


Abstract
Some, perhaps all, G protein-coupled receptors form homo- or heterodimers. We have shown that metabotropic glutamate receptors are covalent dimers, held together by one or more disulfide bonds near the N terminus. Here we report how mutating cysteines in this region affect dimerization and function. Covalent dimerization is preserved when cysteines 57, 93, or 99 are mutated but lost with replacement at 129. Coimmunoprecipitation under nondenaturing conditions indicates that the C[129]S mutant receptor remains a dimer, via noncovalent interactions. Both C[93]S and C[129]S bind [3H]quisqualate, whereas binding to C[57]S or C[99]S mutants is absent or greatly attenuated. The C[93]S and C[129]S receptors have activity similar to wild-type when assayed by fura-2 imaging of intracellular calcium in human embryonic kidney cells or electrophysiologically in Xenopus laevis oocytes. In contrast, C[57]S or C[99]S are less active in both assays but do respond with higher glutamate concentrations in the oocyte assay. These results demonstrate that 1) covalent dimerization is not critical for mGlu5 binding or function; 2) mGlu5 remains a noncovalent dimer even in the absence of covalent dimerization; and 3) high-affinity binding requires Cys-57 and Cys-99.
Many classes of receptors, such as the tyrosine kinase-linked receptors and the ligand-gated ion channels, function as di- or oligomeric assemblies of polypeptides. In contrast, G protein-coupled receptors (GPCRs) have traditionally been thought to exist and operate as monomers. This view has been changing. For example, early indirect evidence, from radiation inactivation target-size analysis (Venter and Fraser, 1983) and gel electrophoresis of receptors (Brett and Findlay, 1979; Avissar et al., 1983), suggested that GPCRs are part of larger multimeric structures. More recently, direct functional and structural studies indicated that at least some GPCRs are multimers, most often homodimers. For instance, chimeric receptors composed of an N-terminal sequence from a muscarinic receptor and C-terminal half from an α-adrenergic receptor neither bind ligand nor activate effector systems, but these functions are restored by coexpression with a chimera composed of an adrenergic N-terminal half and a cholinergic C-terminal half (Maggio et al., 1993). Similarly, expression of the metabotropic γ-aminobutyric acid (mGABA1 or GABA-B1) receptor yields a polypeptide that binds radiolabeled antagonist but does not bind agonist or couple to G protein-coupled inwardly rectifying potassium channels unless the mGABA2 receptor polypeptide is coexpressed (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998; Kuner et al., 1999; Ng et al., 1999). Such complementation experiments indicate that receptor functioning requires interactions between more than one receptor polypeptide. Studies demonstrating that δ- and κ-opioid receptors individually form homodimers and also heterodimerize to form a hybrid with unique properties (Cvejic and Devi, 1997; Jordan and Devi, 1999) further substantiate a dimer/oligomer model.
Diverse structural mechanisms underlie GPCR dimerization. For example, catecholamine receptor dimers are held together by noncovalent protein-protein interactions involving the transmembrane domains (Hebert et al., 1996; Ng et al., 1996), whereas the mGABA (Kuner et al., 1999) and δ-opioid receptors (Cvejic and Devi, 1997) dimerize via noncovalent interactions mapped to regions in the intracellular C-terminal region. We have shown that dimers of mGluRs are covalently linked by disulfide bonds located extracellularly, within ≈17 kDa of the N terminus (Romano et al., 1996). Subsequently, the Ca2+-sensing receptors (CaRs), which are homologous to the mGluRs and share many conserved cysteines, have also been shown to dimerize by this mechanism (Bai et al., 1998). Finally, there is evidence that muscarinic cholinergic receptors may covalently dimerize via extracellular cysteines located in the o2 and o3 loops (Zeng and Wess, 1999).
In this study, we examined the structural basis and functional role of dimerization of mGlu5 using a set of mutant receptors. We demonstrate that Cys-129 is critically important for covalent dimerization but conclude that this interaction is not necessary for agonist binding or receptor function. We propose that Cys-57 and Cys-99 participate in an intramolecular disulfide bond not critical for covalent dimerization, but important for the integrity of the ligand binding site. We also demonstrate that novel noncovalent interactions, probably involving the extracellular domains, participate in mGluR dimerization.
Materials and Methods
Construction of Mutant Receptors.
All of the point mutations introduced into the mGlu5 coding sequence were generated by recombinant polymerase chain reaction (Higuchi, 1990) using sense and antisense primers containing the relevant cysteine to serine mutation. Primers included: 5′-AGAGGAAGTCTGGTGCAG-3′, C[57]S; 5′-CACTTGGCTCTGAGATCA-3′, C[93]S; 5′-AGATTCCTCCTGGCATTC-3′, C[99]S; 5′-TGGTACGCTCTGTAGATG-3′, C[129]S; and their complements. The C[57,93,99,129]S mutant as well as all other multiple mutations were generated by using one of the single mutation constructs as a template together with an additional mutant primer pair. The resulting double mutant then served as a template for further mutagenesis etc. The truncated wild-type or hemagglutinin (HA)-tagged mGlu5 construct was described previously (Romano et al., 1996). The truncated mutant constructs were created by digesting their respective full-length clones with BstEII and NotI then ligating them with a truncated wild-type fragment encoding the first transmembrane region and C-terminal domain cut with the same enzymes. All of the mutant full-length and truncated clones were confirmed by sequencing.
