Article Text

Abstract
Ever since the discovery of insulin and its role in the regulation of glucose metabolism, there has been great interest in the molecule itself, the insulin-like growth factors (IGFs), and their receptors (IR and IGF-R). These receptors form a subfamily of tyrosine kinase receptors which are large, transmembrane proteins consisting of several structural domains. Their ectodomains have a similar arrangement of two homologous domains (L1 and L2) separated by a Cys rich region. The C-terminal half of their ectodomains consists of three fibronectin type 3 repeats, and an insert domain that contains the α–β cleavage site. This review summarises the key developments in the understanding of the structure of this family of receptors and their relation to other multidomain proteins. Data presented will include multiple sequence analyses, single molecule electron microscope images of the IGF-1R, insulin receptor (IR), and IR–Fab complexes, and the three dimensional structure of the first three domains of the IGF-1R determined to 2.6 Å resolution by x ray crystallography. The L domains each adopt a compact shape consisting of a single stranded, right handed β-helix. The Cys rich region is composed of eight disulphide bonded modules, seven of which form a rod shaped domain with modules associated in an unusual manner.
- insulin-like growth factor
- insulin
- receptors
- three dimensional structure
- electron microscopy
Statistics from Altmetric.com
For several years now we have been interested in determining the structure of the insulin-like growth factor receptor (IGF-R) and its complex with ligand to understand the molecular details of IGF induced receptor activation. The subject has been extensively reviewed recently.1 The first evidence for the presence of an IGF-R distinct from the insulin receptor (IR) came in 1974 when 125I labelled insulin and 125I labelled NSILA-s (soluble fraction of non-suppressible insulin-like activity) were used to label distinct proteins in purified rat liver plasma membranes.2, 3 The IGF-1R could be solubilised with non-ionic detergents,4 and was subsequently shown by sodium dodecyl sulphate (SDS) gel electrophoresis to resemble the IR in being a homodimer composed of two α and two β chains held together by disulphide bonds.5–7 The next key discovery was the demonstration that the IGF-1R, like the IR, is a tyrosine kinase that is activated and autophosphorylated after IGF-1 binding.8, 9
The next landmark discovery was the cloning and sequencing of the cDNA for human IR in 1986.10 It encoded a 1367 amino acid precursor, which is cleaved by furin into an α and β chain. The α chain and 195 residues of the β chain comprise the extracellular portion of the IR. There is a single transmembrane sequence and a 408 residue cytoplasmic domain containing the tyrosine kinase. The cDNA for the IR and a third member of the IR subfamily, the insulin receptor related receptor (IRR), have been cloned and sequenced and are similarly organised.11, 12 These receptors are heavily glycosylated. The ligands—insulin and the two IGFs—share a common three dimensional architecture and can bind to each other's receptor in a competitive manner. The IRR ligand is unknown.
The IR subfamily is related to another class of cell surface receptors, the epidermal growth factor receptor (EGFR) subfamily. Like the IR subfamily, EGFR members have a single transmembrane region and a cytoplasmic region containing a tyrosine kinase catalytic domain flanked by regulatory juxtamembrane and C-tail sequences.13 The major feature that separates members of the IR subfamily from most other receptor families is that they exist on the cell surface as disulphide linked dimers and require domain rearrangement on binding ligand, rather than receptor oligomerisation, to initiate signal transduction. The EGFRs do not form disulphide linked dimers and the ligands, such as EGF and transforming growth factor α (TGF-α), are structurally unrelated to insulin and the IGFs.
