


Abstract
Transforming growth factor β1 (TGF-β1) is a potent fibrotic factor responsible for the synthesis of extracellular matrix. TGF-β1 acts through the TGF-β type I and type II receptors to activate intracellular mediators, such as Smad proteins, the p38 mitogen-activated protein kinase (MAPK), and the extracellular signal-regulated kinase pathway. We expressed the kinase domain of the TGF-β type I receptor [activin receptor-like kinase (ALK)5] and the substrate, Smad3, and determined that SB-431542 is a selective inhibitor of Smad3 phosphorylation with an IC50 of 94 nM. It inhibited TGF-β1–induced nuclear Smad3 localization. The p38 mitogen-activated protein kinase inhibitors SB-203580 and SB-202190 also inhibit phosphorylation of Smad3 by ALK5 with IC50values of 6 and 3 μM, respectively. This suggests that these p38 MAPK inhibitors must be used at concentrations of less than 10 μM to selectively address p38 MAPK mechanisms. However, the p38 MAPK inhibitor SB-242235 did not inhibit ALK5. To evaluate the relative contribution of Smad signaling and p38 MAPK signaling in TGF-β1–induced matrix production, the effect of SB-431542 was compared with that of SB-242235 in renal epithelial carcinoma A498 cells. All compounds inhibited TGF-β1–induced fibronectin (FN) mRNA, indicating that FN synthesis is mediated in part via the p38 MAPK pathway. In contrast, SB-431542, but not the selective p38 MAPK inhibitor SB-242235, inhibited TGF-β1–induced collagen Iα1 (col Iα1). These data indicate that some matrix markers that are stimulated by TGF-β1 are mediated via the p38 MAPK pathway (i.e., FN), whereas others seem to be activated via ALK5 signaling independent of the p38 MAPK pathway (i.e., col Iα1).
Numerous reports describe TGF-β1 as a potent regulator of extracellular matrix. Thus, cells can be induced to produce extracellular matrix protein and inhibit protease activity by exogenous TGF-β1 (Nakamura et al., 1992; Ziyadeh et al., 1994). Transgenic mice that overexpress TGF-β1 develop severe glomerulosclerosis and liver fibrosis (Kopp et al., 1996; Kanzler et al., 1999). Finally, neutralizing antibodies against TGF-β1 can prevent the accumulation of extracellular matrix in models of renal disease and lung fibrosis (Border et al., 1990; Sharma et al., 1996;Ziyadeh et al., 2000).
Several signaling pathways have been implicated in mediating TGF-β–induced extracellular matrix production and fibrosis. Transforming growth factor-β signals through two highly conserved single transmembrane receptors with intracellular serine/threonine kinase domains. Specifically, the type II receptor phosphorylates threonine residues in the glycine-serine (GS)-rich domain of the ligand-occupied type I receptor (ALK5), which results in the activation of ALK5 (Wrana, 1998). ALK5, in turn, phosphorylates Smad proteins (Abdollah et al., 1997), which mediate intracellular signaling to the nucleus. TGF-β also activates the p38 mitogen-activated protein kinase (MAPK) through the TGF-β activated kinase, TAK1 (Hanafusa et al., 1999). Thus, the TGF-β receptors can regulate gene transcription directly by activating the Smad transcription factors, indirectly via p38 MAPK, or by a combination of both pathways. Other kinases, such as JNK1 and MKK4, also seem to be involved in TGF-β regulation of fibronectin synthesis (Hocevar et al., 1999). Furthermore, TGF-β1 can activate the extracellular signal-regulated kinase through a protein synthesis requiring a mechanism that may involve basic fibroblast growth factor (Finlay et al., 2000). It is clear that TGF-β can interact with several kinase pathways to influence many components of the extracellular matrix.
