


Abstract
Photoincorporation of ligands into the benzodiazepine site of native γ-aminobutyric acidA (GABAA) receptors provides useful information about the nature of the benzodiazepine (BZ) binding site. Photoincorporation of flunitrazepam into a single population of GABAA receptors, recombinant human α1β3γ2, was investigated to probe further the mechanism and orientation of flunitrazepam and other ligands in the BZ binding site. It was concluded that the receptor is primarily derivatized with the entire, unfragmented, flunitrazepam molecule, which undergoes a conformational change during photolysis and largely vacates the benzodiazepine binding site. Investigation of the BZ site after photoincorporation of [3H]flunitrazepam confirmed that binding of other radioligands was unaffected by incorporation of flunitrazepam. This did not correlate with their efficacy but depended on the presence of particular structural features in the molecule. It was observed that affected compounds have a pendant phenyl moiety, analogous to the 5-phenyl group of flunitrazepam, which are proposed to overlap and interact with the same residue or residues in the BZ binding site. Because the major site of flunitrazepam photoincorporation has been shown to be His102, we propose that this group of compounds interacts directly with His 102, whereas compounds of other structural types have no direct interaction with this amino acid. The orientation of ligands within the BZ binding site and their specific interaction with identified amino acids are not well understood. The data in the current study indicate that His102 interacts directly with the pendant phenyl group of diazepam, and further implications for the pharmacophore of the BZ binding site are discussed.
GABA is the major inhibitory neurotransmitter in the mammalian central nervous system, and it mediates its effects largely through the GABAAreceptors, a family of ligand-gated Cl− ion channels (for reviews, see Smith and Olsen, 1995; Stephenson, 1995;McKernan and Whiting, 1996). Many neuroactive drugs, including the barbiturates, neurosteroids, BZs, and ethanol, exert at least some of their effects through interaction with the GABAAreceptors. Structurally, it is known that the GABAA receptors are pentameric and many are composed of α, β, and γ subunits. The exact stoichiometry has been an area of debate, but the weight of evidence indicates that there are two copies of an α subunit, two copies of a β subunit, and one copy of a γ subunit in a receptor (Chang et al., 1996;Sigel and Buhr, 1997; Tretter et al., 1997). Both the α and γ subunits are necessary for a receptor to exhibit sensitivity to BZs, and it has been proposed that the BZ binding site is formed with contributions from both of these polypeptides (Stephenson et al., 1990; McKernan et al., 1995; Wingrove et al., 1997).
Several amino acids have been identified that are important for binding of ligands to the BZ site, including His102 (Wieland et al., 1992; Duncalfe et al., 1996) and Gly200 (Pritchett and Seeburg, 1991) of the α subunit and Phe77 and Met130 of the γ2 subunit (Buhr and Sigel, 1997; Buhr et al., 1997a; Wingroveet al., 1997). The orientation of any of the benzodiazepine ligands between these residues is currently not known.
It was observed >10 years ago that photolysis of the BZ site agonist flunitrazepam results in irreversible incorporation of the molecule into GABAA receptor proteins (Sieghart et al., 1983). However, this does not prevent subsequent binding of some other BZ site ligands, such as the β-carbolines (Thomas and Tallman, 1983; Gibbs et al., 1985). Consequently, it was proposed that agonists and inverse agonists occupy distinct and possibly nonoverlapping modulatory binding sites on the same receptor molecule. However, these conclusions were drawn before our current understanding of the extent of GABAA receptor heterogeneity and the observation that some receptors, such as those of composition α4βγ2 and α6βγ2, do not bind flunitrazepam with high affinity but retain the ability to bind some BZ ligands of other structures, particularly the β-carbolines. Such receptor heterogeneity would, in retrospect, have confounded the interpretation of studies using brain membranes, where multiple subtypes are present. The preservation of inverse agonist (e.g., β-carboline) binding after photoincorporation with flunitrazepam may be due to binding to the α4 and α6 subtypes rather than binding to a different site on the flunitrazepam-sensitive receptors.
We therefore reevaluated the photoaffinity labeling of the BZ binding site with flunitrazepam and other ligands and have used one GABAA receptor subtype of a specific defined composition, α1β3γ2. The aim of these experiments was to provide further information on whether agonist, antagonist, and inverse agonist BZ site ligands occupy the same binding pocket and, because His102 is modified by flunitrazepam, to investigate whether different chemical classes of BZ ligands interact with the BZ site in the same way and provide some information regarding which part of the BZ site ligands is orientated toward this residue.