Cell Culture and Transfections.
HEK cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum (Life Technologies, Grand Island, NY) and incubated at 37°C with 5% CO2. For experiments requiring Western blotting and/or immunoprecipitation, plasmid DNA was transfected into subconfluent HEK cells with either LT1 (PanVera, Madison, WI) or Fugene6 (Roche Diagnostics, Nutley, NJ) according to the manufacturer's instructions, using a ratio of 3 μl of transfecting reagent per 1 μg of DNA. Cells were harvested 24 h after transfection.
For ratiometric Ca2+ imaging, DNA was introduced into HEK cells via electroporation, by a modification of published protocols (Puchalski and Fahl, 1992; Gomeza et al., 1996; Ishmael et al., 1996). DNA (8 μg) was electroporated into 3 to 4 × 106 HEK cells in 300 μl of Ca2+-, Mg2+-free PBS with a Bio-Rad Gene Pulser (0.4-cm electrode gap cuvette, 2 pulses, 25 μF, 600 ohms, 1 kV). Cells were subsequently kept at room temperature for 5 min, diluted in Dulbecco's modified Eagle's medium enriched with 10% fetal calf serum, and plated on poly-d-lysine-treated dishes with 35-mm grids (MatTek, Ashland, MA). Cells were allowed to recover for 15 h before imaging.
Membranes, SDS-Polyacrylamide Gel Electrophoresis and Western Blotting.
HEK cells transfected with plasmids encoding wild-type (wt) or mutant mGlu5 were washed twice with PBS, then incubated for 10 min at room temperature in PBS containing 12 mMN-ethylmaleimide. This buffer was replaced by ice-cold lysis buffer composed of 2 mM HEPES, 2 mM EDTA, plus protease inhibitors (Complete tablets, Boehringer Mannheim, Germany). Cells were scraped and then disrupted in a tight-fitting, motor-driven, Teflon and glass homogenizer. Nuclei were pelleted (1000g, 5 min) and discarded. Membranes were pelleted by centrifugation (35,000g, 40 min) and usually used without further washes. Membrane proteins were separated by electrophoresis in 7.5% Laemmli gels. The sample buffer for reducing gels contained 20 mM dithiothreitol (DTT) or 5% 2-mercaptoethanol. Samples were incubated at 60°C for 3 min before loading; samples were never boiled. Western blotting was done as described previously (Romano et al., 1996), using polyvinylidene difluoride membranes (Immobilon; Millipore Corp., Milford, MA) and chemiluminescent visualization. Antibody against mGlu5 was an affinity-purified polyclonal raised against the C-terminal 13 amino acids, as described previously (Romano et al., 1996). Antibodies against the HA epitope were from BABCO (Berkeley, CA).
Immunoprecipitations.
For immunoprecipitations, membranes were homogenized in immunoprecipitation buffer (40 mM HEPES, 400 mM NaCl, protease inhibitors as above, pH 7.5) containing either 0.5% SDS (at 60°C for complete, denaturing solubilization) or 0.5% dodecyl maltoside (at room temperature, for gentle, nondenaturing solubilization). The SDS extract was diluted 5-fold into immunoprecipitation buffer containing 0.5% dodecyl maltoside (to sequester free SDS into mixed micelles, thereby permitting immunoprecipitation) and protease inhibitors. The solubilized extract was then centrifuged at 100,000g for 35 min to remove any undissolved material, anti-receptor antibody was added, and the solution was incubated at 4°C overnight. Protein A (or protein G)-Sepharose (Sigma, St. Louis, MO) was added, and the incubation was continued for 2 h at room temperature on a rocking table. The protein A pellets were washed three times with PBS before elution with sample buffer and electrophoresis.
[3H]Quisqualate Binding Assay
To increase the yield of receptors and thereby maximize signal, total cell membranes were used for the [3H]quisqualate binding assay. Cells were washed twice in PBS (without N-ethyl maleimide, which decreased binding), lysed in hypotonic lysis buffer as above, and centrifuged at 17,000g for 35 min. The pellet was resuspended in buffer containing 40 mM HEPES, 2.5 mM Ca2+, and protease inhibitors. Usually, 600 nM [3H]quisqualate was present in a final assay volume of 100 μl. Incubation was for 60 min at 25°C, and bound label was separated from free label by fast filtration over #30 filters (Schleicher & Schuell, Keene, NH). Nonspecific binding was determined in the presence of 1 mM glutamate.
Single Cell Fluorescent Ratiometric Measurements of Intracellular Ca2+.