Although it has been shown that the p38 MAPK inhibitor SB-203580 can inhibit ALK5 autophosphorylation with an IC50of 20 μM (Eyers et al., 1998), more selective inhibitors have not been applied to TGF-β regulation of extracellular matrix. We characterized several compounds for their ability to selectively inhibit ALK5 or p38 MAPK to delineate the relative contribution of Smad and p38 MAPK activation in TGF-β regulation of extracellular matrix. In this study, therefore, we examine the effects of the selective ALK5 inhibitor SB-431542 (Callahan et al., 2002) and the selective p38 MAPK inhibitor SB-242235 (Badger et al., 2000) on TGF-β1–induced fibronectin, collagen Iα1, thrombospondin-1, and plasminogen activator inhibitor-1 mRNA.
Materials and Methods
Expression of Recombinant Protein.
The kinase domain of ALK5, amino acids 200 to the C terminus, which lacks the GS region, and the full-length Smad3 protein were expressed as N-terminal glutathioneS-transferase (GST) fusion proteins in baculovirus expression system. Proteins were purified with glutathione Sepharose beads 4B (Amersham Biosciences, Uppsala, Sweden) (Roshak et al., 2000).
Kinase Assay.
Kinase assays were performed with 65 nM GST-ALK5 and 184 nM GST-Smad3 in 50 mM HEPES, 5 mM MgCl2, 1 mM CaCl2, 1 mM dithiothreitol, and 3 μM ATP. Reactions were incubated with 0.5 μCi of [33P]γATP for 3 h at 30°C. Phosphorylated protein was captured on P-81 paper (Whatman, Maidstone, England), washed with 0.5% phosphoric acid, and counted by liquid scintillation. Alternatively, Smad3 or Smad1 protein was also coated onto FlashPlate Sterile Basic Microplates (PerkinElmer Life Sciences, Boston, MA). Kinase assays were then performed in FlashPlates with same assay conditions using either the kinase domain of ALK5 with Smad3 as substrate or the kinase domain of ALK6 (BMP receptor) with Smad1 as substrate. Plates were washed three times with phosphate buffer and counted by TopCount (Packard Bioscience, Meriden, CT).
Cell Culture.
A498 renal epithelial carcinoma cell line was obtained from American Type Culture Collection (Manassas, VA) and grown in Earle's minimum essential medium (GlaxoSmithKline, King of Prussia, PA) supplemented with 10% fetal calf serum, penicillin (5 units/ml), and streptomycin (5 ng/ml). Cells were serum-starved for 24 h before treatment. Renal proximal tubule epithelial cells (RPTEC) were obtained from Clonetics Corp. (San Diego, CA) and grown in supplied RPTEC medium.
XTT Viability Assay.
A498 cells were seeded at 5,000 to 10,000 cells/well in 96-well plates. The cells were serum-deprived for 24 h and then treated with compounds for 48 h to assess the cellular toxicity. Cell viability is determined by incubating cells for 4 h with XTT labeling and electron coupling reagent according to the manufacturer's directions (Roche Applied Science, Indianapolis, IN). Live cells with active mitochondria produce an orange-colored product, formazan, which is detected using a plate reader at between A 450 nm andA 500 nm with a reference wavelength greater than 600 nm. The absorbance values correlate with the number of viable cells.
Nuclear Translocation.
RPTEC or MG63 osteoblasts were seeded at about 30 to 50% confluence on microscope slides. The cells were serum-starved for 24 h and pretreated for 4 h with the compounds. The cells were treated with 5 ng/ml TGF-β1, BMP-4, or vehicle for 30 min. The cells were fixed in 4% paraformaldehyde for 15 min, washed three times with PBS, permeabilized for 20 min in 0.5% Triton X-100 in PBS, and washed three times with PBS. The slides were blocked for 1 h at room temperature with 10% horse serum in PBS. Slides were incubated with 1:500 dilution of primary antibody raised against full-length Smad3 (Santa Cruz Biotechnology Inc., Santa Cruz, CA) at 4°C in 10% horse serum in PBS overnight. The slides were washed three times with PBS for a total of 30 min. Secondary antibody conjugated with fluorescein isothiocyanate was added at 1:5000 dilution in 3% horse serum in PBS for 1 h. Slides were washed three times in PBS and covered with mounting medium containing 4,6-diamidino-2-phenylindole stain (Vector Laboratories Inc., Burlingame, CA) and coverslipped for viewing. Digital images were captured from 10 fields per condition under identical exposure settings. Average fluorescent intensity was measured for every nucleus in the field and averaged for the field with Kodak Image analysis software (Eastman Kodak, Rochester, NY). Mean nuclear fluorescent intensities were calculated for each condition from the average intensity generated from each field.
mRNA Analysis.