Sequence analyses and structural predictions
There are three approaches to gaining information about the three dimensional structure of proteins, namely: (1) homology searching for protein domains the structure and functions of which are known, (2) de novo structural predictions using a variety of programs, and (3) direct experimentation. All three have been applied to the insulin receptor family. The first important analysis was the observation that approximately the first 120 residues of the IR (and EGFR) ectodomain upstream of the Cys rich region showed sequence similarity with the 120 residues immediately downstream of the Cys rich region.14 These domains were termed L1 and L2 (where the L stands for large) and were shown to contain four internal repeats in the region equivalent to 1–119 of the human IR.14 The repeats ranged in size from 20–42 residues and included a 13 residue repeating motif X–X-(x)X-G-X(x)X, where X is hydrophobic and (x) is often hydrophobic. In addition, they applied several structural prediction methods to conclude that the repeats were composed of an α helix/β strand/turn/β strand secondary structure, with the conserved G residue being part of the turn.14
These authors also suggested, on the basis of sequence alignments, that the Cys rich regions of the IR and EGFR (∼ 150 amino acid residues with 24–26 Cys residues) consisted of three repeats of eight Cys residues. The spacing between the homologous Cys residues and the nature of the amino acid residues in these loops were variable.14 The report on the three dimensional structure of the tumour necrosis factor receptor p55 (TNFR)15 prompted us to investigate whether this structure might represent the first view of the modules that make up the Cys rich regions of the IR and EGFR.16 The TNFR ectodomain has a similar size and Cys content to the IR and EGFR Cys rich regions, and consists of four repeating modules each containing six Cys residues in Cys1-Cys2 (loop 1), Cys3-Cys5, and Cys4-Cys6 (loop 2) disulphide linkages. The Cys3-Cys5 pair is occasionally missing and additional disulphides are found in some repeats.15, 16 When the SwissProt database was searched with profiles generated from multiple sequence alignments of TNFR repeats the first 14 hits were known members of the TNFR family. Five of the next seven highest ranking alignments were members of the IR family, suggesting that the Cys rich region of the IR might be structurally related to the TNFR. The data allowed the disulphide bonds in the Cys rich regions of the IR and EGFR to be predicted, and indicated that these Cys rich regions were not simple repeats of eight Cys residues, but were more complicated combinations of loop 1-type and loop 2-type disulphide linked modules.16 The validity of these alignments took on greater importance with the publication of the three dimensional structure of three consecutive repeats of laminin.17 Laminins A (18th and 21st) and B (27th and 36th) were very highly ranked in the SwissProt searches with the TNFR profiles,16 and the three dimensional structure was consistent with the predictions of disulphide bonded modules obtained from the profile analyses.
Sequence analyses have also been used to show that the C-terminal half of the IR ectodomain contains three fibronectin type III (FnIII) modules, one of the most common structural modules found in many proteins.18, 19 The FnIII domain is relatively small (∼ 100 residues) and has a fold similar to that of the immunoglobulins but with a distinctive sequence motif. The domain consists of a seven stranded β sandwich in a “three on four” (EBA:GFCC`) topology. Its main functions appear to be to mediate protein–protein interactions, including ligand binding, and to act as spacers for the correct positioning of functionally important regions of extracellular proteins. O'Bryan and colleagues20 were the first to describe the existence of FnIII domains in members of the IR subfamily after their cloning and characterisation of the tyrosine kinase axl. Their sequence alignments and descriptions covered the two C-terminal FnIII domains in the IR subfamily, and these were given structural assignments after comparisons21 with the FnIII modules present in the growth hormone receptor.22 Recently, it has been shown that members of the IR subfamily contain an additional FnIII domain in the region referred to previously as the connecting domain.23–25 This first FnIII domain (equivalent to residues 461–579 in the IGF-1R) is 118–122 residues long, whereas the second FnIII domain (equivalent to 580–798 in the IGF-1R) has a major insert of 120 to 130 amino acids. The third FnIII domain (equivalent to residues 799–901 in the IGF-1R) is of normal size.
