Article Text

Abstract
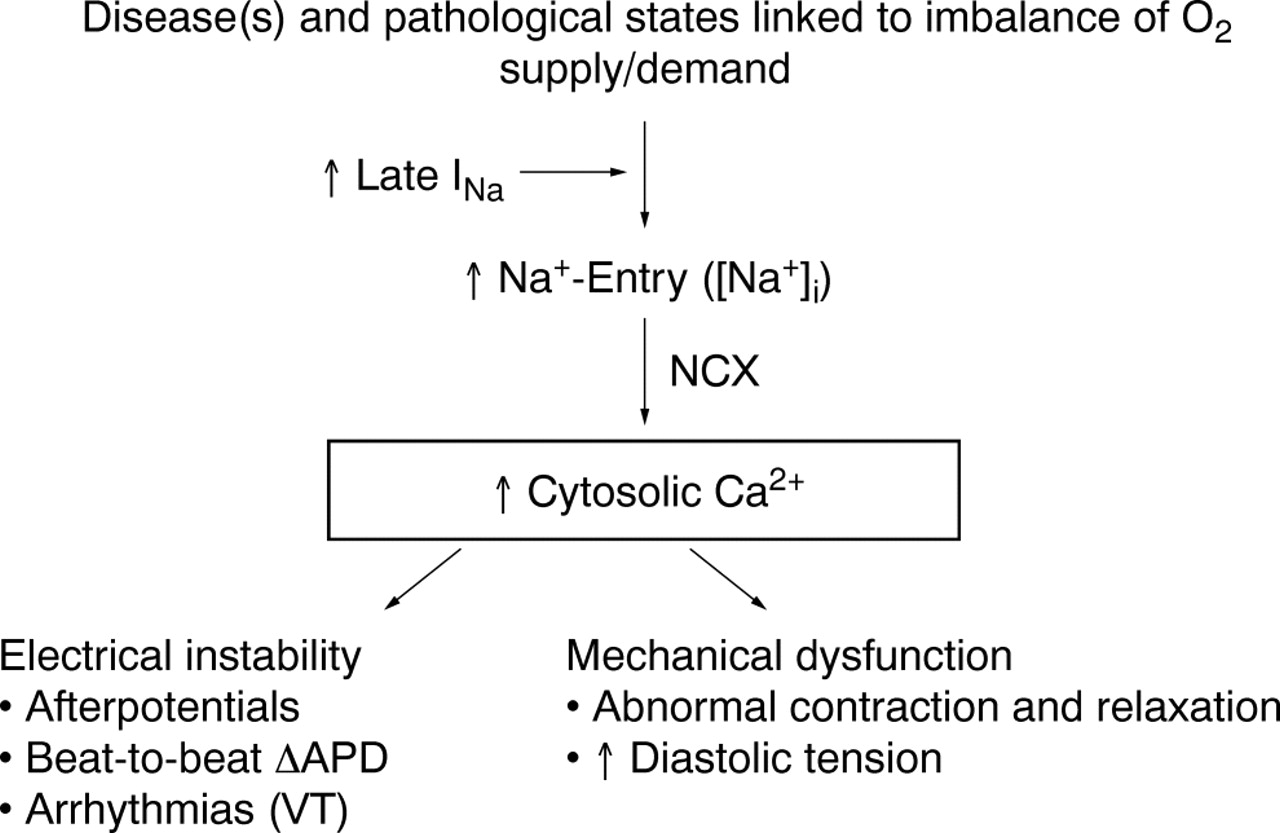
Pathological conditions linked to imbalances in oxygen supply and demand (for example, ischaemia, hypoxia and heart failure) are associated with disruptions in intracellular sodium ([Na+]i) and calcium ([Ca2+]i) concentration homeostasis of myocardial cells. A decreased efflux or increased influx of sodium may cause cellular sodium overload. Sodium overload is followed by an increased influx of calcium through sodium-calcium exchange. Failure to maintain the homeostasis of [Na+]i and [Ca2+]i leads to electrical instability (arrhythmias), mechanical dysfunction (reduced contractility and increased diastolic tension) and mitochondrial dysfunction. These events increase ATP hydrolysis and decrease ATP formation and, if left uncorrected, they cause cell injury and death. The relative contributions of various pathways (sodium channels, exchangers and transporters) to the rise in [Na+]i remain a matter of debate. Nevertheless, both the sodium-hydrogen exchanger and abnormal sodium channel conductance (that is, increased late sodium current (INa)) are likely to contribute to the rise in [Na+]i. The focus of this review is on the role of the late (sustained/persistent) INa in the ionic disturbances associated with ischaemia/hypoxia and heart failure, the consequences of these ionic disturbances, and the cardioprotective effects of the antianginal and anti-ischaemic drug ranolazine. Ranolazine selectively inhibits late INa, reduces [Na+]i-dependent calcium overload and attenuates the abnormalities of ventricular repolarisation and contractility that are associated with ischaemia/reperfusion and heart failure. Thus, inhibition of late INa can reduce [Na+]i-dependent calcium overload and its detrimental effects on myocardial function.