A498 cells were grown to near confluence in 100-mm dishes, serum-starved for 24 h, and pretreated with compounds for 4 h followed by a 10 ng/ml addition of TGF-β1 (R & D Systems, Inc., Minneapolis, MN). Cells were treated for 6 h [plasminogen activator inhibitor-1 (PAI-1) and thrombospondin-1 (TSP-1) mRNA] or 24 h [fibronectin (FN) mRNA] after the addition of TGF-β1. Cellular RNA was extracted by acid phenol/chloroform extraction (Chomczynski and Sacchi, 1987). Total RNA (10 μg) was resolved by agarose gel electrophoresis and transferred to nylon membrane (GeneScreen; PerkinElmer Life Sciences, Boston, MA). Membranes were probed with32P-labeled cDNA probes (Stratagene, La Jolla, CA) for FN, TSP-1, PAI-1, and for control ribosomal protein L32 (rpL32). Collagen Iα1 mRNA was measured by TaqMan real-time RT-PCR (Applied Biosystems Inc., Foster City, CA).
Results
To identify potential inhibitors of ALK5 kinase activity, compounds were tested for their ability to inhibit ALK5 phosphorylation of Smad3 (Callahan et al., 2002). The kinase domain without the GS region was cloned and expressed as a GST fusion protein. Expressing the protein without the GS domain, which has been shown to regulate the kinase activity (Huse et al., 2001), creates a constitutively active kinase that is able to phosphorylate GST-Smad3 (Fig.1). The screening results identified many inhibitors of p38 MAPK, indicating that the ATP binding site of ALK5 and p38 MAPK bind similar pharmacophores. This was further illustrated by a report that SB-203580 inhibited the autophosphorylation of ALK5 (Eyers et al., 1998). Based on analysis of structure-activity relationships, SB-431542 was designed to be a selective ALK5 inhibitor with little activity against p38 MAPK (compound 14; Callahan et al., 2002). SB-431542 was identified as a potent ALK5 inhibitor with an IC50 of 94 nM (Fig.2). Analogs of the p38 MAPK inhibitor SB-203580 were also tested (SB-202190 and SB-242235). SB-203580 and SB-202190 inhibited ALK5 kinase activity with IC50 values of 6 and 3 μM, respectively (Fig.2). SB-242235, which was developed as a more selective p38 MAPK inhibitor (Badger et al., 2000), did not inhibit ALK5 at concentrations up to 50 μM. Selectivity of SB-431542 was further evaluated against other type I receptors, such as ALK4 and ALK2. Although SB-431542 inhibited ALK4 with an IC50 of 140 nM, it had no effect on the kinase activity of ALK2 (Fig.3). This is consistent with the degree of homology between these kinases, such that ALK4 is the closest related kinase to ALK5. These results are further supported by activities against the full-length receptors transfected in NIH 3T3 cells (Inman et al., 2002). These data clearly demonstrate that SB-431542 is a potent and selective inhibitor of ALK5 and ALK4, with slightly higher selectivity for ALK5.