Cytosolic calcium determination upon stimulation of cells with glutamate or carbachol was performed using the fluorescent calcium indicator fura-2 (Grynkiewicz et al., 1985) as described previously (Hyrc et al., 1997). The fura-2 was bath-loaded at 37°C by incubation for 30 min with 6 μM fura-2/AM (Molecular Probes, Eugene, OR) and 0.12% of Pluronic F-127, followed by another 30-min incubation at 37°C to allow for hydrolysis of the AM ester. Calcium measurements were carried out using standard ratio-imaging techniques. Cells loaded with fura-2 were imaged on an inverted microscope (Nikon Diaphot, Nikon Inc., Melville, NY) using a 40×, 1.3 numerical aperture fluorite oil immersion objective (Nikon) and an ICCD camera (Hamamatsu Photonics, Oak Brook, IL). A 75-W xenon arc lamp was used to provide fluorescence excitation. The excitation wavelengths were selected by using band-specific filters (340HT15 and 380HT15; Omega Optical, Brattleboro, VT) in combination with an XF73 dichroic mirror (Omega Optical).
Ratio images were obtained by acquiring pairs of images at alternate excitation wavelengths (340/380 nm) and filtering the emission at >510 nm (EFLP filter, Omega Optical). Image acquisition and processing were controlled by a personal computer connected to the camera and filter wheel (MetaFluor, Universal Imaging Corp., West Chester, PA). Image pairs were captured every 10 s and digitized, and the images at individual wavelengths were averaged over four frames. All imaging experiments were carried out at room temperature in a HEPES- and bicarbonate-buffered salt solution containing 116 mM NaCl, 5.4 mM KCl, 1 mM NaH2PO4, 25 mM NaHCO3, 1.8 mM CaCl2, 1 mM MgSO4, 12 mM HEPES, and 5.5 mMd-glucose, pH 7.45 ± 0.2.
Immunocytochemistry.
After image analysis, cells were analyzed immunocytochemically to confirm receptor expression. Cells were washed with PBS for 5 min, fixed for 15 min at room temperature with 4% paraformaldehyde in PBS, washed with PBS for 5 min, and further processed or stored in 5 ml of PBS at 4°C until analyzed for receptor content the next day. The cells were washed two more times for 5 min per wash; blocked for 30 min with 1% bovine serum albumin, 0.1% Triton, in PBS; incubated 1 h at 37°C in 1% bovine serum albumin, 0.1% Triton in PBS containing the polyclonal rabbit anti-mGluR5; washed three times for 5 min per wash with PBS; incubated in the dark for 45 min at room temperature with goat-anti-rabbit Cy3 (Jackson ImmunoResearch, West Grove, PA) in 1% bovine serum albumin, 0.1% Triton in PBS; washed three times for 5 min per wash with PBS; stored in PBS; and observed and photographed under fluorescence at magnification 200. In the case of N-terminally HA-tagged receptors (see Fig. 5C), surface expression was confirmed by performing the labeling in the absence of detergent, using anti-HA (Covance, Richmond, CA) as primary antibody.

Changes in intracellular free [Ca2+] induced by glutamate in HEK cells expressing full-length wt and cysteine-mutated mGlu5. A, representative traces from two cells for each of six different receptors. The first bar under each of the traces indicates the time of exposure to 1 mM glutamate; the second bar indicates the time of exposure to 1 mM carbachol. B, compiled data from all cells. Each data point represents the maximum response to glutamate in a single cell, and the horizontal bars show the average responses of all cells for each mutant. The number of cells of each type is indicated. The statistical significance of the differences (α = 0.05) was assessed using one-way ANOVA on ranks. ∗, different from those of nontransfected controls; #, different from wild-type. The cells included in the analysis were pooled from 4 to 13 independent experiments (2 for vector controls). C, immunocytochemical labeling under nonpermeabilizing conditions using an anti-HA-antibody demonstrates surface expression of N-terminally HA-tagged mGlu5 (wild-type, C[57]S, or C[99]S).
Activation of Ca2+-Dependent Cl−Channels in Xenopus laevis Oocytes.
Linearized pcDNA3 plasmids containing wild-type or mutant mGlu5 coding sequences were transcribed using an in vitro transcription kit (mMESSAGE mMACHINE, Ambion, Austin, TX). Transcript levels and integrity were verified on formaldehyde RNA gels. Oocytes were harvested from X. laevis under tricaine (0.1%) anesthesia. Oocytes were enzymatically defolliculated in a collagenase (2 mg/ml) saline solution containing 96 mM NaCl, 2.5 mM KCl, 1.0 mM MgCl2, and 10 mM HEPES, pH 7.4 at 37°C for 20 min. Oocytes were maintained at 18°C in the above saline solution with 1.8 mM CaCl2 and supplemented with theophylline (0.5 mM) and sodium pyruvate (0.55 mg/ml). Individual oocytes were injected with 40 ng of wild-type or mutant complementary RNA and were assayed electrophysiologically 2 to 8 days after injection.
Oocyte electrophysiology was performed using an Axoclamp 2-B amplifier (Axon Instruments, Foster City, CA) in the two-electrode voltage-clamp configuration. Recording pipettes filled with 3 M KCl had an open tip resistance of approximately 1 MΩ. For recording, individual oocytes were washed for 15 to 30 min with calcium-containing saline solution lacking the theophylline and pyruvate supplements. Oocytes were placed in a 100-μl chamber and were constantly perfused (2 ml/min) during the experiment with unsupplemented saline solution.