- APD, action potential duration
- [Ca2+]i, intracellular calcium concentration
- EAD, early afterdepolarisation
- ICa, L, L-type calcium current
- IKr, potassium rapid delayed-rectifier current
- INa, sodium current
- INa, Ca, sodium-calcium exchange current
- [Na+]i, intracellular sodium concentration
- LV, left ventricular
- NCX, sodium-calcium exchanger
- NHE, sodium-hydrogen exchanger
- +Vmax, maximum upstroke velocity of phase 0 of the action potential
Statistics from Altmetric.com
- APD, action potential duration
- [Ca2+]i, intracellular calcium concentration
- EAD, early afterdepolarisation
- ICa, L, L-type calcium current
- IKr, potassium rapid delayed-rectifier current
- INa, sodium current
- INa, Ca, sodium-calcium exchange current
- [Na+]i, intracellular sodium concentration
- LV, left ventricular
- NCX, sodium-calcium exchanger
- NHE, sodium-hydrogen exchanger
- +Vmax, maximum upstroke velocity of phase 0 of the action potential
Cardiac function is dependent on homeostasis of the intracellular concentrations of sodium ([Na+]i) and calcium ([Ca2+]i). Pathological conditions such as ischaemia and heart failure are often associated with changes of intracellular concentrations of these ions and subsequent mechanical dysfunction (fig 1).1 An increase of [Na+]i may be the first step in disruption of cellular ionic homeostasis. This step may be followed by increases in sodium–calcium exchange, cellular uptake of calcium, and excessive calcium loading of the sarcoplasmic reticulum.2 Calcium overload of myocardial cells is associated with electrical instability, increased diastolic and reduced systolic force generation, and an increase in oxygen consumption.3 At the same time, the increase of diastolic force causes vascular compression and reduces blood flow and oxygen delivery to myocardium.4 Calcium overload may lead to cell injury and death if it is not corrected.
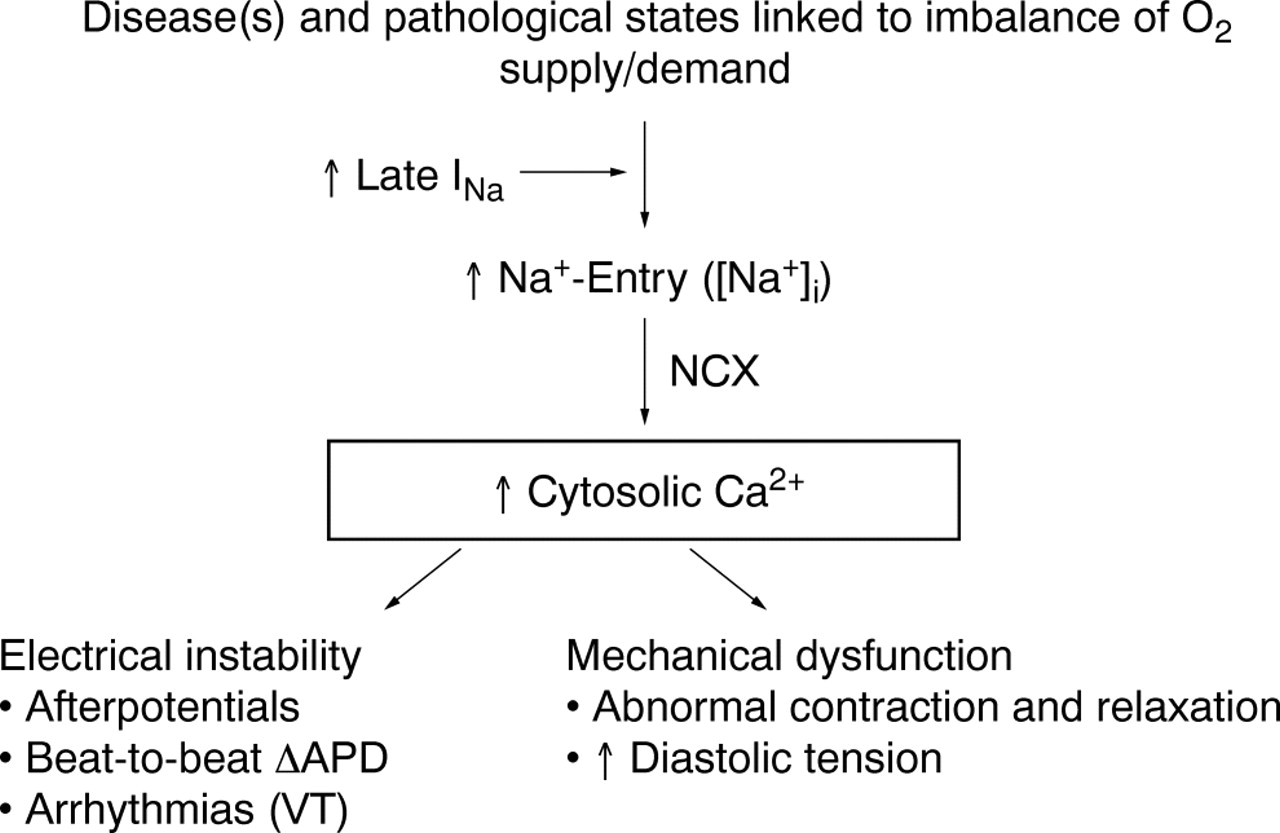
Increase in intracellular sodium concentration ([Na+]i) in pathological conditions linked to imbalances between oxygen supply and demand causes calcium entry through the Na+/Ca2+ exchanger (NCX). A pathologically enhanced late sodium current (INa) contributes to [Na+]i-dependent calcium overload, leading to electrical instability and mechanical dysfunction. APD, action potential duration; VT, ventricular tachycardia.
Cellular sodium and calcium homeostasis is maintained by ion channels, pumps and exchangers. This review summarises the cell processes involved in cardiac sodium and calcium homeostasis, the pathophysiology causing disruption of this homeostasis and the benefits of inhibiting the persistent or “late” sodium current (INa) to maintain ionic homeostasis and reduce cardiac dysfunction. Sodium and calcium ion channels, pumps and exchangers are prime targets for drugs intended to reduce [Na+]i, calcium overload and cardiac dysfunction. In the second half of this review we describe the cardioprotective effects of ranolazine, a selective inhibitor of late INa that is in clinical development for the treatment of angina pectoris.
SODIUM HOMEOSTASIS IN CARDIAC MYOCYTES
Homeostasis of [Na+]i is the result of a balance between the influx and efflux of sodium ions. Sodium influx and efflux occur by multiple pathways (table 1), many of which are subject to regulation.5 Depending on species and conditions, [Na+]i varies from 4–16 mmol/l in normal cardiomyocytes.5 The extracellular sodium concentration is about 140 mmol/l. The large transmembrane sodium concentration gradient, in conjunction with a negative resting membrane potential of about −90 mV, results in a substantial electrochemical gradient that favours sodium influx across the cell membrane. When sodium influx exceeds efflux, [Na+]i rises.