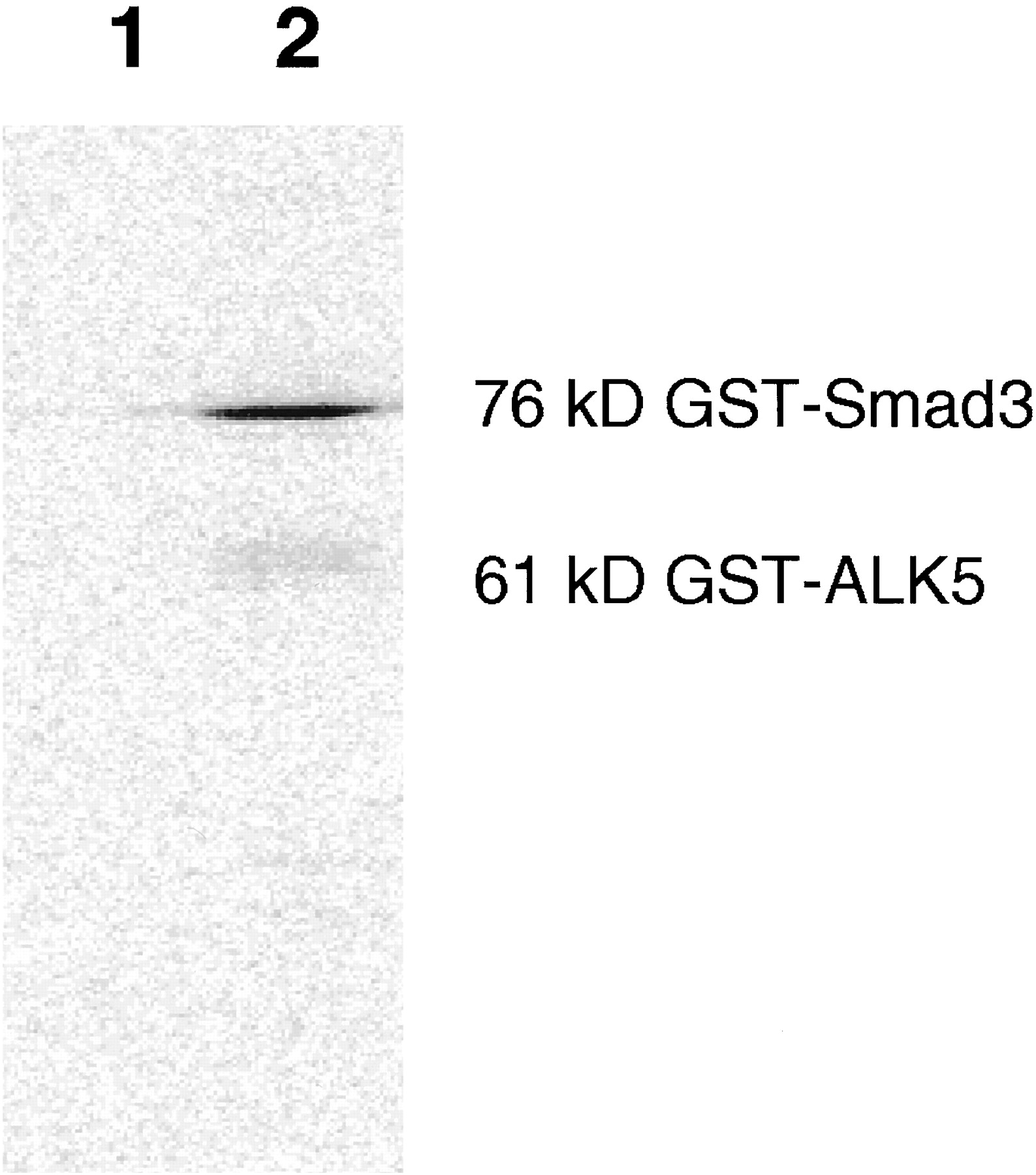
Phosphorylation of GST-Smad3 by purified GST-fused kinase domain of ALK5. The kinase domain lacking the GS region was able to phosphorylate Smad3 in vitro. SDS-polyacrylamide gel electrophoresis of 20-μl reaction of 81 nM ALK5 with 600 nM Smad3 incubated at 30°C for 3 h. Buffer and ATP concentrations are as described underMaterials and Methods. 1, substrate only; 2, substrate plus enzyme.