Experimental Procedures
[3H]CGS 8216 (20.0 Ci/mmol, NET-999E) [3H]Ro 15-4513 (21.7 Ci/mmol, NET-925),N-[methyl-3H]Ro 15-1788 (87 Ci/mmol, NET-757), and [methyl-3H]flunitrazepam (84.5 Ci/mmol, NET-S67) were from DuPont-New England Nuclear (Boston, MA). Flunitrazepam, diazepam, and Ro 15-1788 were from Sigma Chemical (St. Louis, MO). PK 11195, Ro 15-4513, zolpidem, and DMCM were from Research Biochemicals (Natick, MA). CGS 8216 was a gift from Ciba-Geigy (Basel, Switzerland). Abecarnil was a gift from Schering AG (Berlin, Germany). The pyridodiindoles β-CCM and β-CCT were a kind gift from Prof. James Cook (University of Wisconsin, Milwaukee, WI). All other compounds were synthesized by the Chemistry Department at Merck Sharp and Dohme (Essex, UK).
Cells expressing the human α1β3γ2 subtype of the GABAA receptor were grown and harvested as described previously (Hadingham et al., 1992). Cell membranes were resuspended in 50 mmNaKPO4 buffer, pH 7.5, at l mg protein/ml and preincubated for 30 min at 4° with 50 nm Ro 15-4513, 50 nm flunitrazepam, 50 nm desmethyl flunitrazepam, or 500 nm chlordiazepoxide. Membranes then were exposed to UV light (365 nm, 50–60 Hz, 200–220 V; UVP, San Gabriel, CA) for 60 min at 4° at a distance of 25 cm. Control membranes were incubated with flunitrazepam but not exposed to UV light. After UV exposure, membranes were washed extensively to remove unincorporated compound by centrifugation and resuspension eight times in 40 ml of phosphate buffer. Radioligand binding was carried out on membranes resuspended to 1 mg of protein/ml using [3H]flunitrazepam (0.1–20 nm) and [3H]CGS 8216 (0.05–10 nm), [3H]Ro 15-1788 (0.05–10 nm), or [3H]Ro 15-4513 (0.5–10 nm). Assays were carried out in a total volume of 0.5 ml and were incubated for 30 min at 20°. Nonspecific binding was defined with 10 μmRo 15-1788. Incubations were terminated by filtration through GF/B filters followed by three 5-ml washes with cold buffer and scintillation counting. Competition curves were carried out using [3H]Ro 15-1788 as ligand, and compounds were tested over the range of 0.3 nm to 1 μm. The presence of a modulatory interaction between the GABA and BZ binding sites was examined by measuring the ability of GABA (1 mm) to modulate binding of the BZ site ligands [3H]flunitrazepam (3 nm), [3H]Ro 15-1788 (1 nm), and [3H]Ro 15-4513 (3 nm) in the presence and absence of photoincorporated flunitrazepam. Saturation and competition curves were analyzed using RS 1 (Bolt, Beranek and Newman, Cambridge, MA), and statistical analysis was performed using Student’st test.
Preliminary experiments revealed that 60 min of irradiation was required to maximally incorporate flunitrazepam and Ro 15-4513. Using [3H]Ro 15-1788, this level of irradiation reduced the B max value for binding in cell membranes to 77.5 ± 8% of control (unirradiated) levels, consistent with earlier studies using rat or chick brain (Gibbset al., 1985; Borden and Gibbs, 1990). Binding of other radioligands was similarly reduced after irradiation of membranes. Conversely, incubation of membranes with BZ ligands in the absence of UV irradiation had no effect on the number of BZ binding sites at the end of the experiment (B max for [3H]Ro 15-1788 = 99.3 ± 4% relative to binding in control, unirradiated, cell membranes in the absence of BZ ligand) Consequently, all binding results were expressed as a percentage of membranes exposed to UV irradiation in the absence of other ligands.
Results
UV photoincorporation of Ro 15-4513 and flunitrazepam.
The binding of four radioligands, [3H]flunitrazepam (full agonist), [3H]Ro 15-1788 (antagonist), [3H]Ro 15-4513 (partial inverse agonist), and [3H]CGS-8216 (full inverse agonist at α1βγ2), was measured after irreversible photoincorporation of Ro 15-4513 or flunitrazepam.