Main pathways involved in the regulation of intracellular sodium concentration ([Na+]i) in cardiomyocytes
There is general agreement that [Na+]i is raised in some pathological conditions, including cardiac ischaemia and reperfusion, hypertrophy and heart failure.6,7 The rise in [Na+]i during ischaemia and reperfusion appears to involve one or more of the following mechanisms: increased sodium-hydrogen exchange,7,8,9,10 decreased activity of the sodium-potassium ATPase (sodium pump)11 and increased sodium entry through sodium channels.7,8,9,10 The contributions of the sodium-bicarbonate co-transporter, the sodium-potassium-chloride co-transporter and the sodium-magnesium antiporter to the increase of [Na+]i during ischaemia and reperfusion appear to be minimal but have not been thoroughly investigated.5
Sodium-hydrogen exchanger
The sodium-hydrogen exchanger (NHE) is a proton (H+) extruder and an important regulator of intracellular pH and cell volume.12 NHE facilitates an electroneutral, passive exchange of intracellular hydrogen for extracellular sodium. The large transmembrane electrochemical potential gradient for sodium provides the energy that drives proton extrusion. The activity of NHE is very sensitive to small changes in the intracellular proton concentration, being low at the normal intracellular pH of 7.2 but greatly increased when intracellular pH decreases.13 Extracellular acidosis (that is, increased extracellular proton concentration) decreases the concentration gradient favouring hydrogen efflux and inhibits proton extrusion through NHE.
The relative contribution of NHE to total sodium influx at normal intracellular pH in stimulated (1 Hz) rabbit ventricular myocytes is < 5%.5 During acidosis (intracellular pH ⩽ 6.9), however, sodium influx through NHE may be as much as 39% of total sodium influx.5 Cellular depletion of ATP14 and extracellular acidosis are associated with a reduction of NHE. Regardless, inhibition of NHE during an ischaemic episode attenuates the ischaemia-induced rise in [Na+]i,10,15 suggesting that NHE is active during ischaemia. On reperfusion following ischaemia, extracellular pH rises towards normal and NHE increases, further loading the myocardial cell with sodium.9 The NHE-mediated increases of sodium influx and [Na+]i during reperfusion lead to increases in reverse mode sodium-calcium exchange and influx of calcium (see below). This series of events may explain the effect of the NHE inhibitors to protect against reperfusion injury.10
Sodium-potassium ATPase
The sarcolemmal sodium-potassium ATPase ion pump creates and maintains the differences in the extracellular and intracellular concentrations of sodium and potassium that are necessary for cell function.16 The free energy of hydrolysis of ATP is used by the ATPase to translocate three sodium ions from the cell cytoplasm to the extracellular fluid in exchange for two potassium ions that move in the opposite direction. This coupled transport is electrogenic and creates a small outward (repolarising) current. An increase of the [Na+]i increases the activity of the pump. Pump activity is decreased when the free energy of hydrolysis of ATP is less than that needed for ion transport. This may occur during prolonged, severe ischaemia17 when the intracellular concentration of ATP falls and concentrations of ADP and inorganic phosphate increase. The contribution of reduced sodium-potassium ATPase activity during mild ischaemia to increased [Na+]i may not be significant, however,9 and the activity of the sodium pump is not decreased but may be increased in heart failure.18
Sodium channel currents
Voltage-gated sodium channels are transiently activated on depolarisation of the cardiac cell membrane. The INa flowing through these channels is responsible for the upstroke (phase 0) of the action potential.19 It has been estimated that influx of sodium through sodium channels is about 19% of total sodium entry into rabbit ventricular myocytes when myocytes are stimulated at a rate of 1 Hz.5 Depolarisation of the cell membrane leads to a rapid increase of INa that lasts for a few milliseconds before sodium channels inactivate (that is, close). Recovery of each channel from inactivation requires repolarisation of the cell membrane and a change of the conformation of the channel from an inactivated to a resting closed state.
Although most sodium channels are inactivated within a few milliseconds and remain closed and non-conducting throughout the plateau phase of the cardiac action potential, a small percentage of channels either do not close, or close and then reopen.20–22 These channels may continue to open and close spontaneously during the action potential plateau for reasons that are not understood. The late channel openings allow a sustained current of sodium ions to enter myocardial cells throughout systole.23,24 This current has been referred to as late, sustained or persistent to distinguish it from the peak or transient INa. The amplitude of late INa is less than 1% of peak INa25 but it is sufficient to prolong action potential duration (APD).26 Thus, although the amplitude of late INa is small, because it persists for hundreds of milliseconds the influx of sodium by this mechanism may be substantial.2 The magnitude of late INa in canine mid-myocardial cells and Purkinje fibres is greater than that in either epicardial or endocardial myocytes (but see Noble and Noble2).27 The duration of action potentials in mid-myocardial cells and Purkinje fibres is also longer than in other cells of the heart.28 A role for late INa as a contributor to prolongation of APD is suggested by observations that lidocaine and tetrodotoxin shorten APDs of Purkinje fibres and ventricular mid-myocardial myocytes.26 Furthermore, transmural heterogeneity in the magnitude of late INa can cause transmural heterogeneity of APD that may have pathophysiological significance as a mechanism of ventricular tachyarrhythmias such as torsade de pointes.29
In recent years it has become clear that several experimental and pathological conditions can significantly increase the late component of the sodium channel current.30,31 These conditions (table 232–40) include: (1) exposure of myocytes to peptides (for example, the sea anemone toxin, ATX-II),41 chemicals (for example, veratridine)42 and oxygen free radicals that slow the rate of inactivation of the sodium channel; (2) mutations in the sodium channel gene SCN5A that are associated with the long QT-3 syndrome43; and (3) disease states such as hypoxia, heart failure and post-myocardial infarction. It has been shown that hypoxia increases the amplitude of “persistent tetrodotoxin-sensitive sodium currents” (that is, late INa) in rat ventricular myocytes.32 The hypoxia-induced increase of late INa was observed experimentally at both the whole cell and the single channel current levels.32 The amplitude of late INa is reported to be increased 2–4-fold from 50–100 pA during normoxia to 180–205 pA during hypoxia.32 The mechanism of the hypoxia-induced increase of late INa has not been explained. Nevertheless, evidence exists that specific domains in an intracellular loop of the α subunit of the sodium channel play a part in channel inactivation and are targets for phosphorylation by protein kinase C.44,45
