


Abstract
Prostaglandin (PG) E2 is a potent inducer of cortical and trabecular bone formation in humans and animals. Although the bone anabolic action of PGE2 is well documented, the cellular and molecular mechanisms that mediate this effect remain unclear. This study was undertaken to examine the effect of pharmacological inactivation of the prostanoid receptor EP4, one of the PGE2 receptors, on PGE2-induced bone formation in vivo. We first determined the ability of EP4A, an EP4-selective ligand, to act as an antagonist. PGE2 increases intracellular cAMP and suppresses apoptosis in the RP-1 periosteal cell line. Both effects were reversed by EP4A, suggesting that EP4A acts as an EP4 antagonist in the cells at concentrations consistent with its in vitro binding to EP4. We then examined the effect of EP4 on bone formation induced by PGE2in young rats. Five- to 6-week-old rats were treated with PGE2 (6 mg/kg/day) in the presence or absence of EP4A (10 mg/kg/day) for 12 days. We found that treatment with EP4A suppresses the increase in trabecular bone volume induced by PGE2. This effect is accompanied by a suppression of bone formation indices: serum osteocalcin, extent of labeled surface, and extent of trabecular number, suggesting that the reduction in bone volume is due most likely to decreased bone formation. The pharmacological evidence presented here provides strong support for the hypothesis that the bone anabolic effect of PGE2 in rats is mediated by the EP4 receptor.
Prostaglandins, especially PGE2, have multiple effects on bone, including stimulation of both resorption and formation (Raisz at al., 1993; reviewed in Bergmann and Schoutens, 1995). PGE2administered to rats in vivo increases cortical as well as trabecular bone mass (Jee et al., 1985, 1987; Mori et al., 1990; Suponitzky and Weinreb, 1998). PGE1, an alternate agonist with the same activity spectrum as PGE2, was shown to stimulate bone formation and cause hyperostosis in infants (Ueda et al., 1980; Ringel et al., 1982).
Despite extensive documentation of in vivo bone anabolic effects, the cellular and molecular mechanisms that mediate PGE2 action remain unclear. In organ culture of fetal rat calvaria, PGE2 stimulates DNA synthesis in the periosteum, but suppresses collagen production (Raisz and Koolmans-Beynen, 1974). In the mouse MC3T3-E1 osteoblastic cell line, low concentrations of PGE2 increase cell proliferation, and high concentrations stimulate differentiation (Hakeda et al., 1986). These effects correlate with an increase in intracellular calcium and cAMP, respectively. In cultures of adult rat calvaria cells, PGE2 stimulates nodule formation, via a Ca2+ dependent pathway (Kaneki et al., 1999). In rat RP-11 periosteal cells (Machwate et al., 1998), PGE2 increases cell number in vitro by suppressing apoptosis, without affecting proliferation. Similar effects were obtained using PGE1 and forskolin, suggesting mediation via increased cAMP. It is thus unclear which of these biological responses and intracellular signaling pathways are more relevant to the bone anabolic effects of PGE2 in vivo. Local administration of PGE2 or E1 into long bones in rats stimulates new bone formation (Jee et al., 1985), suggesting that PGE2 acts directly on bone tissue to induce osteogenesis. PGEs bind to four subtypes of cell-surface receptors, EP1–4 (reviewed in Narumiya et al., 1999;Sugimoto et al., 2000). These receptors belong to the G protein-coupled seven transmembrane domain family of receptors and activate either adenylate cyclase or phospholipase C (PLC). EP4and EP2 activate adenylate cyclase, EP1 activates PLC, and EP3inhibits adenylate cyclase, although EP3C-terminal splice variants can activate adenylate cyclase or PLC when expressed in recombinant systems. Prostaglandin receptors are expressed in a wide variety of cells and tissues (reviewed in Narumiya et al., 1999; Sugimoto et al., 2000). In MC3T3-E1 osteoblastic cells, PGE2 stimulates both cAMP and phosphatidylinositol signal transduction pathways (Hakeda et al., 1986). Accordingly, both EP1 and EP4 were found to be expressed in these cells (Suda et al., 1996). In addition, EP1, EP2, and EP4 were found to be expressed in preosteoblasts and osteoblasts in fetal bone tissues by in situ hybridization (Kasugai et al., 1995). EP3was found to be expressed in perichondrial cells. Analysis of the role of the individual EP subtypes in PGE2 action on bone has been relatively limited because of the lack of specificity and limited efficacy of available agonists and antagonists for these receptors (Ono et al., 1998; Kozawa et al., 1998). However recent findings (Pan et al., 1998), using mice deficient in EP receptors, showed that those lacking EP2 or EP4 have defects in bone metabolism. Interestingly, the EP4 deficient mice showed a marked decrease in histomorphometric parameters of bone formation as compared with the EP2 deficient mice. In addition, we found, by Northern blot analysis, that only EP4, but not EP2, was detected in adult bone tissue. Together these data suggest that among the EP receptors, EP4 may play a more predominant role in bone anabolic action of PGE2.