Endogenous calcium-dependent chloride currents, activated by 20-s applications of 100 μM glutamate, were used to detect mGluR activation. Oocytes were clamped at −20 mV, near the chloride equilibrium potential to avoid intracellular chloride shifts during mGluR activation. The membrane potential was briefly (20 ms) pulsed to +30 mV once per second. The outward chloride current at the end of this 20-ms voltage pulse was measured before, during, and after perfusion of glutamate.
Results
Mutating Cysteine 129 Disrupts Covalent Dimerization of mGlu5.
Previous results indicated that the disulfide bond(s) responsible for covalent dimerization of mGlu5 involve(s) a cysteine or cysteines located within ≈17 kDa of the N terminus (Romano et al., 1996). There are four cysteines in this region, residues 57, 93, 99, and 129. Three of these, 57, 99, and 129, occupy positions conserved across the entire family of mGluRs and CaRs.
To determine whether these cysteines were involved in disulfide-dependent dimer formation, we adopted the strategy of mutating to serine and examining the consequences of these manipulations for receptor structure. Under nonreducing conditions, receptors mutagenized at Cys-129 migrated at the monomeric molecular weight, whereas wt, C57S, C93S, and C99S migrated as dimers (Fig.1). These data indicate that Cys-129 participates in interactions critical for covalent dimerization.

Western blot analysis of wild-type and cysteine-mutated mGlu5 expressed in HEK cells. Samples were prepared without reduction. The numbers above the gel lanes signify the position of the cysteine that has been changed to serine.
Previous results indicated that a truncated form of mGlu5 (composed of the extracellular N terminus, one transmembrane domain, and the first nine amino acids of intracellular loop 1 in-frame with the final 32 amino acids of the C terminus) was also a covalent dimer, and, when coexpressed, would assemble into heterodimers with the full-length receptor (Romano et al., 1996). When an epitope-tagged version of this truncated receptor (tm5HA) was coexpressed with each of the set of full-length receptors mutated at individual cysteines, heterodimers formed between tm5HA and the wt, C57S, C93S mutants, but not with C99S or C129S (Fig.2).

Western blot analysis of HA-tagged, truncated mGlu5 coexpressed with wild-type and cysteine-mutated full-length mGlu5 in HEK cells. Samples were prepared without reduction. The numbers above the gel are as in Fig. 1. A monoclonal antibody to the HA-epitope was used for visualization of the HA-tagged truncated mGlu5.
We then examined the behavior of single and multiple cysteine mutants of the truncated mGlu5. When electrophoresed under nonreducing conditions, truncated mGlu5with wild-type cysteines migrated as a dimer, but the tetra-mutated tC[53,93,99,129]S migrated as a monomer (Fig.3, wt, t[57,93,99,129]). This is consistent with the hypothesis that one or more of these cysteines participate in the disulfide bond necessary for covalent dimerization. When tCys-57, tCys-93, or tCys-99 were individually mutated to serine, a small fraction of each of these truncated receptors migrated as monomer under nonreducing conditions, but the majority appeared to be intact dimer. The tC[129]S mutant, on the other hand, migrated nearly exclusively as a monomer (Fig. 3). Interestingly, for all the mutants examined, there was often a fraction of the receptor polypeptide that migrated at a molecular weight greater than that of the dimer, although this was variable from experiment to experiment. This suggests that assembly of mutant truncated receptors may be imperfect. When Cys-129 was left intact, but Cys-57, Cys-93, and Cys-99 were altered to serines, covalent dimerization was also lost. In fact, when any combination of three of the first four cysteines was altered, dimerization was lost (Fig. 3). Collectively, these data strongly support the contention that covalent dimerization of mGlu5 critically depends on Cys-129.

Western blot analysis of wild-type and cysteine-mutated truncated mGlu5 expressed in HEK cells. For the top gel, samples were prepared without reduction; for the bottom gel, samples were reduced using 20 mM DTT. Each lane represents a different mutant. The numbers above the gel lanes signify the position of the cysteine or cysteines that have been changed to serines. Under reduced conditions, all receptors migrated as ≈80 kDa monomers. Under nonreduced conditions, the wt, C[57]S, C[93]S, and C[99]S mutants migrate mostly as dimers, whereas the C[129]S and the multiply mutated receptors, migrate primarily as monomers.
Noncovalent Dimerization of Truncated mGlu5 Is Present in tC[129].