Pathological and physiological conditions in which late sodium current is increased
Sodium influx through late INa appears to be a major contributor to the rise of [Na+]i that is observed during ischaemia15 and hypoxia.32 Exposure of hearts to ischaemia is known to increase lysophosphatidylcholine, palmitoyl-l-carnitine and reactive oxygen species (for example, hydrogen peroxide), and these substances are themselves reported to increase late INa.31,35 Sodium channel blockers (for example, tetrodotoxin and lidocaine) have been shown to reduce the rise in [Na+]i in rat ventricular myocytes and isolated hearts during hypoxia and ischaemia, respectively.9,32,46,47 This action of sodium channel blockers is associated with an improvement of contractile function and with reduction of the hypoxia/ischaemia-induced increase in [Ca2+]i. Late INa is more sensitive to block by tetrodotoxin than is peak INa, and is also reduced by lidocaine, mexiletine, R-56865, flecainide, amiodarone, and ranolazine.31
Sodium-calcium exchanger
The major fraction (perhaps two thirds) of sodium influx into contracting myocytes is facilitated by the cell membrane sodium-calcium exchanger (NCX) and is thus in exchange for calcium efflux.5 The NCX is a facilitated diffusion whereby the electrochemical potential gradients of sodium and calcium are the source of energy to drive the transport. NCX generates an electrical potential because three sodium ions are exchanged for each calcium ion. In the “forward” mode, NCX facilitates the influx of sodium and efflux of calcium, and generates a net inward (that is, depolarising) transmembrane current. In the “reverse” mode, NCX facilitates the efflux of sodium and influx of calcium, and generates a net outward (that is, repolarising) current. The net transport of sodium by NCX is therefore sensitive to changes in the cellular concentrations of both sodium and calcium and the transmembrane electrical potential. During diastole, sodium influx and calcium efflux (the forward mode of NCX) are favoured. During the plateau of the action potential, calcium influx and sodium efflux (the reverse mode of NCX) are favoured. Conditions that increase the duration of the action potential plateau (as a portion of the cardiac cycle) are therefore associated with an increase of the reverse mode of NCX and increased net calcium influx.
Sodium-dependent calcium accumulation
A rise in the [Na+]i leads to an increased exchange of intracellular sodium for extracellular calcium through the reverse mode of NCX. There is general agreement that the cellular calcium overload that occurs during ischaemia and reperfusion is a result of a combination of decreased efflux of calcium ions through the forward mode of NCX and increased influx of calcium ions through the reverse mode of NCX.5,48,49 A relative increase of activity of NCX in the reverse mode (sodium efflux and calcium influx) is a predictable outcome of both a rise in [Na+]i and an increase in APD. As noted above, an increase of late INa causes both an increase of [Na+]i and a prolongation of APD, and thus increased activity of NCX in the reverse mode, and calcium influx. Direct evidence in support of the critical role of the reverse mode of NCX in intracellular calcium overload during reperfusion or reoxygenation after ischaemia is derived from the observations that inhibitors of NCX, and antisense inhibition of NCX, greatly decrease the rise in [Ca2+].50,51 Furthermore, a reduction of the rise in [Na+]i (caused by sodium channel blockers or inhibitors of sodium-hydrogen exchange) can greatly decrease intracellular calcium overload.7–9,46,47,50 This in turn reduces the cellular dysfunction and injury associated with ischaemia and reperfusion.
PROPOSED MECHANISM FOR THE CARDIOPROTECTIVE ACTIONS OF RANOLAZINE
Ranolazine has been reported to reduce the effects of ischaemia or simulated ischaemia on animal hearts in vivo52–56 or isolated, in vitro cardiac preparations,57–60 respectively. The cardioprotective effects of ranolazine are observed at concentrations that have minimal or no effect on heart rate, coronary blood flow and systemic arterial blood pressure.61 Likewise, in patients with chronic ischaemic heart disease (angina pectoris), ranolazine is an effective anti-ischaemic and antianginal drug at concentrations that cause minimal or no changes in heart rate and blood pressure.62,63 These observations led to the hypothesis that ranolazine exerts its antianginal and cardioprotective effects through a primary mode of action distinct from that of typical antianginal drugs such as calcium channel blockers, β adrenoceptor antagonists and nitrates.62,63
The mechanism of action of ranolazine has been difficult to elucidate and has only recently begun to be clarified. Results of early studies suggested that ranolazine altered myocardial energy metabolism to reduce fatty acid oxidation and increase glucose oxidation, although a reduction by ranolazine of fatty acid oxidation was observed only under selected experimental conditions.64–67 The inhibition by ranolazine of fatty acid oxidation appears to require relatively high concentrations of ranolazine (12% inhibition at 100 μmol/l),60 whereas cardiac function can be seen to improve in the presence ⩽ 20 μM ranolazine.57,68,69 Furthermore, ranolazine was found to improve heart function after exposure to hydrogen peroxide,69 ischaemia and reperfusion57,60 or palmitoyl-l-carnitine68 and during heart failure70 in the absence of fatty acids in the heart perfusion solution 57,69 and changes in fatty acid β oxidation,60 carbohydrate metabolism68 and fatty acid/glucose uptake,70 respectively. No specific step or enzyme in a metabolic pathway has been identified as a site of ranolazine action at concentrations that are similar to those at which ranolazine has beneficial effects in vitro and in vivo. In contrast, evidence is increasing that ranolazine is an inhibitor of late INa at concentrations ⩽ 10 μmol/l and that ranolazine reduces the electrical and mechanical dysfunction associated with conditions known to cause [Na+]i-dependent calcium overload. This evidence is reviewed below.