After photoincorporation with Ro 15-4513, binding of all three ligands was reduced by >68% (Fig. 1 and Table1). The reduction in binding was independent of the ligand used to measure the remaining number of sites (i.e., there was no statistically significant difference in the number of remaining sites measured by [3H]Ro 15-1788, [3H]CGS 8216, or [3Hlflunitrazepam). When binding was carried out with [3H]Ro 15-4513 after photoincorporation of the unlabeled compound, it was not possible to determineB max andKd values by saturation analysis because the remaining number of specific counts was too low (Table1). A different pattern was observed when flunitrazepam was used as the photoaffinity ligand. After photoaffinity labeling with flunitrazepam, the subsequent binding of [3H]flunitrazepam was reduced by 84.1 ± 4.9%. Surprisingly, however, when [3H]CGS-8216, [3H]Ro 15-1788, or [3H]Ro 15-4513 was used as radioligand to quantify the number of receptors remaining, the reduction in binding was only 20–25%, and thus the majority of the binding sites were unaffected by photoincorporation with flunitrazepam (Table 1). The number of binding sites remaining as measured by [3H]CGS- 8216, [3H]Ro 15-1788, or [3H]Ro 15-4513 were not significantly different from each other but were significantly more than those remaining when [3Hlflunitrazepam was used as the ligand. There was no reduction in the affinity (Kd ) of any of the radioligands for the receptor after photoincorporation with either Ro 15-4513 or flunitrazepam, indicating that the decreased binding was due to a reduction in the number of receptor sites (B max) and not due to incomplete removal of unreacted ligand (Table 1).

Scatchard plot of the binding of [3H]flunitrazepam, [3H]Ro 15-4513, [3H]Ro 15-1788, and [3H]CGS-8216 before and after photoincorporation of Ro 15-4513 and flunitrazepam. Data shown are representative of three to five separate experiments. Membranes expressing receptor of composition α1β3γ2 were incubated with no addition (▪) flunitrazepam (50 nm, •), or Ro 15-4513 (50 nm, ▴) for 20 min at 4° before UV exposure for 1 hr. Membranes then were washed extensively before saturation binding and Scatchard analysis using [3H]flunitrazepam, [3H]Ro 15-4513, [3H] Ro 15-1788, and [3H]CGS-8216 as indicated. The control cells expressed 2300–4400 fmol receptor sites/mg of protein.
Reduction in benzodiazepine binding after photoincorporation of Ro 15-4513 or flunitrazepam
The observation that photoincorporation with flunitrazepam does not prevent subsequent binding of other radioligands confirms previous observations from the literature in which a heterogeneous population of receptors, such as rat or chick brain membranes, has been used (Brown and Martin, 1984; Gibbs et al., 1985).
Pharmacology of the BZ site of the GABAA receptor α1β3γ2 labeled by four different radioligands.
If [3H]flunitrazepam does indeed bind to a site that is distinct from the other ligands tested, then it may be possible to detect differences in the pharmacology of the BZ binding site when labeled by these different ligands. This was investigated by labeling the BZ site of the α1β3γ2 GABA receptor with four different ligands and comparing the pharmacology of the BZ binding site, as shown in Table 2. The affinity and rank order of potency of a series of structurally diverse compounds were very similar when [3H]flunitrazepam was used as the ligand compared with the other three radiolabeled compounds, and no large differences in affinity (>3–4-fold) were observed. The observation that the affinity (Ki ) for each compound at the BZ binding site is the same independent of the radioligand used is consistent with a competitive interaction between all four radioligands and seems not to support the hypothesis that flunitrazepam labels a site that is in some way distant from the site labeled by the other ligands. In addition, the Hill coefficients measured were not significantly different from 1 (data not shown), which is consistent with the hypothesis that all compounds tested, whether agonist, antagonist, or inverse agonist, compete for the same binding site.
Affinity of benzodiazepine site ligands for the BZ receptor measured with radioligands of different structures
Pharmacology of the BZ binding site after UV incorporation of flunitrazepam.