Effect of SB-431542 and the p38 MAPK inhibitors SB-203580, SB-202190, and SB-242235 on ALK5 kinase activity with GST-Smad3 as substrate. IC50 values are 94 nM and 6, 3, and >50 μM, respectively. Graphs show means ± S.D. (n = 3). ▾, SB-202190; ♦, SB-242235; ▪, SB-203580; ●, SB-431542.

Effects of SB-431542 on activin type I receptor kinase activities (ALK2 and ALK4). IC50 values are >10 μM and 140 nM, respectively. Graphs show means ± S.D. (n = 3).
A concern with kinase inhibitors is nonspecific toxicity caused by inhibition of important but unrelated kinases. To assess potential toxicity of these kinase inhibitors, SB-431542, SB-203580, SB-202190, and SB-242235 were examined by XTT assay, which measures mitochondrial activity as an index of cell viability. Exposure of these compounds to A498 renal epithelial carcinoma cells for 48 h resulted in LD50 values of >30, 15, 30, and >30 μM, respectively (data not shown). Therefore, cells can be treated with SB-431542 for up to 48 h at concentrations up to 10 μM without impact on cell viability.
TGF-β1 has been shown to cause the translocation of Smad proteins from the cytoplasm to the nucleus (Hoodless et al., 1996). To evaluate whether ALK5 activity is required for TGF-β1–induced translocation of Smad3, Smad proteins were visualized in A498 cells by indirect immunofluorescence using antibodies raised against Smad3. SB-242235 at a concentration of 20 μM was unable to prevent the TGF-β–induced translocation of Smad protein (data not shown). However, SB-431542 at a concentration of 1 μM significantly reduced the TGF-β–induced nuclear accumulation of Smad proteins. To confirm that primary renal epithelial cells also respond to TGF-β1 with increased nuclear Smad, a dose-response curve using 0, 50, 250, 500, and 700 nM SB-431542 in primary RPTEC was performed. The IC50 for inhibiting TGF-β1–induced nuclear fluorescence is approximately 50 nM (Fig. 4). BMP-stimulated Smad1 nuclear fluorescence in MG63 cells was unaffected by SB-431542 (data not shown). Thus, SB-431542 selectively inhibits TGF-β1–induced Smad translocation without affecting BMP-induced Smads. Furthermore, p38 MAPK activity is not required for TGF-β1–induced Smad translocation.

Fluorescent immunohistochemistry of primary renal proximal tubule epithelial cells with antibody raised against full-length Smad3. A, cells received vehicle (■); B, TGF-β1 (5 ng/ml; ●) or TGF-β1 plus increasing concentrations of SB-431542 (50, 250, 500, and 700 nM). C, image of cells treated with TGF-β1 plus 700 nM SB-431542. Nuclear fluorescence intensity was quantified for all cells in 10 fields per slide (n = 1).