In this study, we examined the effect of an EP4specific antagonist, EP4A, on bone formation induced by PGE2 in young rats. We found that EP4A suppresses the increase in bone mass induced by PGE2. This effect is accompanied by a reduction in the extent of calcein-labeled surface and trabecular number. Our data suggest that EP4 is the main receptor through which PGE2 induces bone formation in rats.
Materials and Methods
Prostanoid Receptor Radioligand Binding Assays.
EP4A [4′-[3-butyl-5-oxo-1-(2-trifluoromethyl-phenyl)-1,5-dihydro-[1,2,4]triazol-4-ylmethyl]-biphenyl-2-sulfonic acid (3-methyl-thiophene-2-carbonyl)-amide] was synthesized in Merck Research Laboratories. Prostanoid receptor radioligand binding assays were conducted as described previously for the human (Abramowitz et al., 2000) and rat receptors (Boie et al., 1997).
cAMP Measurements.
RP-1 periosteal cells, like the RP-11 cells (Machwate et al., 1998), are spontaneously immortalized from primary cultures of periosteal cells from 4-week old Sprague-Dawley rat tibia and are cultured in DMEM (Life Technologies, Gaithersburg, MD) with 10% fetal bovine serum (FBS) (JRH Biosciences, Lenexa, KS). These cells do not express osteoblast phenotypic markers in early culture, but upon confluence, they express several osteoblast markers: type I collagen, alkaline phosphatase, and osteocalcin.
RP-1 cells were plated at 50,000 cells/cm2 in 96-well plates (Costar, Cambridge, MA) and were cultured for 2 days in DMEM supplemented with 10% FBS. Cells were pretreated with 3-isobutyl-1-methylxanthine (1 μM) in DMEM for 10 min. Cells were treated for 10 min with PGE2 (0.1 μM) (Biomol, Plymouth Meeting, PA) in the presence or absence of increasing concentrations of EP4A (0–10 μM). Cells were lysed and processed for cAMP measurement by radioimmunoassay according to the manufacturer's recommendations (Amersham Pharmacia Biotech, Piscataway, NJ).
Apoptosis.
RP-1 cells were plated at 50,000 cells/cm2 in 24-well plates (Costar, Cambridge, MA) and were cultured for 2 days in DMEM supplemented with 10% FBS. Cells were cultured for 24 h in DMEM supplemented with 2% FBS in the presence or absence of PGE2 (0.1 μM), or in the presence of a combination of PGE2 (0.1 μM) and EP4A (10 μM). For analysis of apoptosis, the cells were trypsinized (0.25% trypsin, 1 mM EDTA) and single cell suspensions (1–2 million cells/well) were prepared. The cells were washed twice in Ca2+/Mg2+-free phosphate-buffered saline (PBS) and fixed in ethanol/PBS (3: 1 v/v) for 30 min. After centrifugation, cells were washed in PBS and processed for terminal deoxynucleotidyl transferase dUTP nick-end labeling staining according to the manufacturer's recommendations (Oncor, Gaithersburg, MD). Briefly, cells were incubated with nucleotide terminal transferase in the presence of dioxygenin-11-dUTP. Labeled cells were identified using an anti-digoxigenin, phycoerythrin-conjugated antibody. As control, the samples were exposed to the same mixture excluding the terminal transferase. Staining for annexin-V was analyzed with a FACScan flow cytometer (Becton Dickinson, San Francisco, CA). The red fluorescence was excited at 488 nm by the Argon laser beam. The data acquisition and analysis were performed using cellQuest software (Becton Dickinson, San Francisco, CA).