After photoincorporation with flunitrazepam, the majority of the BZ binding sites still recognize [3H]Ro 15-1788 with high affinity (Table 1). Therefore, this radioligand was used to characterize the pharmacology of the photoaffinity-labeled sites. As shown in Table3, photoincorporation with flunitrazepam reduced the affinity of the receptor for flunitrazepam, midazolam, and diazepam by ∼30-fold, which is in accord with the observed lack of binding for [3H]flunitrazepam and in agreement with previous studies carried out in rat cerebral cortex (Mohler, 1982). Consistent with the unchanged binding of the radioligands [3H]CGS 8216 and [3H]Ro 151788, the affinity of the unlabeled compounds, CGS 8216 and Ro 15-1788, for the flunitrazepam-photolabeled binding site also was unchanged. The loss of binding affinity is not observed with all compounds with a core BZ structure because the affinities of Ro 15-1788, Ro 15-4513, and the α5-selective imidazobenzodiazepineL-655,708 (Quirk et al., 1996) were not reduced after photoincorporation of flunitrazepam. Three further compounds, chlordiazepoxide and the naphthyridones, L-764,488 [6-benzyl-3-(5-methoxy-[1,3,4]oxadiazol-2-yl)-5,6,7,8-tetrahydro-1H-[1,6]naphthyridin-2-one] and L-763,673 [6-benzyl-3-(5-thiophen-2-yl-[1,3,4]oxadiazol-2-yl)-5,6,7,8-tetrahydro-1H-[1,6]naphthyridin-2-one], were identified whose binding affinity was greatly reduced after photoincorporation of flunitrazepam. Compounds of several other structural classes were unaffected by photoincorporation of flunitrazepam, including the β-carbolines β-CCM and DMCM, the thienylpyrazoloquinoline CGS-8216, and the imidazopyridine zolpidem. Previous studies have concluded that only the binding of agonist BZ site ligands is reduced after photoincorporation of flunitrazepam and that the photoincorporated receptor discriminates between agonists, which lose ∼30-fold in affinity, and antagonists, whose affinity remains unchanged (Mohler, 1982; Brown and Martin, 1984). In the more extensive studies presented here, the discrepancy between compounds that lose affinity after photoincorporation of flunitrazepam cannot be explained in terms of differing binding sites for agonists and antagonists because zolpidem, abecarnil, and zopiclone, all full agonists at the BZ site of α1β3γ2 (Hadingham et al., 1995; Wafford KA and Thompson SA, personal communication), show no loss of affinity for receptors after flunitrazepam incorporation.