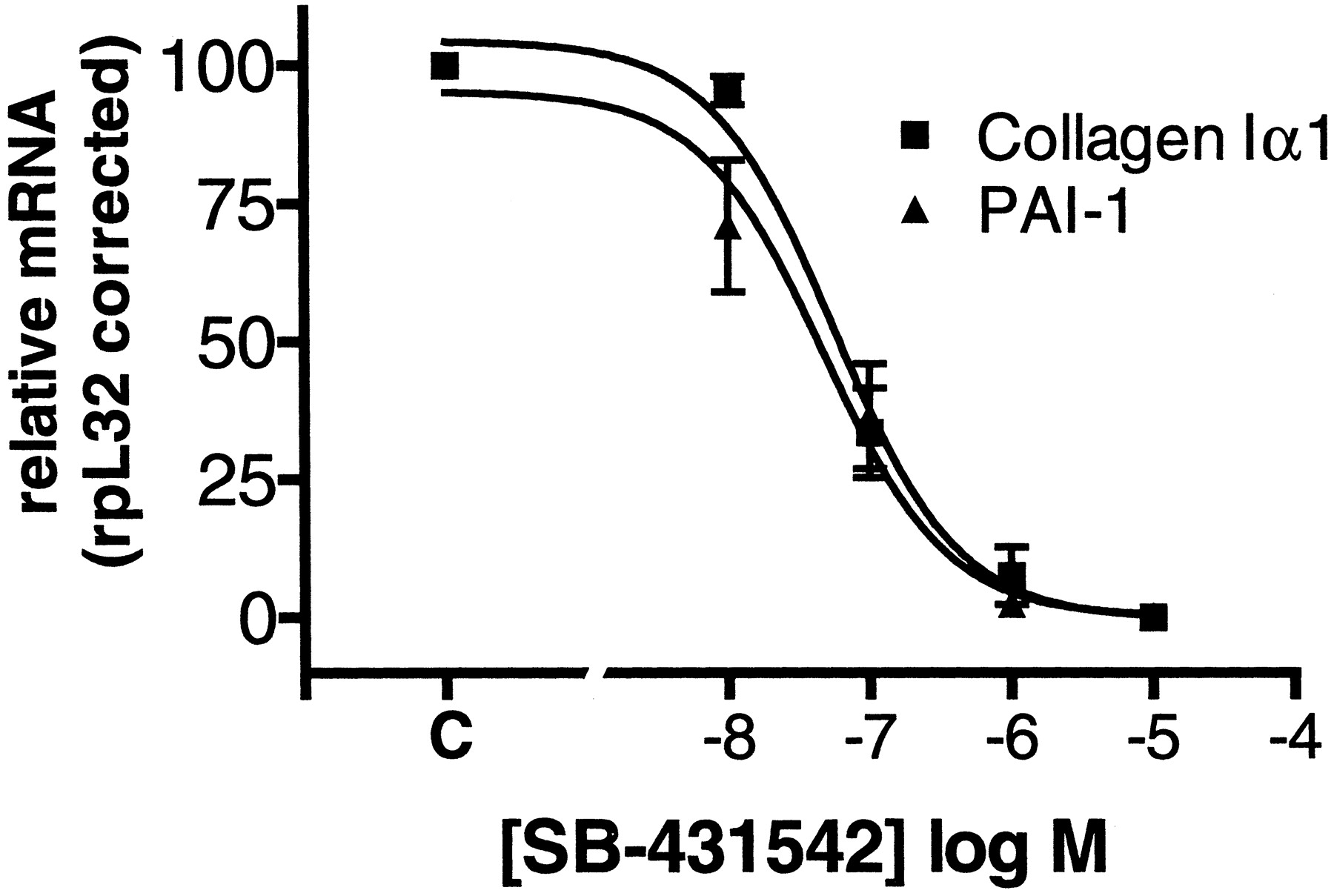
To evaluate the effect of ALK5 kinase inhibition on TGF-β1–induced matrix production, the effect of SB-431542 on TGF-β1–induced collagen Iα1 and FN mRNA was examined in A498 cells. SB-431542 inhibited TGF-β1–induced collagen Iα1 and PAI-1 mRNA with IC50 values of 60 and 50 nM, respectively (Fig.5). In addition, SB-431542 inhibited TGF-β1–induced fibronectin mRNA and protein with IC50 values of 62 and 22 nM, respectively (Fig.6). These data demonstrate for the first time that ALK5 activity is required for TGF-β1 regulation of extracellular matrix markers FN, collagen Iα1, and PAI-1 mRNA.

Inhibition of TGF-β1–induced extracellular matrix markers. SB-431542 inhibits TGF-β1–induced collagen Iα1 and PAI-1 mRNA levels in A498 cells (means ± S.E.M., n= 3).

SB-431542 inhibits TGF-β1–induced FN, mRNA levels, and protein levels in A498 cells. A, representative Northern blot for FN and rpL32. B, representative Western blot for fibronectin. C, percentage maximum TGF-β1 (10 ng/ml)-induced fibronectin mRNA or protein (means ± S.E.M., n = 3).