The absence of disulfide-mediated covalent dimerization does not preclude the possibility that mutant receptor polypeptides may exist as dimers held together by noncovalent protein-protein interactions. To test for noncovalent interactions dependent on the N-terminal region of the receptor, coimmunoprecipitation experiments were performed using the truncated receptors under conditions that would either preserve or disrupt noncovalent interactions. Cells were cotransfected with the HA-tagged truncated wild-type mGlu5, together with truncated receptors containing the wild-type C-terminal epitope but mutated at selected cysteines. Membranes were prepared from these cells, then solubilized under gentle (room temperature, buffer containing 0.5% dodecyl maltoside) or denaturing (0.5% SDS, 3 min heating at 60°) conditions. An aliquot of each membrane preparation was used to confirm receptor expression by Western blotting (Fig.4, top two panels). The remainder of each extract was immunoprecipitated with antibody against the wild-type C terminus, and the presence of HA-tagged mutant receptor in the immunoprecipitate was determined by Western blotting. Despite denaturation, the HA tagged-truncated receptor was coimmunoprecipitated when it was expressed in the presence of truncated wild-type receptor, as expected for a covalent heterodimer (Fig. 4C). Similar results were observed with C[57]S, C[93]S, and C[99]S (data not shown) as expected, because these receptors also appear to be covalent dimers (Fig. 3). Mutation of the first four cysteines, or of Cys-129 individually, led to the loss of coimmunoprecipitation under the denaturing conditions (Fig. 4C). This is consistent with the results presented in Figs. 1 through 3 demonstrating the absence of covalent dimerization in these mutants. In contrast, when gentler, nondenaturing conditions were used to solubilize the membranes, HA-tagged receptor was present in the immunoprecipitate when either t-wt or tC[129]S, but not tC[57,93,99,129]S, were coexpressed (Fig. 4D). These data indicate that the tC[129]S mutant exists as a noncovalent dimer with the receptor containing wild-type cysteines. This noncovalent dimerization is not present when all of the first four cysteines have been mutated.

Coimmunoprecipitation of the epitope-tagged receptor together with the cysteine-mutated receptors demonstrates covalent and noncovalent dimerization. HEK cells were transfected with truncated mGlu5 containing the HA epitope at the C terminus (−/HA) or cotransfected with this receptor and a truncated mGlu5containing the wt C terminus and wt or mutated cysteines as indicated. The top panel demonstrates expression of the receptors bearing the wt C terminus and the cysteine alterations. The second panel demonstrates expression of HA-tagged receptor in each cotransfected cell type. The third panel demonstrates that, upon solubilization under denaturing conditions (0.5% SDS, 60°C), only receptors containing all wt cysteines will coprecipitate the HA-tagged receptor. The fourth panel illustrates that, with gentle solubilization (0.5% dodecyl maltoside, room temperature), the C[129]S mutant will also coprecipitate the HA-tagged receptor but the multple mutant will not.
Effect of Cysteine Mutations on [3H]Quisqualate Binding to mGlu5.
To determine whether elimination of covalent dimerization in mGlu5 alters the properties of the receptor binding site, binding studies using [3H]quisqualate as a radioligand were performed. Relative to the full-length wt receptor, the C[57,93,99,129]S mutant had greatly reduced detectable binding of [3H]quisqualate (Table1). When the individual cysteines were mutated, binding was preserved with C[93]S and C[129]S but effectively eliminated with C[57]S or C[99]S. Similar results were obtained when analogous mutations of the truncated receptor were analyzed (Table 1). Receptor expression was confirmed by Western blotting. Because covalent dimerization is lost in the tC[129]S mutants but binding is preserved, agonist binding is not critically dependent on covalent dimerization.
Binding of [3H]quisqualate to mGlu5 and mutants in HEK cell membranes
Accurate values for dissociation constants are very difficult to obtain by saturation analysis using a relatively low affinity radioligand such as [3H]quisqualate. Nonetheless, saturation analysis was performed with full-length versions of the wild-type and C[129]S mutant receptors. Wild-type receptor bound [3H]quisqualate with aK D value of 514 nM (±187 nM,N = 3 ± S.E.M.), whereas the C[129]S mutant bound with a K D value of 583 nM (±57 nM,N = 3 ± S.E.M.). Thus, the binding affinities for [3H]quisqualate are similar between the wild-type and C[129]S mutant, although subtle differences may exist that will be more readily assessed when higher affinity radioligands are available.
Effect of Cysteine Mutations on mGlu5-Induced Calcium Mobilization in HEK Cells.
Glutamate-induced mobilization of intracellular calcium was analyzed by fura-2 fluorescence microscopy in HEK cells transiently transfected with wt or mutant full-length mGlu5. Representative responses of two individual cells transfected with the indicated mutant are shown in the upper part of Fig. 5; a compilation of the peak responses from all cells are shown below. All the results shown reflect the responses of cells that both responded to carbachol (which activates an endogenous muscarinic receptor on HEK cells) and had confirmed expression of transfected mGlu5 (via post hoc fixation, immunocytochemistry, and field relocation).
Cells transfected with a noncoding vector never showed a calcium response to added glutamate. In contrast, almost all cells expressing wild-type mGlu5 responded to glutamate with an increase in intracellular calcium (Fig. 5). In most cells, the response was oscillatory, and the oscillations persisted for many minutes after washout of glutamate. Similar responses were seen with C[93]S (not shown) and with the dimerization-defective mutant C[129]S (Fig. 5). Many of the cells transfected with the C[57]S mutant had a calcium response, but persistent oscillations were rare. A much smaller fraction of cells transfected with the other mutants responded, but never strongly, or with persistent oscillations. Taken together, these results demonstrate that covalent dimerization is not essential for receptor functioning but alterations in several individual or multiple cysteines disrupt receptor-mediated calcium responses.