In Vivo Studies.
A total of 40 male Sprague-Dawley rats (Taconic, Germantown, NY), 5–6 weeks old, weighing an average of 135 g at the start of the experiment, were randomly assigned to four groups (n = 10). One group was vehicle-treated (10% ethanol in sterile water), one group was treated with PGE2 (6 mg/kg/day), one group with the EP4 antagonist (EP4A, 10 mg/kg/day), and the last group was treated with PGE2 in combination with the EP4 antagonist (EP4A was given 45 min before PGE2). All animals were treated for 12 days by daily intraperitoneal (i.p.) injection. Two days before sacrifice, all animals were given calcein (i.p., 10 mg/kg BW) to label the sites of active mineralization. At sacrifice, the animals were weighed then euthanized by CO2 inhalation. Blood was collected by cardiac puncture, and tibiae were dissected and processed for histomorphometric analysis. The internal animal experimentation committee approved all protocols.
Plasma Biochemistry.
Blood samples were obtained by cardiac puncture and plasma was immediately frozen. The plasma content of osteocalcin was determined by radioimmunoassay using a commercially available kit, according to the manufacturer's recommendations (Immunotopics International, San Clemente, CA).
Histomorphometric Analysis.
Tibiae were dissected free from soft tissue, fixed in 10% phosphate-buffered formaldehyde, dehydrated in ethanol, and embedded undecalcified in methylmethacrylate (Baron et al., 1983). Longitudinal sections (5 μm) were cut with a Polycut S microtome (Reichert Jung, Heidelberg, Germany) and examined without further staining for dynamic histomorphometry, or stained with Masson's trichrome for static histological measurements. All histomorphometric measurements were carried out in cancellous bone with a semiautomatic image analysis system (System IV; Bioquant, Nashville, TN). Histomorphometric indices were measured in the proximal metaphyseal area (4 mm2) at a distance of 500 μm from the growth plate as described previously (Parfitt et al., 1983). Trabecular bone volume is expressed as the amount of bone within the spongy space. The mineralizing surface (MS/BS) is calculated as the sum of length of calcein labels and expressed in percent of the bone surface. Trabecular number is the number of bone trabeculae present in the proximal metaphysis within the area of measurement.
Statistical Analysis.
Statistical analyses of the data were performed using the statistical package Statview (Abacus Concepts Inc., Berkeley, CA). Differences between treatment groups were tested by one-way ANOVA and unpaired two-tailed Student's t test.P values less than 0.05 at 95% confidence level were considered significant.
Results
EP4A Binds Selectively to EP4 and Antagonizes the Effects of PGE2 on RP-1 Periosteal Cell Line.