Inhibitions of late and peak INa by ranolazine
Ranolazine causes a concentration-, voltage- and frequency-dependent inhibition of late INa in canine71 and guinea pig41 ventricular myocytes. The potency of ranolazine to inhibit late INa varies from 5–21 μmol/l,31,71 depending on the experimental preparation, conditions and species, and possibly the sodium channel isoform (for example, brain or cardiac). Ranolazine has notably less effect on peak than on late INa.31 In isolated ventricular myocytes of dogs with chronic heart failure, ranolazine was found to inhibit peak INa and late INa with potencies (50% inhibitory concentrations) of 244 and 6.5 μmol/l, respectively.31 Hence, ranolazine was about 38-fold more potent in inhibiting late INa than peak INa.31 Amiodarone was about 13-fold more potent in inhibiting late than peak INa in the same preparation.72 The velocity of the upstroke (phase 0) of the action potential is proportional to the magnitude of peak INa. Thus, it is not surprising that the maximum upstroke velocity of phase 0 of the action potential (+Vmax) of canine Purkinje fibres is reduced by ranolazine only at high concentrations (⩾ 50 μmol/l).71 Additional evidence that ranolazine is a weak inhibitor of peak INa comes from a study of transepicardial activation times that used a high-resolution optical mapping system to measure action potentials.73 Ranolazine (⩽ 30 μmol/l) had no effect on transepicardial activation times, whereas lidocaine (75 μmol/l) increased total activation time from 15 to 24 ms.73 This finding suggests that the effect of ranolazine on peak INa at concentrations as high as 30 μmol/l is of little functional consequence for action potential +Vmax and impulse propagation, and that ranolazine is a selective blocker of late INa compared with peak INa at concentrations ⩽ 10 μmol/l.
The effects of ranolazine on various ion currents in ventricular potassium myocytes have been investigated.71,74 Table 371,75 summarises the potencies of ranolazine to affect these ion conductances in canine ventricular myocytes. Of note, ranolazine inhibits the potassium rapid delayed-rectifier current (IKr), with a potency of 11.5 μmol/l.71 This action of ranolazine is likely responsible for its effect to prolong the ventricular APD and the QT interval.62,76 In comparison, ranolazine is a very weak inhibitor of either the peak L-type inward calcium current (ICa,L) or the current generated by the NCX. The potencies of ranolazine to inhibit peak ICa,L and sodium-calcium exchange current (INa/Ca) were 296 and 91 μmol/l, respectively.71 This suggests that neither current is likely to play a part in the mechanism(s) underlying the therapeutic effects of ranolazine. There is no evidence that ranolazine inhibits the reverse mode of NCX. Reduction of the [Na+]i by ranolazine, however, could be expected to reduce both sodium efflux and calcium influx through the reverse mode of NCX.
Potencies of ranolazine to inhibit transmembrane ion channel currents in canine ventricular myocytes
The results of studies showing that ranolazine did not depress ventricular contractility or slow either heart rate in the conscious dog or atrioventricular nodal conduction in the rabbit heart77 are consistent with the results of studies of effects of ranolazine on cellular ion currents. Ranolazine at a concentration of 20 μmol/l had no effect on the NHE in MDCK cells.77 In summary, the transmembrane ion conductances most sensitive to ranolazine are the late INa and IKr. Therapeutic concentrations of ranolazine appear to have no effect on ICa,L, INa/Ca (NCX) or NHE, three important contributors to [Ca2+]i and [Na+]i homeostasis.