Although we only counted responses from individuals cells that had been confirmed immunocytochemically to express the wild-type or mutant receptors, another experiment was performed to better document that the poorly responding single mutants were reaching the cell surface. An HA-epitope tag was added to the N terminus, immediately after the presumed signal sequence, of receptors with all wt cysteines or the C[57]S or C[99]S mutations. Cells were incubated with anti-HA followed by fluorescent secondary antibody without permeabilization and visualized. Similar numbers of cells were labeled with a similar intensity (Fig. 5C). Therefore, mutation of Cys-57 or Cys-99 does not prevent receptor transport and expression on the plasma membrane. The possibility of misfolding leading to altered surface expression or function of the triple mutants has not yet been explored.
Effect of Cysteine Mutations on mGlu5 Activation of Endogenous Ca2+-Dependent Cl− Channels inX. laevis Oocytes.
When expressed in X. laevis oocytes, wild-type Group I mGluRs robustly activate the endogenous Ca2+-dependent Cl− channels. Responses of mGlu5 wild-type and mutant receptors to the addition of 100 μM glutamate were similar to those seen with the calcium-imaging studies (Fig. 6, A and B). Specifically, mutation of nonconserved Cys-93, or covalent dimerization-disrupting Cys-129, maintained an activity level comparable with wild-type, whereas C[57]S, C[99]S, or C[57,93,99,129] gave no, or greatly attenuated, responses. Interestingly, C[57]S and C[99]S respond similarly to wild-type when exposed to 20 mM glutamate (Fig. 6C).
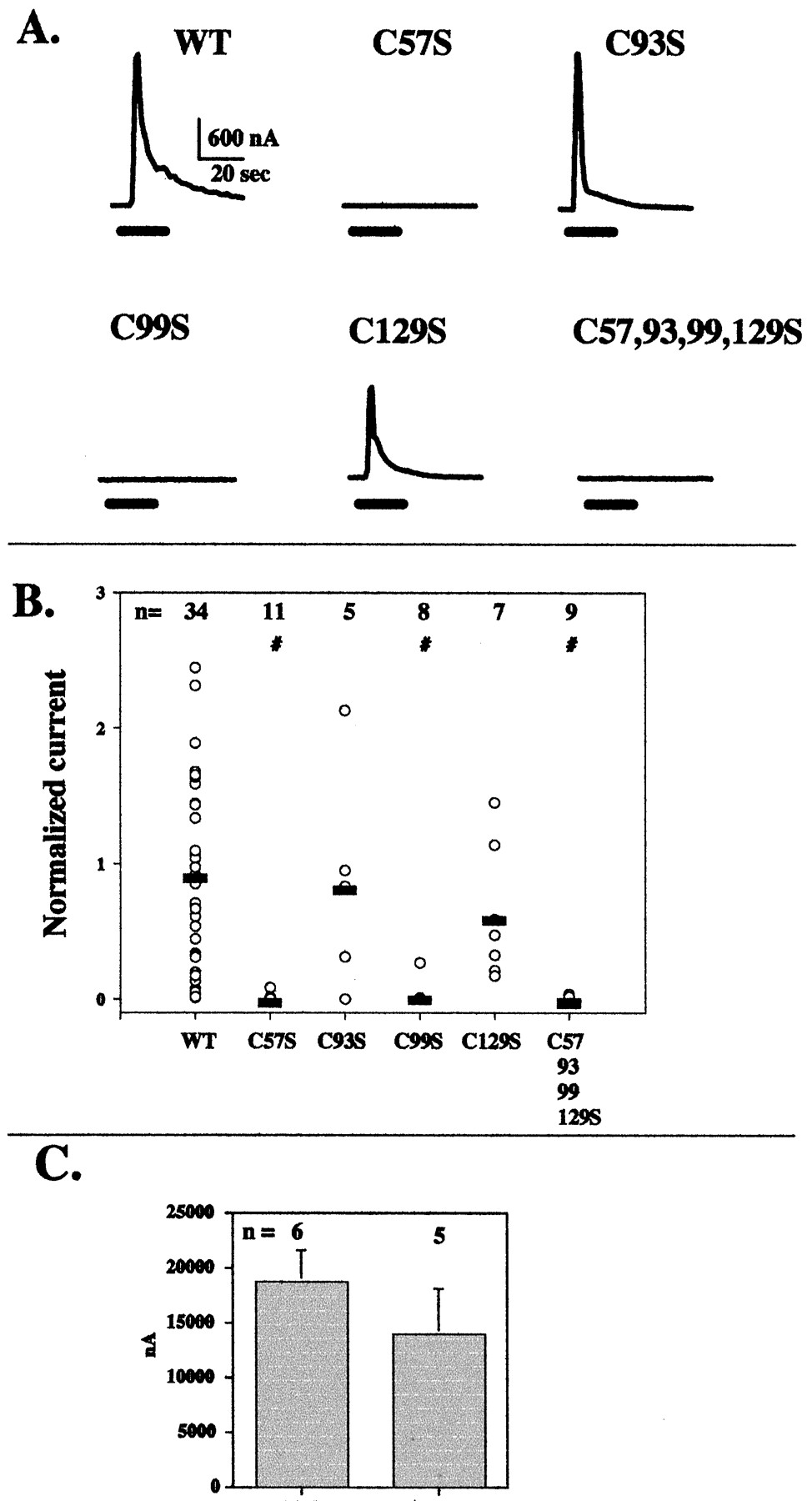
Calcium-dependent chloride currents induced by glutamate in X. laevis oocytes expressing full-length wt and cysteine-mutated mGlu5. A, representative traces for each of five different receptors. The bar under each trace indicates the time of exposure to 100 μM glutamate. Note that wt, C[93]S, and C[129]S responded, whereas the others did not. B, compiled data from all oocytes tested at 100 μM glutamate. Each data point represents the maximum response to glutamate in a single oocyte, and the horizontal bars show the average response of all cells for each mutant. The number of cells tested is indicated. The statistical significance of the differences (α = 0.05) was assessed using one-way ANOVA on ranks. #, different from wt. C, responses to 20 mM glutamate by oocytes injected with C[57]S or C[99]S.