EP4A (Fig.1A) is a high-affinity EP4 prostanoid receptor selective antagonist. It effectively competes with [3H]PGE2 binding to both human and rat recombinant EP4, withK i values of 0.024 and 0.032 μM, respectively (Table 1 and Fig. 1B). EP4A is selective for human EP4 over all other members of the human prostanoid receptor family (EP1, EP2, EP3, DP, FP, and IP). In addition, EP4A is at least 200-fold more selective for rat EP4 than the rat EP1, EP2, and EP3 subtypes (Table 1). To determine whether EP4A acts as an antagonist at rat EP4, we used RP-1 periosteal cells, which express EP4 protein (Weinreb et al., 2001) and in which PGE2 increases cAMP intracellular levels. RP-1 periosteal cells were treated for 10 min with PGE2 alone or in combination with increasing concentrations of EP4A, then lysed and intracellular cAMP was measured by radioimmunoassay. As shown in Fig.2A, PGE2 (0.1 μM) increases intracellular cAMP more than 6-fold in this cell line. Treatment with EP4A inhibits PGE2-induced cAMP increases with an IC50 value of 0.1 μM. To determine whether EP4A interferes with PGE2-independent accumulation of cAMP, we tested the effect of EP4A on intracellular cAMP increased by forskolin. Figure 2B shows that EP4A has no effect on intracellular cAMP induced by forskolin, suggesting that the antagonistic effect of EP4A on PGE2-mediated increase of intracellular cAMP is EP4 mediated. These data are in agreement with previous results from Schild analysis demonstrating that EP4A is a high-affinity competitive antagonist (K B of 3–4 nM) opposing EP4-induced increases in cAMP in HEK 293 cells expressing recombinant human EP4. EP4A did not antagonize cAMP increases induced by forskolin in EP4-expressing or EP4-deficient HEK 293 cells (data not shown). To further document the efficacy of EP4A in antagonizing the PGE2 effect, we tested the effect of EP4A on apoptosis in these cells. Figure 2C shows that, as previously reported for the related RP-11 cells (Machwate et al., 1998), PGE2 (0.1 μM) suppresses apoptosis measured by annexin-V binding. Cotreatment with EP4A (10 μM) completely reverses the antiapoptotic effect of PGE2. Together with the receptor binding data, these functional data suggest that EP4A acts as a specific antagonist for prostanoid receptor EP4.