The role of p38 MAPK in mediating TGF-β1 regulation of extracellular matrix was evaluated with the selective p38 MAPK inhibitor SB-242235 in A498 cells. Although SB-242235 was unable to affect TGF-β1–mediated increase in collagen Iα1 mRNA levels, it inhibited TGF-β1–induced FN mRNA with an IC50 of 16 nM (Fig.7). This clearly indicates that TGF-β1 regulation of collagen Iα1 mRNA is independent of p38 MAPK whereas TGF-β1 regulation of FN mRNA is dependent on p38 MAPK activity. Higher doses of SB-242235 further inhibit FN mRNA and also inhibit TGF-β1–induced TSP-1 mRNA and, to a lesser degree, PAI-1 mRNA (Fig.8). Moreover, the other p38 MAPK inhibitors, SB-203580 and SB-202190, also inhibit TGF-β1–induced fibronectin mRNA with IC50 values of 0.5 and 1 μM, respectively (Fig. 9A). However, these compounds also inhibited collagen Iα1 mRNA at 10-fold higher concentrations (Fig. 9B). This clearly shows that these compounds are less selective than SB-242235 in their ability to distinguish between fibronectin and collagen Iα1 activation pathways and, thus, p38 MAPK dependence.

Effect of p38 MAPK inhibition (SB-242235) on TGF-β1–induced col Ια1 and FN mRNA in A498 cells. mRNA levels were determined by TaqMan RT-PCR and corrected with ribosomal protein L32 mRNA (means ± S.E.M., n = 3).

Effect of p38 MAPK inhibition (SB-242235) on TGF-β1–induced TSP-1, PAI-1, and FN mRNA in A498 cells. mRNA levels were determined by Northern blot and corrected with ribosomal protein L32 mRNA (means ± S.E.M., n = 3–4).

Effect of p38 MAPK inhibitors SB-203580 and SB-202190 on TGF-β1–induced fibronectin (A) and collagen Iα1 (B) mRNA levels in A498 cells as determined by fluorescent real-time RT-PCR (means ± S.E.M., n = 3).
Discussion
TGF-β1 is one of the most potent profibrotic cytokines released and activated after tissue injury (Border and Noble, 1994) and has been implicated in a variety of human fibrotic diseases, including glomerulonephritis (Yoshioka et al., 1993), diabetic nephropathy (Yamamoto et al., 1993), liver cirrhosis (Nagy, 1991), and lung fibrosis (Anscher, 1993). Several studies support a causal relationship between TGF-β1 and fibrosis. Renal cells can be induced to produce extracellular matrix protein and inhibit protease activity by exogenous TGF-β1 in vitro (Nakamura et al., 1992; Ziyadeh et al., 1994). Furthermore, neutralizing antibodies against TGF-β1 can prevent the accumulation of extracellular matrix in nephritic rodents (Border et al., 1990; Sharma et al., 1996; Ziyadeh et al., 2000) and bleomycin-treated mice (Giri et al., 1993). Finally, TGF-β1 transgenic mice or in vivo transfection of the TGF-β1 gene into normal rats resulted in the rapid development of glomerulosclerosis and liver fibrosis (Isaka et al., 1993; Imai et al., 1994; Kopp et al., 1996; Kanzler et al., 1999). Because TGF-β is a potent stimulus for extracellular matrix synthesis, inhibition of ALK5 activity and p38 MAPK may be beneficial in fibrotic disorders. Therefore, we examined kinase inhibitors for their ability to inhibit the TGF-β type I receptor ALK5.
To identify small molecules able to inhibit ALK5, in vitro kinase assays were performed with GST fusion proteins of Smad3 and the kinase domain of ALK5. It must be noted that the kinase assays used the truncated ALK5 kinase domain that lacks the regulatory GS region. This is a relatively weak kinase construct, because an incubation of more than 2 h is required to measure robust phosphorylation signal compared with other constructs using the hyperphosphorylated GS region (Huse et al., 2001). In the intact type I receptor, this region must be phosphorylated for kinase activity (Wrana et al., 1994). The phosphorylation theoretically causes the displacement of the GS region, allowing opening of the ATP binding site and thus phosphorylation of substrate to occur (Huse et al., 2001). By expressing the kinase domain without this regulatory region fused at the N-terminal to glutathioneS-transferase, however, an active kinase is formed, further supporting the hypothesis that the GS region is involved in maintaining the ATP binding site in the inactive state.