The pharmacology of the benzodiazepine binding site after photoincorporation of flunitrazepam
We further investigated whether the efficacy of compounds such as zolpidem might be altered after irreversible incorporation of flunitrazepam, even though their binding affinity was unchanged.
Functional properties of the BZ binding site after photoincorporation of flunitrazepam.
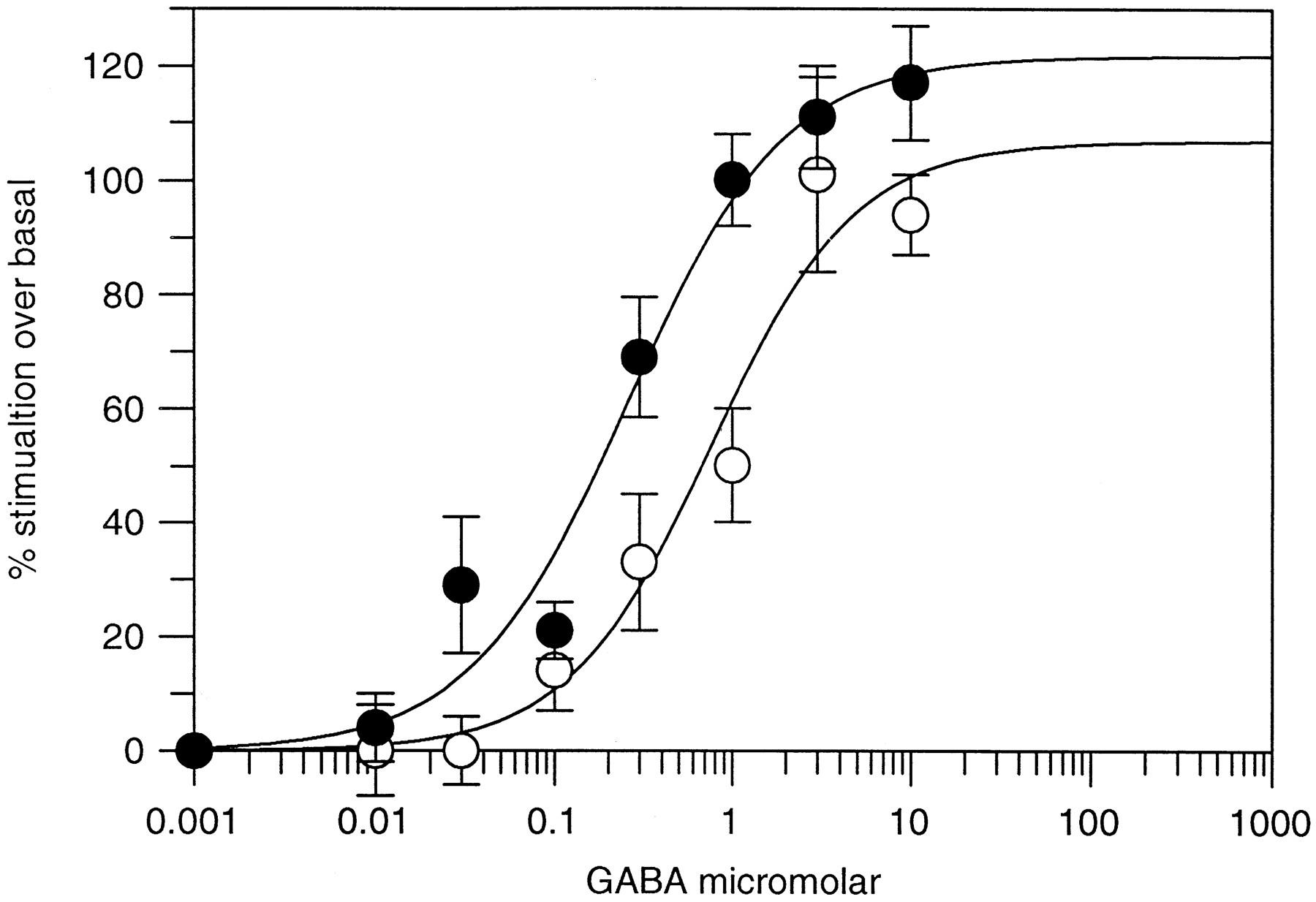
The affinity of BZs for the BZ binding site is increased in the presence of GABA. This allosteric modulation can be observed in a binding assay in which specific binding of an agonist ligand such as [3H]flunitrazepam or [3H]zolpidem is increased in the presence of GABA. The presence of these allosteric interactions after photoincorporation of flunitrazepam into the BZ site was investigated (Fig. 2). [3H]Zolpidem was used as the agonist ligand, and GABA potentiation of binding was measured in the presence and absence of photoincorporated flunitrazepam.

Efficacy of GABAA receptors after flunitrazepam photoincorporation. Dose-dependent potentiation of [3H]zolpidem binding by GABA to α1β3γ2 receptors in the absence (○) and presence (•) of photoincorporated flunitrazepam. The EC50 value for potentiation of [3H]zolpidem binding was 1.0 ± 0.2 μmfor control and 0.4 ± 0.01 μm for flunitrazepam-derivatized receptors. Data shown are mean ± standard error for three separate determinations.
As shown in Table 4, the modulatory ability of the BZ binding site was unchanged after photoincorporation of flunitrazepam. [3H]Zolpidem binding was potentiated by GABA to the same extent before and after photoincorporation of flunitrazepam. Furthermore, the EC50 value for potentiation of [3H]zolpidem binding by GABA was little changed after incorporation of flunitrazepam (EC50 value before flunitrazepam = 1.0 ± 0.2 mm, EC50 value after flunitrazepam = 0.4 ± 0.01 mm). It is concluded, therefore, that irreversible derivatization of the BZ receptor affects neither the affinity of many compounds for the receptor nor the functional efficacy of the receptor.
Allosteric interaction between the GABA and benzodiazepine sites is maintained after photoincorporation of flunitrazepam
Mechanism of UV incorporation of flunitrazepam.