Reversal by ranolazine of ventricular repolarisation abnormalities in disease models and in the presence of drugs that increase late INa
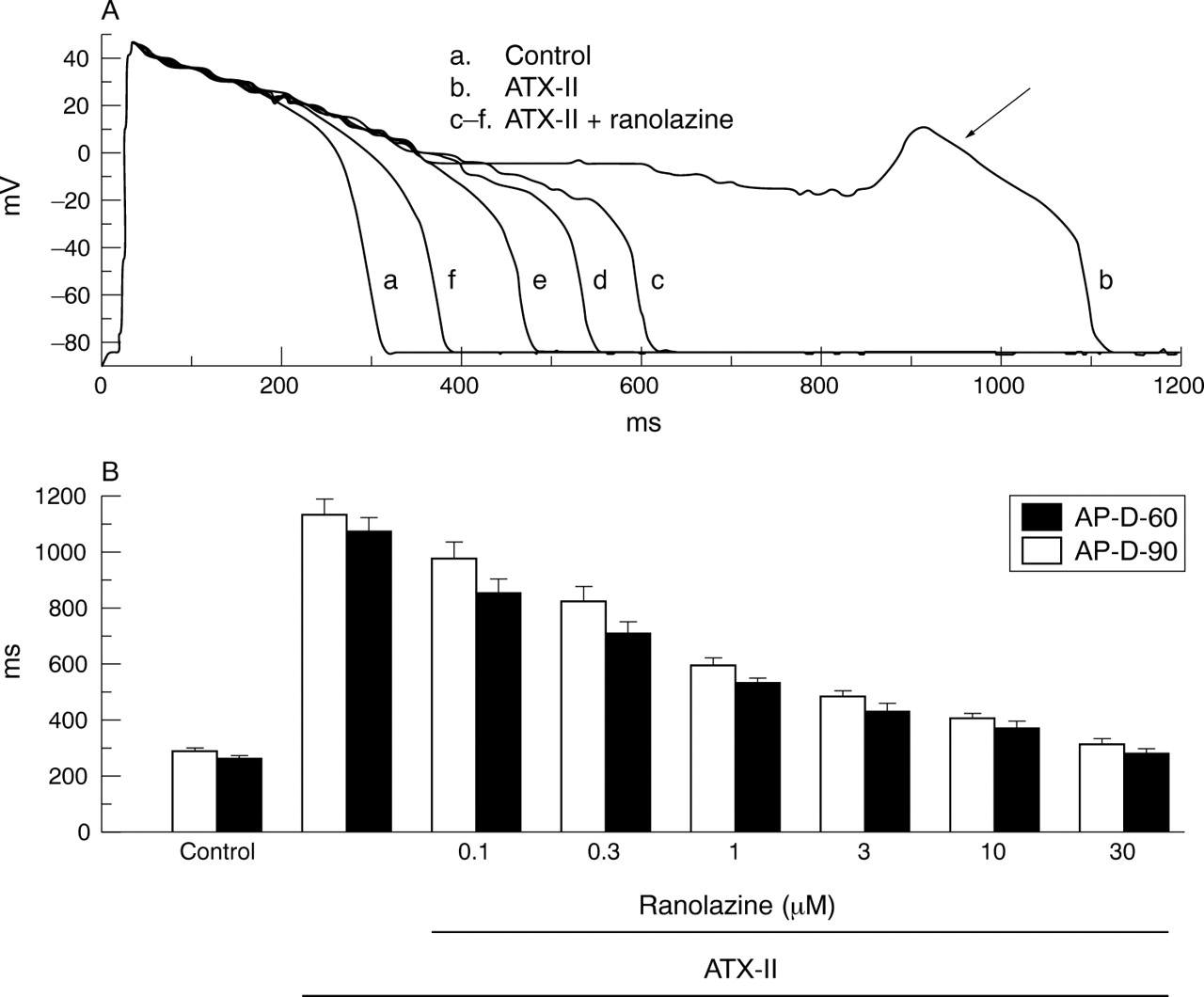
Cellular calcium overload can lead to spontaneous release of calcium from the sarcoplasmic reticulum, which in turn may cause repolarisation abnormalities.2,78,79 Raised [Ca2+]i activates [Ca2+]i-dependent ion currents (for example, INa/Ca; calcium-dependent chloride current; calcium-activated non-specific cation current) that can give rise to afterdepolarisations80 and an increase of beat-to-beat variability of APD. Both are harbingers of arrhythmias.81,82 An augmentation of late INa is expected to increase [Na+]i-dependent calcium entry through NCX, and thereby calcium loading of the sarcoplasmic reticulum, spontaneous calcium release and electrical instability. Consistent with this, some conditions known to increase late INa (table 2) and [Ca2+]i have been shown to cause arrhythmias. As summarised below, ranolazine reverses the ventricular repolarisation abnormalities associated with conditions and drugs known to increase late INa. Thus, the following findings are in keeping with the observation that ranolazine inhibits late INa of ventricular myocytes: (1) ranolazine decreases APD, measured at either 50% or 90% of repolarisation (APD50 or APD90, respectively), and abolishes early afterdepolarisations (EADs) of guinea pig ventricular myocytes treated with ATX-II41 (fig 2); (2) the potency of ranolazine to reverse these effects of ATX-II is 0.41 μmol/l41; and (3) tetrodotoxin, which reverses the increase in late INa caused by ATX-II, similarly reverses the ventricular repolarisation abnormalities (for example, APD prolongation and EADs) induced by ATX-II.41

Ranolazine attenuates the effects of the Anemonia sulcata toxin (ATX-II) on action potential duration (APD) and early afterdepolarisations (EADs) in guinea pig ventricular myocytes. (A) Recordings of action potentials from a ventricular myocyte in the absence of drug (control, (a)), in the presence of 10 nM ATX-II (b), and in the presence of ATX-II and increasing concentrations (1, 3, 10 and 30 μM) ranolazine (c–f). An EAD is indicated by the arrow in recording (b). (B) Concentration–response relationship for ranolazine to decrease APD in the presence of 10 nM ATX-II. Bars indicate the mean (SEM) of measurements from five to 10 cells. All values of APD in the presence of ranolazine are significantly different from ATX-II alone (p < 0.01). Reproduced with permission from Song et al.41
Ranolazine has been shown to have antiarrhythmic effects in a guinea pig in vitro model of the long QT-3 syndrome.83 Ranolazine (5, 10, and 30 μmol/l) attenuated the effect of 20 nM ATX-II to prolong the duration of the monophasic action potential in the guinea pig isolated perfused heart and suppressed the formation of EADs and occurrences of ventricular tachycardias.83 Ranolazine similarly antagonised the proarrhythmic actions of combinations of ATX-II and either the IKr blocker E-4031 or the slow delayed-rectifier current (IKs) blocker chromanol 293B.83 In the same long QT-3 model, ranolazine was found to suppress spontaneous and pause-triggered ventricular arrhythmic activity caused by a diverse group of IKr blockers (that is, moxifloxacin, cisapride, quinidine and ziprasidone).84 Thus, although ranolazine itself is an inhibitor of IKr (table 3), it appears not to potentiate (but may inhibit) the effects of other inhibitors of IKr.41,76,83,84 An effect of ranolazine (10 and 20 μmol/l) to reduce the incidence of ventricular fibrillation was also shown in a study of the rabbit isolated heart exposed to hypoxia and reperfusion in the presence of 2.5 mM external potassium concentration and the KATP channel opener pinacidil.59