Chemical structure of the EP4 antagonist EP4A and associated radioligand binding data. A, chemical structure of EP4A. B, competition curves for EP4A binding to HEK 293 cell membranes expressing recombinant human or rat EP4. Equilibrium competition binding assays were performed as described under Materials and Methods. Curves are representative of independent experiments with each data point a mean of duplicates.
Competition for radioligand binding to HEK 293 cell membranes expressing recombinant human (h) or rat (r) prostanoid receptors by EP4A
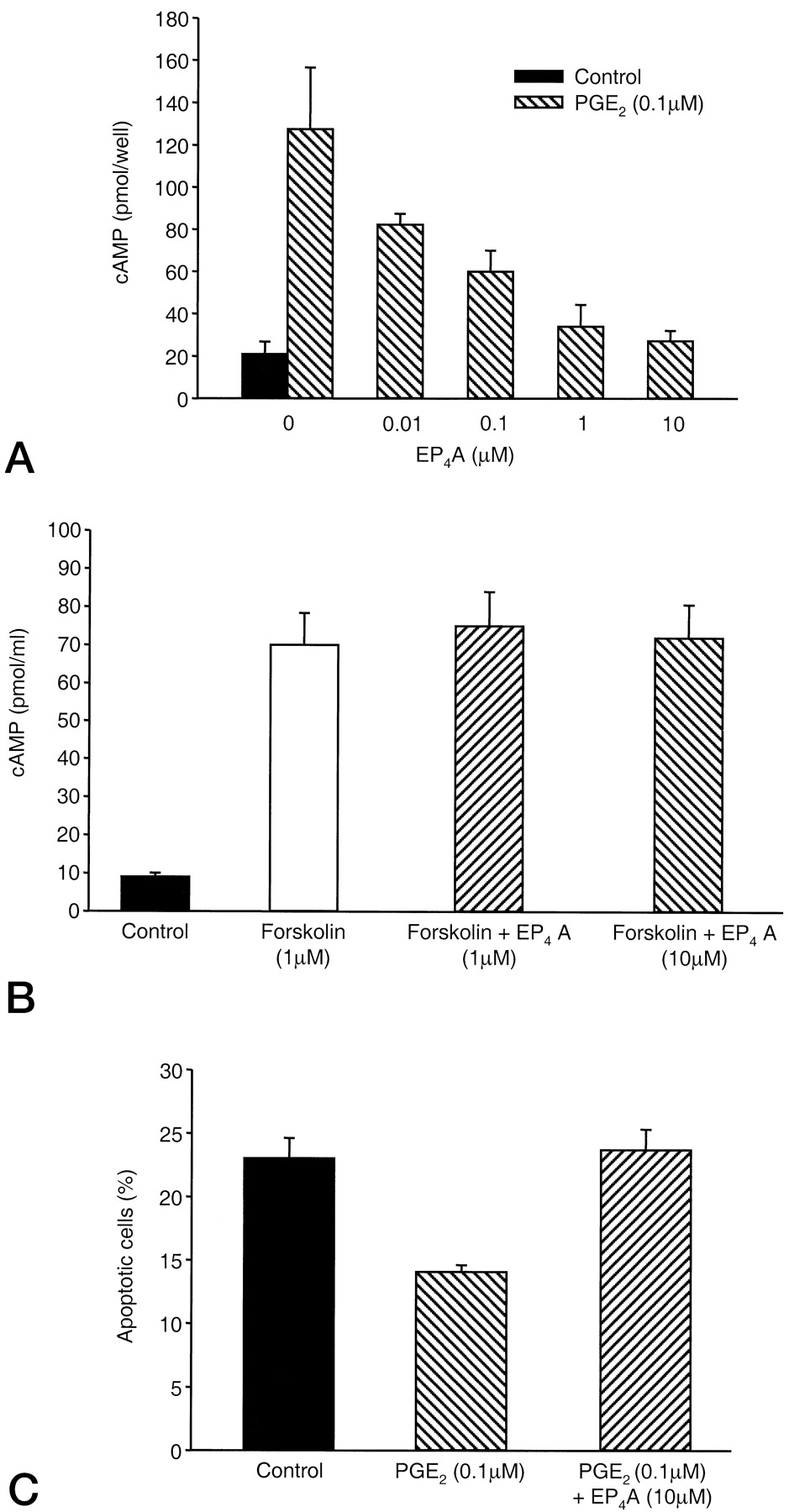
EP4A suppresses the effects of PGE2 on intracellular cAMP accumulation and apoptosis in RP-1 periosteal cells. RP-1 periosteal cells were treated with 0.1 μM PGE2 (A) or 1 μM forskolin (B) in the presence or absence of EP4A (0.01–10 μM). Intracellular cAMP was measured by radioimmunoassay after 10 min of treatment. C, RP-1 periosteal cells were cultured in medium containing low FBS (2%, v/v) in the presence or absence of 0.1 μM PGE2 alone or in combination with 10 μM EP4A. RP-1 periosteal cells were trypsinized and exposed to annexin-V. The percentage of cells bound to annexin-V was quantitated by fluorescence activated cell-sorting (FACS) to determine the percentage of apoptotic cells. Data are reported as mean ± S.D. and are from three experiments.
EP4A Reverses PGE2-Inceased Bone Formation in Rats.
PGE2 was administered at 6 mg/kg/day as described under Materials and Methods. PGE2 has been known to induce diarrhea and decrease body weight. We therefore monitored the animals and evaluated if EP4A interfered with this effect. We observed that diarrhea occurred in the groups treated with PGE2 regardless of the presence of EP4A. Furthermore, we found that treatment with EP4A does not effect body weight loss induced by PGE2, probably because of the diarrhea induced by PGE2 acting on the intestine (Fig.3). These data suggest that prostaglandin receptor EP4 may not play a major role in this effect.