Given the extensive scrutiny of the IR family sequences, it is surprising that this third FnIII domain had been missed by us26 and others,21 including the detailed “pastiche” model of the IR/IGF-1R developed27 after the publication of the three dimensional structure of the IR kinase domain.28 Although it was not discussed by O'Bryan et al,20 those authors appear to have identified this additional FnIII domain because it is illustrated unequivocally in their figure 4. However in that paper, the main focus was on comparisons with axl (which contains only two FnIII domains) and other tyrosine kinases, and their sequence alignments were confined to the two C-terminal FnIII repeats.20
Secondary structure
The ectodomain of the IGF-1R contains 41 cysteine residues in each monomer, 38 in the α chain and three in the β chain. Some of the disulphide bonds have been established by chemical analysis. Cys8 is linked to Cys26 in the L1 domain26 and Cys435 is linked to Cys468 in the L2 domain.29 Sequence alignments suggest that the homologous pairs of Cys residues at the end of the L1 domain (Cys126 and Cys155) and the start of the L2 domain (Cys312 and Cys333) are also disulphide bonded,16 as confirmed in the three dimensional structure.30 None of the disulphide bonds in the Cys rich region has been established chemically except for the additional disulphide bond Cys266-Cys274, in the large loop of the sixth Cys rich module of the human IR.31 Cys524, in the first FnIII domain of the human IR, was shown to form a dimer disulphide bond with Cys524 in the second monomer.29 However, mutation of Cys524 to Ala32–34 resulted in an IR that was still dimeric, indicating that more than one disulphide bond is involved in dimer formation. The additional dimer disulphides of IR were shown, by chemical analysis, to involve the triplet of Cys residues at positions 682, 683, and 685 in the insert domain of IR.26 It was not possible to determine whether one or all three of these Cys residues is involved in dimer disulphides. The sequence around this triplet resembles that found in the hinge region of antibodies,35 where multiple disulphide bonds occur. It is interesting to note that the IGF-1R has an additional Cys, seven residues upstream of the Cys triplet in the insert domain, and lacks the Cys residue equivalent to 884 in the human IR.