In ventricular myocytes from dogs and humans with chronic heart failure (in which late INa is augmented), the APD is prolonged30,36,85 and EADs, aftercontractions and abnormal [Ca2+]i transients are common.85 Ranolazine inhibits late INa in ventricular myocytes from dogs with chronic heart failure with a potency of 6.4 μmol/l75 and, as expected, shortens APD and suppresses EADs in these myocytes at concentrations of 5 and 10 μmol/l.75 The sodium channel blockers tetrodotoxin, saxitoxin and lidocaine have likewise been shown to shorten the APD and suppress EADs in ventricular myocytes from failing hearts.36,37
Dispersion and/or beat-to-beat variability of APD (also referred to as instability of APD) are often observed in myocytes from failing dog hearts, in ischaemic preparations and in myocytes exposed to either ATX-II or to drugs that prolong the QT interval. An increased dispersion of repolarisation is associated with electrical (T wave) and mechanical alternans and is proarrhythmic.86 The role of late INa in increasing beat-to-beat variability of APD and the suppression of this variability by tetrodotoxin, saxitoxin and lidocaine has been reported.36,37,87 Ranolazine (5 and 10 μmol/l) also reduces the variability of APD in single ventricular myocytes from dogs with heart failure75 and in myocytes exposed to ATX-II (fig 3).41 Thus, inhibition of late INa with ranolazine and other sodium channel blockers suppresses arrhythmogenic abnormalities of ventricular repolarisation (that is, EADs and increased dispersion of repolarisation) that are associated with abnormal intracellular calcium homeostasis and with the occurrence of torsade de pointes ventricular tachycardias.41,76,83,84
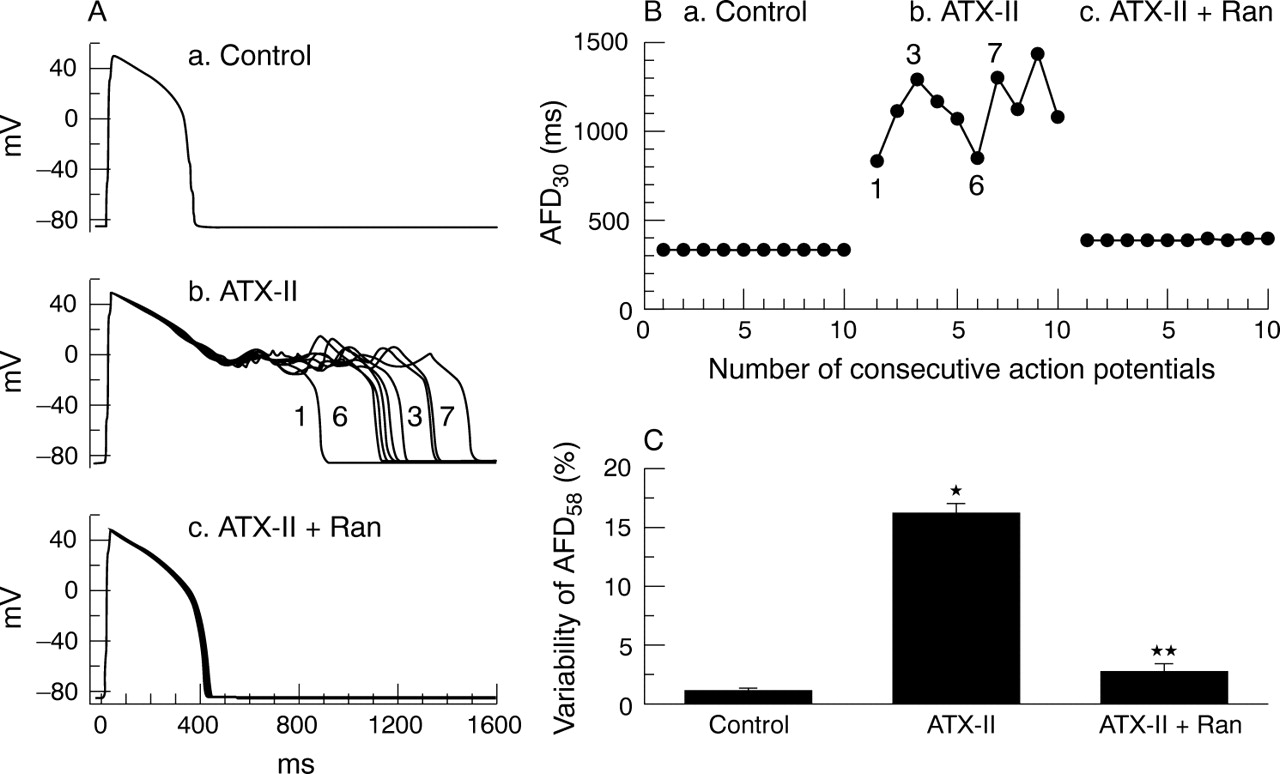
Effect of ranolazine (Ran) on variability of action potential duration (APD) in guinea pig ventricular myocytes exposed to Anemonia sulcata toxin (ATX-II). (A) Superimposed recordings of 10 consecutive action potentials from a myocyte in the absence of drug (a), in the presence of 10 nM ATX-II (b) and in the presence of 10 nM ATX-II and 10 μM ranolazine (c). (B) Graphic summary of the experiment shown in panel A. Each point represents the duration of a single action potential from a series of 10 action potentials recorded consecutively. (C) Summary of the results of all experiments similar to that shown in panels A and B. *p < 0.001 versus control; **p < 0.001 versus ATX-II. Reproduced with permission from Song et al.41
Reversal by ranolazine of mechanical dysfunction in disease models and in the presence of drugs that increase late INa
Intracellular calcium homeostasis has an important role in the regulation of left ventricular (LV) mechanical function.78 A significant increase in [Ca2+]i has been reported under a variety of experimental conditions involving exposures of cardiac tissues to ischaemia, hypoxia, oxygen free radicals, ischaemic metabolites, toxins and drugs. The excessive accumulation of intracellular calcium has been suggested to explain the ischaemic (and post-ischaemic) diastolic and systolic dysfunction and myocardial cell injury.1,3,49 As discussed above, this calcium overload is coupled to an increase in [Na+]i caused in part by an enhanced late INa. Hence, inhibition by ranolazine of this increased late INa should attenuate the LV mechanical dysfunction associated with several pathological conditions (see below).
