 represents SX13 or NEO cells treated with E2 for 48 hours.27
DISTAL SIGNALLING MECHANISMS: CELL CYCLE COMPONENTS
Both IGF-1 and E2 induced the expression of cyclin D1 and cyclin E in MCF-7 cells; once again, co-exposure to both compounds resulted in the potentiation of expression of the cyclin D1 gene, whereas the effect on cyclin E was simply additive. Potentiation of the phosphorylation of RB was also seen. In SX-13 cells, the effects of E2 were similar to those seen in MCF-7 cells, whereas the effects of IGF-1 were reduced, and the combination of both agents was additive. Thus, the potentiation of IGF-1 induced effects by E2 was also observed in several components of the cell cycle and was dependent on the degree of expression of the IGF-1R. We also studied the effect of IGF-1 and E2 on two CDKIs, p21 and p27. In MCF-7 cells, both IGF-1 and E2 reduced the expression of p27, thereby allowing cell cycle progression. E2 had its effects by inhibiting gene transcription, whereas the effects of IGF-1 were post-transcriptional. Our data suggest the possibility that IGF-1 might induce the ubiquitination of p27. Indeed, several studies have shown that the p27 protein can undergo degradation by the ubiquitin–proteasome pathway.42 Surprisingly, p21 expression was enhanced by IGF-1, as seen by an increase in p21 mRNA values. Thus, p21 might act as an inducer of cell cycle progression, rather than as an inhibitor. This has been suggested previously and was explained by the fact that low amounts of p21 might promote the assembly of active kinase complexes, whereas at higher concentrations it may inhibit CDK activity.43 To confirm the effect of p21 on the IGF-1 biological response, we stably transfected a p21 antisense construct into MCF-7 cells, and demonstrated that a reduction in p21 protein concentrations significantly inhibited IGF-1 induced cell proliferation (J Dupont et al, 2001, unpublished data).
Conclusion
The role of the IGF-1R in cellular proliferation is gradually becoming clearer after many years of work by numerous investigators. What is also becoming obvious is that the effects of growth factors such IGF-1 are not a simple linear cascade of events within the cell. The IGF-1R activates several pathways, all of which contribute to the final biological outcome for that cell. These cascades interact with each other, often potentiating one another. Similarly, IGF-1R induced intracellular events are not isolated phenomena; they are greatly affected by many other signalling events occurring concurrently within the cell. Thus, “crosstalk” between these cascades has become a topic of current research by many investigators.
In our study we, like other groups, have investigated the crosstalk between a classic growth factor activating a tyrosine kinase receptor and a classic steroid receptor pathway. Because, in general, our studies have been performed after two to three days incubation with E2, we have interpreted our studies to reflect genomic responses to the oestradiol receptor, a classic concept. On the other hand, we are aware of the recent evidence for the non-genomic effects of E2 and the oestrogen receptor, which may also be crucial in the crosstalk between these two pathways. Indeed, our current research involves such studies.
In the light of current and past studies, future studies will undoubtedly advance our understanding of the interactions of various hormonal responses of cancer cells and hopefully bring new treatments for common cancers, such as breast cancer.