There is only a single α–β disulphide link between Cys647 in the first FnIII repeat and Cys872 (exon 11+ isoform) in the second FnIII repeat of the human IR,26 which is consistent with the mutagenesis data34, 36 and the predictions of Ward et al.16 It differs from the suggestion made by Schaefer and colleagues21 that the disulphide links between the four β chain Cys residues 798, 807, 872, and 884 (exon 11+ numbering) were similar to those in the growth hormone receptor.22 Such an arrangement would confine the two disulphide bonds to the β chain and would not allow for the presence of the known α–β disulphide linkage. The structural implications of the disulphide bond between Cys647 and Cys872 are that the two FnIII domains are aligned side by side,16 not end to end, as is the more common configuration.19 Finally, there is an intrachain disulphide linkage between the β chain residues Cys798 and Cys807 (exon 11+ numbering) in the predicted F and G strands of the third FnIII domain.26 The remaining Cys residue in human IR, Cys884 (exon 11+ isoform), which has no counterpart in the human IGF-1R or human IRR, has been shown to exist as a buried free thiol.26
Three dimensional structure of the L1/Cys rich/L2 domains of the IGF-1R
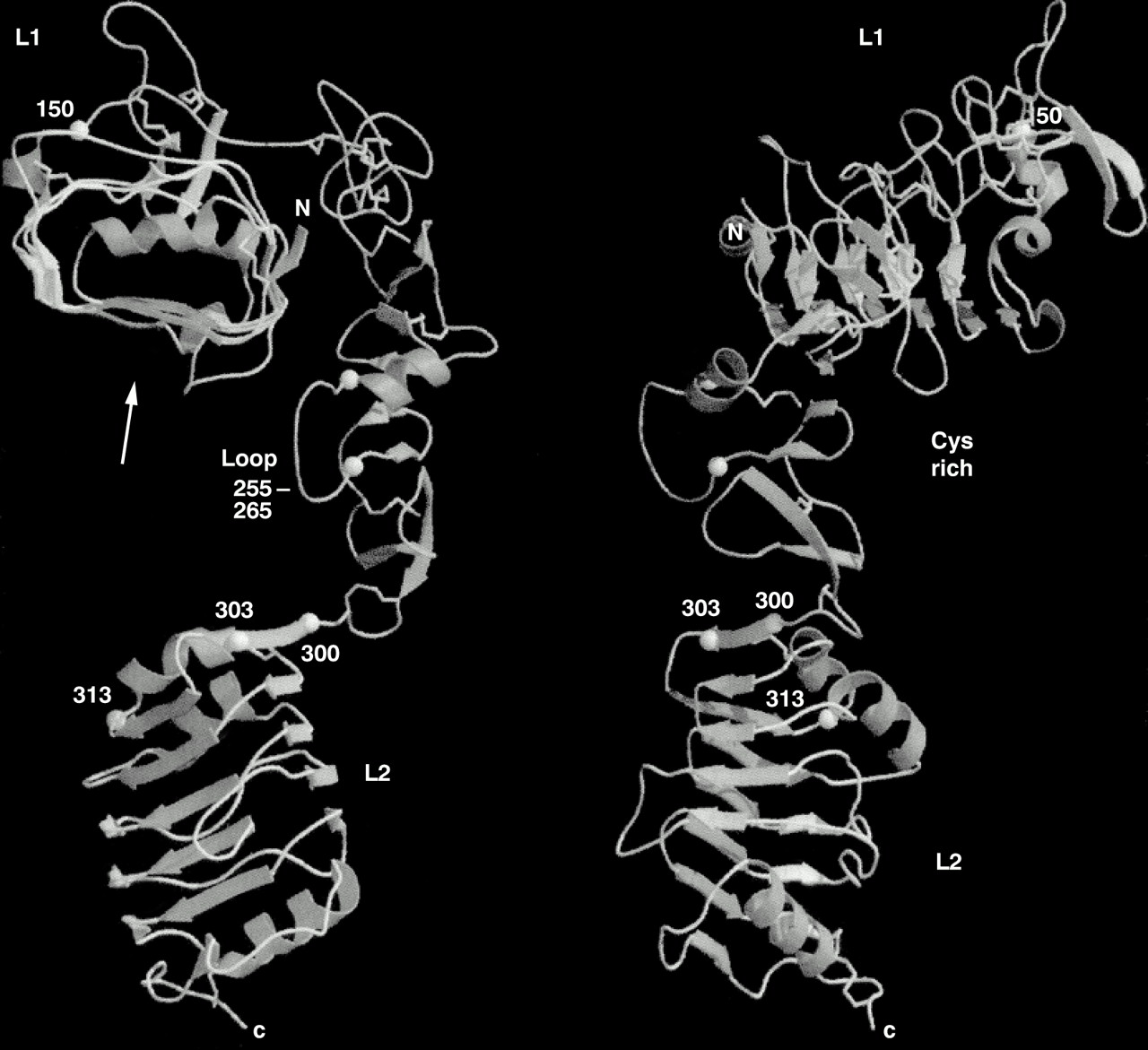
Recently, we solved the three dimensional structure of the L1/Cys rich/L2 domain fragment of the IGF-1R.30 The molecule adopts an extended bilobal structure (∼ 40 × 48 × 105 Å) with the L domains at either end (fig 1). The Cys rich region runs for two thirds of the length of the molecule, making contact along the length of the L1 domain, but having very little contact with the L2 domain. This leaves a space at the centre of the molecule of approximately 24 Å diameter and of sufficient size to accommodate the ligands, IGF-1 or IGF-2. The space is bounded on three sides by the regions of IGF-IR that are known to contribute to ligand binding based on studies of chemical crosslinking, receptor chimaeras, and natural or site specific mutants (see Adams et al and Garrett et al for references1, 30).