The observation that after photoincorporation of flunitrazepam the BZ binding site can still accommodate ligands of many other structural types is unexpected, and further experiments were carried out to understand the process by which flunitrazepam incorporates into the receptor. Irradiation with flunitrazepam can result in fragmentation of the molecule, as well as incorporation of the intact species into the target binding site (Givens et al., 1986; Busker et al., 1987). One proposed major route of decomposition of flunitrazepam is loss of the N-methyl group, which, in [3H]flunitrazepam, is the site of tritiation. It therefore could be possible that UV irradiation results in minor modification of the BZ binding site by N-methylation of a specific residue in the binding pocket, which might still allow other compounds to bind. Alternatively, the entire molecule might become incorporated into the receptor, and during the reaction, a conformational change might occur such that flunitrazepam vacates the binding site, leaving it available for subsequent occupation by other ligands. To investigate the first of these possibilities, derivatives of flunitrazepam were incorporated into the receptor. As shown in Table5, UV photoactivation withN-desmethyl flunitrazepam prevents subsequent binding of flunitrazepam but not Ro 15-1788 or CGS 8216 (i.e., the same pattern of radioligand binding is observed after incorporation of flunitrazepam and N-desmethyl flunitrazepam). N-Desmethyl flunitrazepam has equivalent affinity for the BZ binding site, is photolabile, and incorporates into the receptor, but it has noN-methyl moiety. Therefore, the mechanism of incorporation by flunitrazepam cannot be via transfer of the N-methyl group to the BZ binding site but suggests that in both cases, the complete flunitrazepam molecule, or a significant part of it, is incorporated into the receptor. Neither desmethyl, desnitro flunitrazepam, nor chlordiazepoxide is incorporated into the BZ binding site via photolysis, as is demonstrated by the subsequent binding of both [3H]flunitrazepam and [3H]Ro 15-1788 (data not shown). In the case of desmethyl desnitro flunitrazepam, this is probably caused by the removal of the nitro group, the photoreactive species. In the case of chlordiazepoxide, which is reported to be photolabile and for which a pathway of decomposition has been proposed, there are several possible interpretations. Either the compound is not photosensitive when bound to the receptor or has greatly reduced affinity when photolysed such that it does not attack the receptor protein or it may be photolysed and incorporate at a position that does not interfere with the subsequent binding of any radioligand.
Binding of [3H]flunitrazepam and [3H]Ro 15-1788 after photoincorporation of chlordiazepoxide, flunitrazepam, and analogues
Furthermore, after photoincorporation of flunitrazepam or desmethyl flunitrazepam, the affinities of flunitrazepam, L-764,488,L-763,673, and chlordiazepoxide were similarly reduced, confirming that flunitrazepam and desmethyl flunitrazepam modify the receptor similarly.
Discussion
The observation that photoaffinity labeling with Ro 15-4513 of a single homogeneous GABAA receptor subtype prevents subsequent binding of all BZ site ligands is consistent with previous observations in rat brain where [3H]Ro 15-4513 irreversibly labels virtually all BZ binding sites (Mohleret al., 1984). After photoaffinity labeling of a single homogeneous GABAA receptor subtype with flunitrazepam, many BZ site ligands still bound without any change in affinity, despite irreversible incorporation of the molecule (Tables1-3). This also is consistent with previous observations using heterogeneous GABA receptors from rat brain (e.g., Thomas and Tallman, 1983; Gibbs et al., 1985). It is concluded that the BZ site of a single homogeneous GABAA receptor (α1β3γ2) can accommodate concurrently both flunitrazepam, irreversibly incorporated, and other BZ ligands. This has been regarded previously as evidence that agonists and inverse agonists bind to different sites on the BZ receptor. However, even though the same observation has been made using brain membranes or a homogeneous recombinant receptor preparations, several lines of evidence seem to argue against the agonist and inverse agonist sites being separate.
First, all the literature to date shows the interaction among agonists, inverse agonists, and antagonists at the BZ site to be competitive, and the rank order of potency of a series of structurally different compounds for a single population of GABAAreceptors (α1β3γ2) as labeled by four different ligands of different structural classes is very similar (Table 2).
Second, the residues involved in binding of BZ site ligands have been much studied since the receptor subunits were cloned. Some compounds, such as DMCM and Ro 15-4513, for example, are agonists at one subtype, α6β3γ2, and inverse agonists at another, α1β3γ2 (Waffordet al., 1994; Puia et al., 1991). Because these receptors are structurally very similar, with only a few amino acid residues in the α subunit varying between them, and there is no significant difference in binding affinity (Hadingham et al., 1995), it is likely that DMCM and Ro 15-4513 bind to the same pharmacophore in the α1- and α6-containing receptors and that the binding sites for agonists and inverse agonists are not separate entities. Third, these studies demonstrate that zolpidem, abecarnil, and zopiclone, all full agonists at the BZ site of α1β3γ2, show no loss of affinity for receptors after flunitrazepam incorporation.