Discussion
The structural bases and functional consequences of GPCR dimerization are just beginning to be explored. Previously, we demonstrated that mGluRs covalently dimerize via extracellular disulfide bonds. The present findings demonstrate that covalent dimerization of mGlu5, mediated by disulfide bonds, is not critical for the functioning of this receptor. This conclusion is based on the identification of a mutant that differs from wild-type by a single amino acid, C[129]S, in which covalent dimerization is absent, yet the receptor is synthesized, exported to the plasma membrane, binds agonist, and serves to mobilize intracellular calcium in a manner indistinguishable from wild-type. The presence of noncovalent dimerization was also identified. Thus, covalent and noncovalent bonds participate in mGluR dimerization.
A model of mGluR structure was previously proposed by O'Hara et al. (1993). These investigators identified sequence and secondary structure homologies between the ionotropic and metabotropic glutamate receptors and a structurally well defined class of bacterial proteins known as the periplasmic binding proteins (PBPs). Based on these similarities, as well as receptor mutagenesis, they proposed a bi-lobed “Venus Flytrap” model for the extracellular ligand-binding domain of mGluRs. Further confirmation of this model, in particular the extracellular location of the binding site in mGluRs, was found in studies showing that binding is preserved when the truncated extracellular domains of mGlu1 or mGlu4 were expressed as secreted, soluble proteins (Okamoto et al., 1998; Han and Hampson, 1999). Additionally, recent chimeric and point mutation studies of the closely related CaR (Brauner-Osborne et al., 1999) and the more distantly related mGABA receptor (Malitschek et al., 1999) demonstrated that the extracellular domain of these receptors also could fold and bind agonists in a manner analogous to the mGluRs. Finally, molecular modeling studies of the mGABA1 receptor suggest an even closer fit to the PBP-like structure than exhibited by mGluRs or CaRs (Galvez et al., 1999).
Our previous work suggesting that the disulfide bond responsible for dimerization was located within 20 kDa of the N terminus, together with the model inferred from the PBP crystallography data (O'Hara et al., 1993). This permitted us to predict a role for Cys-129 in dimerization. Specifically, of the four possible cysteines in this region, Cys-93 is not conserved, and Cys-57 and Cys-99 probably form an intramolecular disulfide bond as described below. Only Cys-129 would be located on the outer edge of the “Venus Flytrap” in a region removed from the core of the binding pocket and not predicted to be near other free cysteines. Inasmuch as the mutant C[129]S failed to covalently dimerize, the proposed model received further confirmation.
High resolution crystallographic studies of at least six different PBPs have established the presence of an intramolecular disulfide bond ensuring that one side of the ligand-binding pocket is covalently held in place (Sack et al., 1989). An analogous pair of cysteines has been conserved in the mGABA receptor, and mutation of these residues leads to the loss of binding. However, DTT treatment of membranes containing wild-type mGABA receptor did not prevent binding, leading Galvez et al. (1999) to suggest that this disulfide bond is important for the correct initial folding, but not maintenance, of the active conformation of the binding site. Interestingly, mGluRs and CaRs, which are less homologous to the mGABA receptors or PBPs and more similar to each other, have not conserved one of the pivotal cysteines involved in the PBP-like intramolecular disulfide. Rather, mGluRs and CaRs have a conserved cysteine located in the insert between the region homologous to the first β-sheet and the first α-helical region of the PBP [see (Brauner-Osborne et al. (1999) and Galvez et al. (1999) for alignments]. The alignment of O'Hara et al. (1993) of the secondary structure elements would place this cysteine (Cys-57 of mGlu5) in close proximity to the cysteine that is completely conserved throughout the known PBPs, mGABAs, CaRs, and mGluRs (Cys-99 of mGlu5). Therefore, given the spatial proximity predicted by the molecular modeling studies (O'Hara et al., 1993; Brauner-Osborne et al., 1999; Galvez et al., 1999), it seems reasonable to propose that Cys-57 and Cys-99 of mGlu5 form an intramolecular disulfide bond that would be in approximately the same position as the disulfide bond of the PBPs. Mutation of these residues would be expected to disrupt function. Indeed, negligible binding of [3H]quisqualate to either C[57]S or C[99]S (or any of the triple mutations, including these residues, data not shown) was observed (Table 1). Moreover, there was no functional response to 100 μM glutamate when either of these mutants were expressed in X. laevis oocytes but the C[57]S mutant did exhibit a transient response to glutamate in the HEK cell Ca2+ mobilization assay. Both C[57]S and C[99]S responded in X. laevis oocytes when tested at 20 mM glutamate. Presumably, this reflects an extant, but greatly decreased affinity of agonists for the C[57]S and C[99]S mutant receptors. Taken together, these results are consistent with important roles for Cys-57 and Cys-99 in the structure of the agonist binding site.