Polypeptide fold for residues 1–459 of the human insulin-like growth factor 1 (IGF-1) receptor. In the left hand view, the L1 domain is at the top viewed from the N-terminal end. In the right hand view, the model has been rotated clockwise 90°. Helices are depicted as curled ribbons and β strands as broad arrows. Based on Garrett et al.30
THE L DOMAINS
Each L domain of the human IGF-1R (residues 1–150 and 300–460) adopts a compact shape (∼ 24 × 32 × 37 Å), being formed from a single stranded, right handed β helix, capped on the ends by short α helices and disulphide bonds. The body of each domain looks like a loaf of bread with three flat sides and an irregular top (fig 1). The two domains are superimposable with a root mean squared deviation (rmsd) in position of 1.6 Å for 109 Cα atoms.30 The repetitive nature of the β helix is reflected in the sequence where a fivefold repeat, centred on a conserved glycine, had been identified by sequence analyses.14 The structure, however, revealed that the L domains comprised six helical turns and a fold that was quite unexpected.30 A notable difference between the two domains is found at the C-terminal end. For L1, the indole ring of Trp176 from the Cys rich region is inserted between the last two turns of β helix into the hydrophobic core of the domain, and the C-terminal α-helix of L1 becomes vestigial. The sequence motif of residues that form the Trp pocket in L1 does not occur in L2 of the IR family.30 However, in the EGFR, which has an additional Cys rich region after the L2 domain, the motif can be found in both L domains and the Trp residue is conserved in both Cys rich regions.30
THE CYS RICH DOMAIN
As anticipated from the TNFR profile analyses,16 the Cys rich domain is composed of modules with disulphide bond connectivities resembling parts of the TNFR15 and laminin17 repeats (fig 2). The first module sits at the end of L1, whereas the remaining seven form a curved rod running diagonally across L1 and reaching to L2 (fig 1). The strands in modules 2–7 run roughly perpendicular to the axis of the rod, in a manner more akin to laminin than to the TNF receptor, where the strands run parallel to the axis (fig 2). The modular arrangement of the IGF-1R Cys rich domain is different to other Cys rich proteins for which structures are known (fig 2). The first three modules of the IGF-1R have a common core, containing a pair of disulphide bonds, but show considerable variation in the loops. These modules are referred to here as C2 (two disulphide bonds). The connectivity of the cysteines is the same as the first part of an EGF motif (Cys1–3 and 2–4) but their structures do not appear to be closely related to any member of the EGF family. Modules 4 to 7 have a different motif, a β finger, seen previously in residues 2152–2168 of fibrillin.37 Each is composed of three polypeptide strands, the first and third being disulphide bonded and the second and third forming a β ribbon. These are referred to here as C1, because of the single disulphide bond. The β ribbon of each β finger (or C1) module lines up antiparallel to form a tightly twisted eight stranded β sheet (fig 2). Module 6 deviates from the common pattern, with the first segment being replaced by an α helix, followed by a large loop that is implicated in ligand binding.30 Because modules 4–7 are similar it is possible that they arose from a series of gene duplications. The final module is a disulphide linked bend of five residues.

The first Cys rich region in the EGFR ectodomain has the same eight modules in the same order, C2-C2-C2-C1-C1-C1-C1-C1`, as in the IR family.16, 38 In contrast, the second Cys rich region of the EGFR has the C1 and C2 modules arranged in a different combination [C2-C1-C1]2-C2,16, 38 where the C2-C1-C1 repeat is similar to that seen in laminin17 and the furin-like proteases.16 The fact that the C1 and C2 Cys rich modules are grouped according to type implies that these are the minimal building blocks of the EGF-like Cys rich domains found in many proteins. Although it can be as short as 16 residues, the β finger (C1) motif is clearly distinct and capable of forming a regular extended structure. Thus, Cys rich domains such as laminin17 and fibrillin37 can be thought of not as modified EGF repeats but as a series of repeat units each composed of a small number of C1 and C2 modules. The EGF repeats in proteins such as fibrillin can be viewed as [C2-C1] repeats, whereas the laminin repeat is [C2-C1-C1].