There are, however, some mechanistic differences in the binding of the agonist flunitrazepam and the inverse agonist Ro 15-4513. For example, Ro 15-4513 requires an intact disulfide bridge for high affinity binding, whereas flunitrazepam does not (Duncalfe and Dunn, 1993), and the two ligands differ in their sensitivity to the sulfhydryl modifying reagent N-ethyl maleimide (Duncalfe and Dunn, 1993), suggesting differential contributions of amino acids containing a free sulfhydryl group to their binding interaction. Furthermore, binding of [3H]flunitrazepam is reduced at pH ≤ 7, whereas binding of [3H]Ro 15-4513 is unaffected by low pH. This has been interpreted as the involvement of a histidine residue in binding of [3H]flunitrazepam but not [3H]Ro 15-4513 (Davies et al., 1996). Further evidence that these two compounds interact differently with the receptor binding site comes from experiments where photoaffinity labeled receptor is cleaved with hydroxylamine and radioactivity associated with photoincorporated [3H]Ro 15-4513 is associated with a fragment of the receptor between residues 104 and the carboxyl terminus of the αl subunit. In contrast, radioactivity from [3H]flunitrazepam is associated with residues 1–102 of the bovine α1 subunit (Duncalfe and Dunn, 1996), and more recent experiments have revealed that the major site of [3H]flunitrazepam incorporation is His102 of the bovine α1 subunit (Duncalfe et al., 1996). All these differences in the binding characteristics are consistent with flunitrazepam and Ro 15-4513 binding in the same binding pocket but with different amino acid residues.
Our current understanding of the structure of the α1βγ2 subtype proposes that there are two copies each of an α subunit, two copies of a β subunit, and only one copy of a γ subunit (i.e., 2× α1, 2× β2, and 1× γ2; Chang et al., 1996; Tretter et al., 1997 and references therein). Furthermore, as the BZ binding site is formed at the interface of the α subunit and the γ subunit (Buhr et al., 1997b; Wingrove et al., 1997; Sigel and Buhr, 1997), and because there is only one γ subunit, there can be only one BZ site per receptor monomer.
It is concluded that photoaffinity labeling of the benzodiazepine site of the GABAA receptor with flunitrazepam occurs first by binding flunitrazepam into the BZ site reversibly (i.e., between the α and γ subunits), followed by an irreversible incorporation on exposure to UV light. It is concluded that the entire flunitrazepam molecule is incorporated at His102 of the α subunit for two reasons. First, photoincorporation of a major photometabolite desmethyl flunitrazepam produces the same changes in binding observed with flunitrazepam (Table 5). Second, antibodies raised against flunitrazepam recognize and are able to immunoprecipitate the flunitrazepam-derivatized receptor (Davies et al., 1996), and therefore at least the majority of the flunitrazepam molecule is still present.
Surprisingly, incorporation of flunitrazepam does not prevent binding of other ligands at the BZ site. Receptor heterogeneity or the presence of multiple binding sites have previously been proposed as explanations for observations of this type. However, the experiments reported here were carried out using one homogeneous population of receptors with one BZ binding site per receptor. It is therefore necessary to propose an alternative mechanism by which the entire flunitrazepam molecule can be incorporated and the ability to bind other ligands is retained. It is possible that either the histidine residue or the photoincorporated flunitrazepam molecule undergoes a structural change, after photoincorporation such that flunitrazepam is removed from the binding pocket but remains irreversibly incorporated to the protein through a flexible bond. There is some precedent for such a mechanism in the literature. For example, a flexible histidine residue in the active site of carbonic anhydrase has been exploited in the design of more potent inhibitors (Greer et al., 1994).