EP4A does not affect PGE2-induced decreased rat body weight. Five- to 6-week-old rats were randomized and treated with PGE2 (6 mg/kg/day) or EP4A (10 mg/kg/day) either alone or in combination. Animals were weighed after 12 days of treatment. Data are reported as mean ± S.E.M. *p < 0.01 versus vehicle and EP4A-treated groups
As expected, all the bone formation parameters were increased by treatment with PGE2. Serum osteocalcin, which is a systemic marker for bone formation activity, increased by about 17% (Fig. 4). Treatment with EP4A completely suppresses the increase in serum osteocalcin (Fig. 4). Trabecular bone volume in the tibial metaphysis increased by 66% (Fig. 5, A & C). These effects were associated with increases in structural and dynamic histomorphometric indices of bone formation. Indeed, both the trabecular number (Fig. 5D) and the extent of calcein-labeled surface (Fig. 5B) increased by 28 and 70%, respectively. Treatment with EP4A alone has no significant effect on all the above markers. However, when EP4A was given in combination with PGE2, it reversed the increase in bone formation markers induced by PGE2 and maintained them at control levels. Relative to PGE2 treated animals, EP4A decreased trabecular bone volume by 81% (Fig. 5C), trabecular number by 93% (Fig. 5D), and the calcein-labeled surface by 54% (Fig. 5B). These data show that an EP4 antagonist suppresses the bone anabolic effect of PGE2 and suggest that the EP4 receptor mediates PGE2 effects on bone formation.
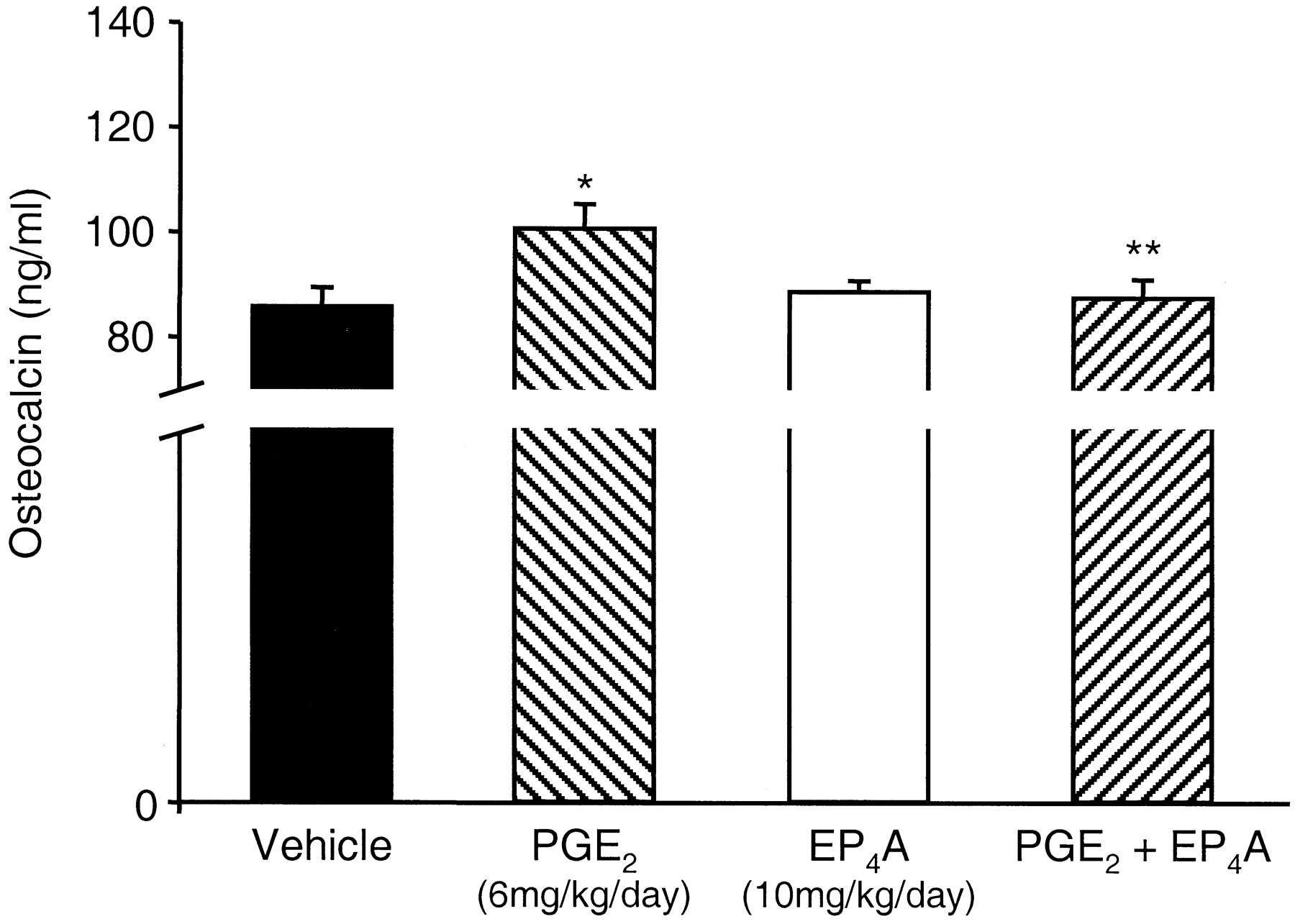
EP4A reverses PGE2- induced increased serum osteocalcin. Five- to 6-week-old rats were treated with PGE2 (6 mg/kg/day) or EP4A (10 mg/kg/day) or in combination for 12 days. Osteocalcin levels were determined in serum by radioimmunoassay. Data are reported as mean ± S.E.M. *p < 0.01 versus vehicle-treated group; **p < 0.05 versus PGE2-treated group