The insulin receptor ectodomain dimer
A remaining issue is how are these various building blocks (domains) organised in the dimeric, native receptor. The first clues have come from single molecule images of the IR ectodomain and its complexes with three Fabs obtained by electron microscopy.39 These images show that the IR ectodomain dimer resembles a U shaped prism of approximate dimensions 90 × 80 × 120 Å. The images show clearly the dimeric structure of the IR ectodomain. Measurement of the images yields a length of approximately 80 Å along each bar, and a width of approximately 90 Å across the two bars. The width of the cleft (assumed to be membrane distal) between the two side arms is approximately 30 Å, sufficient to accommodate ligand.39 Fab molecules from the monoclonal antibody 83-7, which recognises the Cys rich region of the IR, bound half way up one end of each side arm in a diametrically opposite manner, indicating a twofold axis of symmetry normal to the membrane surface.39 Fabs 83-14 and 18-44, which have been mapped to the first FnIII repeat (residues 469–592) and residues 765–770 in the insert domain, respectively, bound near the base of the prism at opposite corners.39 The single molecule images, together with the three dimensional structure of the first three domains of the IGF-R1,30 suggest that the IR dimer is organised into two layers, with the L1/Cys rich/L2 domains occupying the upper (membrane distal) region of the U shaped prism and the fibronectin type III domains, the insert domains (and the disulphide bonds involved in dimer formation) located predominantly in the membrane proximal region.39
High resolution data are required to establish the precise arrangement of the 14 modules that make up the ectodomain dimer and the way they interact with the respective ligands insulin and the IGFs to generate signal transduction. Recently, whole receptors solubilised from human placental membranes have been examined by electron cryomicroscopy and three dimensional reconstruction has been performed using a library of 700 images.40 Gold labelled insulin was used to locate the insulin binding domain. The images seen were compact and globular, measuring 150 Å in diameter. Some domain-like features became evident at intermediate density thresholds, which indicated a strong twofold vertical rotational symmetry. When this symmetry was applied to the reconstruction some structural features became evident. Their overall model showed the L1/Cys rich/L2 domains arranged in an antiparallel manner and at an angle to each other when viewed from the side. The six FnIII domains and the two L2 domains are placed in a central band, with the two tyrosine kinase domains at the base of the model.40 The images described in these studies39, 40 are substantially different from the “T”, “X”, or “Y” shaped objects reported for recombinant ectodomain,21 detergent solubilised, or vesicle reconstituted whole receptors.41–43
Ligand binding
Information on ligand binding has come from chemical crosslinking with derivatised insulin or IGFs and from the analysis of receptor chimaeras and receptors with point mutations (see Adams et al for detailed review1). The results of the studies with IR–IGF-1R chimaeras are summarised (fig 3), and indicate that the determinants of specificity for insulin and IGF-1 binding reside in different regions of the two receptors. In the studies with whole receptor chimaeras,44, 45 three fragments from the N-terminal region of the human IR and human IGF-1R were joined in different combinations in human IR and human IGF-1R backgrounds. The data (fig 3) show that residues 131–315 in the IGF-1R (Cys rich plus flanking regions from L1 and L2) are a prime requirement for IGF-1 binding. In contrast, residues 1–137 in the L1 domain of the human IR and residues 325–524, comprising most of the L2 domain and part of the first fibronectin III domain of the human IR, are important determinants of insulin binding. The importance of the N-terminal region in insulin binding was confirmed by examining a series of IGF-1R based chimaeric ectodomains,46 where the N-terminus contained decreasingly smaller proportions (191, 83 and 68 residues, respectively) of human IR derived sequences. All showed similar binding affinities (fig 3), binding insulin with comparable affinity to the wild-type human IR, while retaining relatively high (10–20%) binding affinity for IGF-1.46, 47