Results of in vitro and in vivo studies of various cardiac preparations show that ranolazine can either prevent or reverse contractile and biochemical dysfunction in the ischaemic57 and failing heart,88 as well as in hearts exposed to the ischaemic metabolites palmitoyl-l-carnitine68 and hydrogen peroxide.69 Relevant to the hypothesis that ranolazine exerts its cardioprotective effect through inhibition of late INa, and consequently by reducing the sodium-dependent rise in [Ca2+]i is the substantial literature showing that sodium channel blockers are cardioprotective.1,6–8,42,46,47,89–91 The concept that the cytoprotective activity of sodium channel blockers to reduce ischaemia/reperfusion injury is principally mediated by inhibition of late INa has been proposed.1,9,47
Ranolazine attenuates contractile and biochemical dysfunction associated with ischaemia/reperfusion and anoxia/reoxygenation. Ischaemia/reperfusion and anoxia/reoxygenation increase LV diastolic pressure, a phenomenon attributed to calcium overload triggered by a rise in [Na+]i. The increases in LV diastolic pressure and tension can be mimicked by ischaemic metabolites such as palmitoyl-l-carnitine, lysophosphatidylcholine and reactive oxygen species.33,48,68,69 Common to all these conditions is an increase in late INa (table 2). The consequent rises in LV diastolic pressure and tension are associated with an increased rate of ATP hydrolysis, increased [Ca2+]i, increased release of creatine kinase, and histological evidence of cell damage.47,57,68,69,89
In rabbit and rat isolated perfused hearts, ranolazine (5–20 μmol/l) has been shown to significantly reduce ischaemia/reperfusion-, palmitoyl-l-carnitine- and hydrogen peroxide-induced increases in LV diastolic pressure and creatine kinase release, and decreases in tissue levels of ATP57,68,69 (figs 4 and 5). The reduction in ischaemia/reperfusion injury by ranolazine was associated with electron microscopic evidence of preservation of ultrastructural cell integrity.57

Effect of ranolazine on the left ventricular end diastolic pressure (LVEDP) and creatine kinase release of rabbit isolated Langendorff-perfused hearts. Hearts were subjected to 30 min of global ischaemia followed by 60 min of reperfusion. Hearts were treated with either ranolazine (10 μmol/l) or drug vehicle (dimethylsulfoxide plus saline) 10 min before ischaemia and throughout the period of ischaemia and reperfusion. Each value represents the mean (SEM).* Values significantly different (p < 0.05) from drug vehicle. Modified with permission from Gralinski et al.57

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of ranolazine to attenuate hydrogen peroxide- (H2O2; left graph) and palmitoyl-l-carnitine- (PalmCarn; right graph) induced increases of left ventricular end diastolic pressure (LVEDP) of rat isolated Langendorff-perfused hearts. Each value represents the mean (SEM) from six to eight hearts. *Values significantly different (p<0.05) from drug vehicle. Modified and reproduced with permission from Maruyama et al68 and Matsumura et al.69
The observation that ranolazine decreases post-ischaemic contracture (that is, increase of LV end diastolic pressure on reperfusion) has been recently confirmed.31 The post-ischaemic increase in LV end diastolic pressure, decrease in the rate of LV pressure development (LV+dP/dt), and decrease in the rate of LV pressure decline (LV−dP/dt), which are indices of contracture, contractility and relaxation, respectively, were significantly less in hearts treated with 5.4 μM ranolazine than in vehicle-treated hearts.31 Ranolazine, as well as lidocaine and mexiletine, increase the time to onset and reduce the rate of development of contracture of isolated rat left atria that are exposed to ATX-II.92
The most plausible explanation for the cardioprotective effect of ranolazine is its action to inhibit late INa, and consequently to reduce pathological increases of [Na+]i and [Ca2+]i. Two recent studies support this hypothesis.93,94 Ranolazine was found to attenuate significantly the increases in diastolic and systolic [Ca2+]i caused by the sea anemone toxin ATX-II. ATX-II increases late INa and thereby mimics the effects of ischaemia/reperfusion to increase [Na+]i and [Ca2+]i. The concentration-dependent attenuation by ranolazine (4.4 and 8.5 μmol/l) of the rise in [Ca2+]i caused by ATX-II was accompanied by a reduction of the ATX-II-induced (a) decrease in LV minute work (LV mechanical function), (b) decrease in LV systolic pressure and rise in LV end diastolic pressure, (c) decreases in both peak LV+dP/dt and LV−dP/dt, and (d) increase of myocardial lactate release.93,94 In addition, ATX-II decreased coronary flow and coronary vascular conductance (caused by the increase in LV end diastolic pressure, and hence diastolic stiffness), and this effect was also reversed by ranolazine (CV Therapeutics, unpublished data). This finding shows that ranolazine reverses the ATX-II-induced increase in extravascular compression and may account for the effect of ranolazine to maintain coronary flow near normal levels during exposure to ATX-II. An increase in extravascular compression is an important contributor to the decrease in myocardial blood flow when LV wall tension is increased during demand-induced ischaemia.4 Thus, ranolazine significantly reduces the LV mechanical dysfunction due to the sustained rises in diastolic and systolic [Ca2+]i caused by ATX-II.94 These data are consistent with the findings that ranolazine attenuates LV diastolic dysfunction caused by [Na+]i-dependent calcium overload during ischaemia/reperfusion57 in the presence of ischaemic metabolites,68 in the presence of reactive oxygen species69 and in ventricular myocytes from dogs with ischaemic heart failure.75
CONCLUSION
Impaired inactivation of INa increases late INa, leading to increased [Na+]i. The increase of [Na+]i leads to cellular calcium overload. Calcium overload is arrhythmogenic and causes abnormal LV relaxation and diastolic dysfunction. Strong evidence supports the hypothesis that ranolazine suppresses late INa and by this action reduces intracellular calcium overload and [Na+]i-dependent calcium-mediated arrhythmias and LV diastolic dysfunction. Ranolazine improves diastolic function without decreasing systolic function, because ranolazine does not reduce either the peak inward sodium current or the peak inward calcium current.
REFERENCES
Footnotes
-
All authors are full-time employees of CV Therapeutics, Inc, which has ownership of intellectual property rights for ranolazine