EP4A suppresses PGE2-induced increased trabecular bone volume, extent of calcein-labeled bone surface and trabecular number. Five- to 6-week-old rats were treated with PGE2 (6 mg/kg/day) or EP4A (10 mg/kg/day) or in combination for 12 days. The proximal tibial metaphysiswas embedded undecalcified. Frontal sections (5 μm) were stained with Masson's trichrome and subjected to quantitative histologic analysis. A, photomicrographs representative of the area of secondary spongiosa from which data were collected. The bone is stained blue, and the marrow stroma stained red. Scale bar, 0.1 mm. B, calcein-labeled surface, MS/BS (calcein labeled surface as a percentage of total bone surface). C, trabecular or cancellous bone volume, BV/TV (cancellous bone volume as a percentage of total bone volume). D, trabecular number, Tb. N, trabecular number per square millimeter of total tissue volume). Data are reported as mean ± S.E.M. *p < 0.01 versus vehicle-treated group; **p < 0.05 versus PGE2-treated group
Discussion
The present study demonstrates that pharmacological inactivation of prostanoid receptor EP4 with EP4A suppresses PGE2-induced bone formation in vivo. PGE2 and its analog PGE1are potent inducers of osteogenesis in humans (Ueda et al., 1980;Ringel et al., 1982) and animals (Jee et al., 1985, 1987; Mori et al., 1990; Suponitzky and Weinreb, 1998); however, the EP receptor that mediates osteogenic effects of PGE2 has not been identified previously. In vitro studies did not provide conclusive evidence as to which EP subtype (EP1, EP2, EP3, or EP4) mediates the anabolic effects of PGE2. This is mainly because PGE2 has variable in vitro effects, depending on the osteoblastic cell type used (Raisz and Koolmans-Beynen, 1974;Hakeda et al., 1986; Kaneki et al., 1999), as stated in the introduction. In addition, the agonists and/or antagonists used so far to study PGE effects on bone were not sufficiently selective for the individual EP subtypes (Kozawa et al., 1998; Ono et al., 1998).