{kind=link}
{kind=link}
{kind=link}
Schematic summary of insulin receptor (IR)–insulin-like growth factor 1 receptor (IGF-1R) chimaeras. The domain organisation of the receptors is shown at the top. The approximate locations of the fragment boundaries exchanged in the whole receptor chimaeras are shown, as are the α–β cleavage sites for the human IR and human IGF-1R. The fragment composition of each chimaera is shown by solid and open boxes. The residues substituted in the ectodomain IR based or IGF-1R based chimaeras are indicated above each construct. The relative binding affinities of the parent receptors and the various chimaeras are shown opposite each construct on the right hand side. Reproduced from Adams et al.1
There are at least two determinants in the N-terminal 68 residues of the human IR responsible for the 200 fold increase in insulin binding because the 1–27 and 28–68 chimaeras displayed eightfold and 20 fold increases in insulin affinity, respectively, compared with IGF-1R.47 Enhanced insulin binding was retained in a chimaera in which only residues 38–43 corresponded to the human IR.47, 48 Residues 38–43 are predicted to lie in the second rung of the L1 β helix domain,30 at the edge of the putative binding pocket (fig 1). The region 223–274 in the IGF-1R, implicated in IGF-1 specificity, contains major sequence differences when compared with the human IR.10, 11 It corresponds to modules 4–6 in the Cys rich region and includes a large and somewhat mobile loop (residues 255–263, mean B(Cα atoms) = 57 Å2), which extends into the central space. In the human IR, this loop is four residues bigger, differs totally in sequence, and is stabilised by an additional disulphide bond.16, 31 The improvement in IGF-1 binding by the human IR Cys loop exchange chimaera, hIR_CLX, suggests that the larger loop of the human IR might exclude IGF-1 from the hormone binding site but allow the smaller insulin molecule to bind.49 It is interesting to note that the mosquito IR homologue, which has a loop two residues larger than the mammalian IRs, also appears to bind insulin but not IGF-1.50 The third region of residues implicated in insulin binding (326–524) starts in the middle of the first rung of the L2 β helix domain30 and extends to Cys514 in the middle of the first FnIII domain.
Because chimaeras are only useful for investigating residues that differ between the two receptors, a more precise analysis of binding determinants can be obtained from single site mutants. Most of these studies have been carried out on the human IR and include analyses of mutant receptors from patients, as well as receptors generated by site specific mutagenesis. The naturally occurring mutations of the human IR fall into five classes: impaired biosynthesis of full length receptor (class 1), impaired transport to the plasma membrane (class 2), decreased binding affinity (class 3), defective tyrosine kinase (class 4), and accelerated receptor degradation (class 5). These mutations have been described in detail.51–54
In Ala replacement studies, four regions of L1 important for insulin binding were identified.55 The first three are at similar positions on successive turns of the β helix and the fourth lies on the conserved bulge on the large β sheet.30 These residues, along with the naturally occurring mutants Asp59 and Leu62,54 form a footprint for insulin binding on the first half of the second β sheet that faces into the central cavity formed by the L1/Cys rich/L2 domains. Residues further along the sheet, which are conserved in the IGF-1R and were not subjected to Ala mutagensis, could also be important.30 The two leprechaun associated mutations, Ile119Met and the Lys121 deletion, involve residues in a similar location on the fifth rung of the L1 β helix.
Ala scanning mutagenesis has been carried out in the L1 domain of the IGF-1R57 at residues equivalent to 10 of the 14 amino acids identified as important contributors to insulin binding in the human IR.55 Mutations of Asp8, Asn11, and Phe58 in the human IGF-1R resulted in a three to four fold reduction in affinity for IGF-1, whereas the other seven mutations had no effect.56 This is in contrast to the effects of these mutations on insulin binding to the human IR.55 Of the 10 IGF-1R mutations, only one, Arg10Ala, had a significant effect on insulin binding as measured by insulin displacement of labelled IGF-1 or labelled insulin super analogue X92.56