Covalent, disulfide-dependent dimerization has also been reported in a human CaR (Bai et al., 1998), although there are conflicting reports concerning the effects of mutating cysteines on receptor dimerization and function. In one study (Pace et al., 1999), mutating the residue equivalent to mGlu5 Cys-129 (hCaR Cys-131) did not disrupt dimerization, whereas individual mutations at positions homologous to Cys-99 and Cys-240 (hCaR Cys-101 and Cys-236) partially disrupted dimerization, and mutating both residues prevented dimerization. In contrast, Ray et al. (1999) noted that there are two cysteines in this region (hCaR Cys-129 and Cys-131), and covalent dimerization of the receptor is prevented when both are mutated to serine. Pace et al. (1999) did not examine the properties of this double mutant. Interestingly, introduction of a new cysteine between the mutagenized sites restored covalent dimerization (Ray et al., 1999). Thus, either Cys-129 or Cys-131 (or both) of the hCaRs participate in disulfide-dependent dimerization. Functionally, this dimerization-deficient double mutant is properly glycosylated, is expressed on the cell surface, and actually has a higher affinity for Ca2+ than the wild-type receptor (31). These results are quite analogous to the findings presented here.
Our results indicate that mGluRs dimerize via noncovalent bonds as well as disulfide bonds, although neither the mechanism nor the functional role of this noncovalent dimerization has been determined. We have identified some mutants in which noncovalent as well as covalent dimerization has been disrupted (data not shown), however, poor expression or misfolding make straightforward interpretation of such results difficult at present. Because the tetramutated tC[57,93,99,129]S lost the ability to coimmunoprecipitate under nondenaturing conditions, this may indicate that the extracellular domain contains moieties critical to noncovalent dimerization. We cannot rule out, however, the possibility that the single remaining transmembrane portion of this truncated receptor affects this process. Additional experiments will be required to address this model. Because each functional mutant we have studied is either a covalent or noncovalent dimer, dimerization may be required for functioning. This has not been adequately tested however. Localization of the specific residues involved in noncovalent dimerization should help elucidate the role that the dimerized state plays in the functioning of the mGluRs.
Several different types of protein-protein interactions have been described as responsible for the noncovalent bonds between GPCR polypeptides. For example, D2 and β-adrenergic receptor dimers appear to involve interactions between transmembrane domains (Hebert et al., 1996; Ng et al., 1996), whereas dimerization of mGABA receptors involves a region of the intracellular C-terminal domain (Kuner et al., 1999). Our experiments demonstrate that a site of noncovalent interaction between mGlu5 monomers is likely to be in the extracellular domain. Dimerization of the bradykinin B2 receptor has recently been shown to involve the extracellular N terminus (AbdAlla et al., 1999).
Given the presence of noncovalent dimers of mGlu5, and the activity of the C[129]S mutant, it is not clear what the precise function of disulfide-mediated covalent dimerization of mGluRs may be. Because it is not required for agonist binding or signal transduction measured in the standard ways, it must serve a more elusive role. Mechanistically, covalent dimerization might “lock-in” noncovalent interactions, thereby contributing to dimer specificity and/or stability. An additional speculation is that covalent dimerization is related to modulation of signal transduction, perhaps involving receptor desensitization or the kinetics of ligand binding or second-messenger formation. Alternatively, covalent dimerization may be important for a function not directly related to signal transduction, perhaps involving interaction with an extracellular target on the plasma membrane of the pre- or postsynaptic neuron, a glial cell, or the extracellular matrix.
Footnotes
- Received March 29, 2000.
- Accepted September 25, 2000.
-
Send reprint requests to: Carmelo Romano, Ph.D., Department of Ophthalmology & Visual Sciences, Campus Box 8096, Washington University School of Medicine, 660 South Euclid Ave., St. Louis, MO 63110. E-mail: romano{at}vision.wustl.eduemail
-
This work was supported by National Institutes of Health Grants MH57817 and EY02687, an unrestricted grant from Research to Prevent Blindness, and the McDonnell Center for Cellular and Molecular Neuroscience.
Abbreviations
- GPCR
- G protein-coupled receptor
- mGluR
- metabotropic glutamate receptor
- HA
- hemagglutinin epitope
- HEK
- human embryonic kidney
- AM
- acetoxymethyl ester
- t
- truncated
- h
- human
- wt
- wild-type
- DTT
- dithiothreitol
- mGABA
- metabotropic γ-aminobutryic acid
- CaR
- Ca2+-sensing receptor
- PBP
- periplasmic binding protein
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}