The genetic inactivation of EP subtypes in mice has provided evidence that prostanoid receptor EP4 mediates PGE2-induced bone resorption in mice (Ono et al., 1998; Miyaura et al., 2000; Suzawa et al., 2000). Indeed, studies of osteoclast formation in vitro showed that induction of this process depends on the presence of EP4 in osteoblastic cells. These data are supported by recent in vivo findings showing that PGE2-increased bone resorption is abrogated in these EP4 deficient mice (Perry et al., 2000). On the other hand, another study, using EP2deficient mice, showed that EP2 mediates, at least partially, the induction of bone resorption induced by thyroparathyroidectomy (Tomita et al., 1999). As mentioned above, PGE2 can stimulate both bone resorption and formation in vivo. These effects are species specific, which should be considered when interpreting the data from gene deletion studies in mice. In mice, PGE2 is a strong stimulator of bone resorption compared with rats and humans, in which PGE2 predominantly increases bone formation. A pharmacological approach aimed at specifically targeting EP subtypes, therefore, is better suited for identifying which receptors mediate PGE2 effects on bone in rats.
EP4A, the EP4 antagonist used in this study, is highly selective for EP4. This compound displays a K i value for binding to rat EP4 that is at least 225-fold lower than the K i values determined at rat EP1, EP2, and EP3. We also showed pharmacologically that EP4A acts as a PGE2antagonist, in that it dose-dependently inhibited PGE2-induction of intracellular cAMP formation in a responsive cell line, RP-1. In addition, EP4A reverses a cAMP-mediated biological effect of PGE2 in these cells, the suppression of apoptosis. Pharmacokinetic studies (data not shown) showed that EP4A reaches 1 μM in the blood 1 h after intraperitoneal injection with an estimated half-life of 3 h. EP4A, therefore, is a highly selective antagonist for EP4 with appropriate pharmacokinetic properties for in vivo studies evaluating pharmacologically the function of EP4 in PGE2 action on bone formation. Treatment with EP4A suppressed PGE2-induced increases in trabecular bone volume, suppressed serum osteocalcin and reduced the extent of calcein-labeled bone surface and trabecular number. These findings indicate that the reduction in bone volume is most likely a result of decreased bone formation.
The cellular mechanisms that mediate the bone anabolic effects of PGE2 are still unclear and require further study. We have previously shown that PGE2 increases periosteal cell number in vitro by suppressing apoptosis, without affecting proliferation. Similar effects were obtained using PGE1 and forskolin, indicating cAMP mediation (Machwate et al., 1998). Interestingly, parathyroid hormone, which also stimulates bone formation and intracellular cAMP accumulation, was found to increase osteoblast number in vivo without increasing proliferation (Dobnig and Turner, 1995; Jilka et al., 1999). PGE2, which may act via a mechanism similar to that of parathyroid hormone, may prolong the life span of bone forming cells and thereby increase their number. The role of apoptosis and cellular life span in the anabolic effect of PGE2in vivo remains to be documented. Future in vivo studies, potentially using EP4A, will be necessary to determine the extent to which regulation of osteoblast apoptosis plays a role in PGE2 bone anabolic effects.
This is the first study demonstrating the use of a selective PGE2 receptor antagonist, targeting EP4, to elucidate the role of EP4 in in vivo effects of PGE2 on bone. Further studies using a similar approach with an EP4 agonist and ligands that target the other EP receptors are necessary to evaluate whether EP4 is the only or major prostanoid receptor that mediates the bone anabolic effects of PGE2 in rats.
Acknowledgments
We thank Drs. Dwight Towler and Donald Kimmel for their critical reading of this manuscript. We also thank all the other members of the bone biology group at Merck for the many helpful discussions. We thank Chantal Rochette, Claude Godbut, and Nathalie Tremblay for technical support.
Abbreviations
- PG
- prostaglandin
- PLC
- phospholipase C
- EP4A
- EP4 receptor antagonist [4′-[3-butyl-5-oxo-1-(2-trifluoromethyl-phenyl)-1,5-dihydro-[1,2,4]triazol-4-ylmethyl]-biphenyl-2-sulfonic acid (3-methyl-thiophene-2-carbonyl)-amide]
- DMEM
- Dulbecco's modified Eagle's medium
- FBS
- fetal bovine serum
- PBS
- phosphate-buffered saline
- HEK
- human embryonic kidney
- Received February 23, 2001.
- Accepted March 26, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}