Irreversible modification of His102 of the α1 subunit of the bovine GABAA receptor by flunitrazepam has been observed (Duncalfe et al., 1996). Therefore, although other explanations may be plausible, the simplest would be that irreversible incorporation of flunitrazepam at His102 of the human α1 subunit produces a conformational change in the receptor, removing flunitrazepam from the binding pocket, allowing other ligands to enter, and reducing the affinity only of those that interact specifically with His102. Such compounds would include flunitrazepam and the two naphthyridones L-764,488 and L-763,673. A consistent feature of compounds that lose affinity after photoincorporation of flunitrazepam into the BZ site is the presence of a pendant phenyl group, such as the 6-benzyl ring inL-764,488 and L-763,673 and the 5-phenyl ring in molecules of the diazepam group. Consistently, L-816,993 [3-(5-thiophen-2-yl-[1,3,4]oxadiazol-2-yl)-5,6,7,8-tetrahydro-1H-[1,6]naphthyridin-2-one], the desbenzyl analogue of L-763,673, shows no loss of affinity at flunitrazepam-derivatized receptors. A proposed system for overlapping of the molecules discussed is presented in Fig.3. We propose that only compounds that possess a phenyl ring that can interact directly with His102 lose affinity for the BZ binding site after photoincorporation of flunitrazepam. A possible mechanism for this interaction is hydrogen bonding or π-π interaction of the phenyl ring with the histidyl side chain; interactions between a phenyl ring and a histidine side chain are supported by analysis of phenylalanine/histidine side chain/side chain interactions in proteins (Singh and Thornton, 1992).1 Furthermore, we propose that compounds of other structural types, including the imidazopyridazines, cyclopyrrolones, and β-carbolines, do not derive any binding energy through interaction with His102 and probably do not occupy this area of the binding pocket. In addition, data presented here support the hypothesis that His102 is not involved in transducing the efficacy of agonists or inverse agonists because derivatization of this amino acid has no effect on the ability of GABA to potentiate or inhibit binding of agonist or inverse agonist radioligands.

Modeling of the BZ pharmacophore based on the reduced affinity of selected compounds after UV photoincorporation of flunitrazepam. Top, flunitrazepam (green carbon atoms; other atoms in CPK colors) superimposed onL-764,488. Bottom, as in thetop but with benzyloxybetacarbolines (orange carbon atoms; other atoms in CPK colors) as taken from theZhang et al. (1995) pharmacophore. Dotted spheres, coincide approximately with the hydrogen-bonding sites described by Zhang et al. (1995) as H1 (top edge of figures) and H2 (bottom).
Data derived from these studies also allow further evaluation of the BZ site pharmacophore. We propose that compounds that dramatically lose affinity after photoincorporation of flunitrazepam share a common binding area, whereas those that show no affinity shift do not occupy this space in the receptor. It is possible to model how these molecules might overlay each other as described in Fig. 3. In the comprehensive model of the BZ receptor pharmacophore developed by Zhang et al. (1995), the pendant phenyl group of flunitrazepam is proposed to interact with a lipophilic pocket in the receptor, termed L3. The results of work presented here would put that in close association to, and potentially in direct interaction with, His102 of the α subunit.Zhang et al. (1995) predict that this part of the binding pocket is occupied by the pendant phenyl ring of diazepam, flunitrazepam, chlordiazepoxide, and other 5-phenyl BZs. We propose that the 6-benzyl ring of the naphthyridones also occupies this space. However, the model proposed by Zhang et al. (1995) predicts that the phenyl substituent on the agonist β-carboline abecarnil overlays the 5-phenyl substituent of flunitrazepam. Our experimental data do not support this orientation because no difference in affinity was observed between the control and flunitrazepam-derivatized membranes. All other structures studied, including the phenyl pyrazoloquinolinones, pyridodiindoles, and BZs, were consistent with this published pharmacophore, in contrast to a number of published overlays, such as that of Villar et al. (1989) or Martinet al. (1993). Both of these latter groups suggest that the space occupied by the 5-phenyl ring of the benzodiazepines would be occupied not only by the benzyl group of the benzyloxy β-carbolines [in agreement with Zhang et al. (1995)] but also by at least one other series of ligands that were studied in this work and found not to be affected by flunitrazepam incorporation.
Acknowledgments
We thank Prof. J. Cook for the gifts of pyridoindole 1, pyridoindole 2, ZK93423, and β-CCM and β-CCT.
Footnotes
- Received December 26, 1997.
- Accepted March 13, 1998.
-
Send reprint requests to: Dr. Ruth McKernan, Department of Biochemistry, Merck Sharp and Dohme Research Laboratories, Terlings Park, Eastwick Road, Harlow, Essex CM2O 2QR, UK. E-mail:ruth_mckernan{at}merck.com
-
↵1 http://www.biochem.ucl.ac.uk/bsm/sidechains/His/Phe/sindex.html
Abbreviations
- GABA
- γ-aminobutyric acid
- BZ
- benzodiazepine
- DMCM
- methyl-6,7-4-dimethoxy-4-ethyl-β-carboline-3-carboxylate
- β-CCM
- 3-carbomethoxy-β-carboline
- β-CCT
- 3-carbo(t-butyloxy)-β-carboline
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}