Certain similarities seem to exist in the ATP binding pocket between ALK5 and the serine-threonine kinase, p38 MAPK, because the p38 MAPK inhibitor, SB-203580, was also able to inhibit ALK5 autophosphorylation with an IC50 of 20 μM (Eyers et al., 1998). We have shown that SB-203580 and its analog SB-202190 inhibit ALK5 phosphorylation of Smad3 at 6 and 3 μM, respectively. This class of imidazoles led to the discovery of a selective ALK5 inhibitor, SB-431542 (Callahan et al., 2002).
We have characterized SB-431542 as a potent (nanomolar) inhibitor of ALK5 with greater than 100-fold selectivity against p38 MAPK and 25 other kinases (Inman et al., 2002), which can prevent TGF-β1–induced elevation of FN, PAI-1, and collagen Iα1 mRNA. The selectivity of SB-431542 seems to be greater for ALK5 than for the closely related activin type I receptor ALK4 (94 versus 140 nM). This is supported by findings by Inman et al. (2002), where cells transfected with constitutively active type I receptors show that SB-431542 preferentially inhibits ALK5 and, to a lesser degree, ALK4, and, further, ALK7 (Inman et al., 2002). Moreover, this study, as well as the study by Inman et al. (2002), shows that SB-431542 has no significant activity against the BMP-activated receptors ALK2, ALK3, and ALK6 (Inman et al., 2002). Thus, SB-431542 is a useful tool to evaluate TGF-β–regulated cellular mechanisms.
Although SB-203580 and SB-202190 are potent p38 MAPK inhibitors (Beyaert et al., 1996; Feoktistov et al., 1999), they were also able to inhibit the kinase activity of ALK5 at micromolar concentrations. It should be noted that earlier reports using SB-203580 to describe the role of p38 MAPK in TGF-β signaling are unlikely to distinguish p38 MAPK from ALK5 activity, due to lack of selectivity at concentrations greater than 10 μM (Ravanti et al., 1999; Gruden et al., 2000). It is therefore imperative that SB-203580 and SB-202190 be used at concentrations of less than 10 μM to avoid nonselective inhibition of other kinases.
Several studies have shown that TGF-β signaling requires both ALK5 and the TGF-β type II receptor and is mediated via the cytoplasmic mediators Smad2 and Smad3. The type II receptor phosphorylates serine and threonine residues in the GS domain of the ligand-occupied type I receptor, which results in the activation of the type I receptor (Wrana, 1998). ALK5 then phosphorylates Ser465and Ser467 of the conserved C-terminal end of Smad2 (Abdollah et al., 1997; Souchelnytskyi et al., 1997), and this phosphorylation of Smad2 is required for its nuclear accumulation and signaling (Macias-Silva et al., 1996). Smad3 behaves similarly upon TGF-β stimulation. Consistent with these data, SB-431542, but not SB-242235, prevented the TGF-β1–induced nuclear accumulation of Smad proteins at concentrations similar to those needed to block TGF-β1–induced matrix markers. Because SB-242235 had no effect on Smad translocation, the p38 MAPK pathway is unlikely to be involved in modulating nuclear accumulation of Smad proteins.
Because the p38 MAPK selective inhibitor SB-242235 (Badger et al., 2000), which did not affect ALK5 kinase activity up to 50 μM, did not significantly affect TGF-β1–induced collagen Iα1 mRNA production, it is likely that TGF-β1 regulation of collagen Iα1 mRNA is mediated primarily through ALK5 and Smad activation. However, SB-242235 did inhibit TGF-β1–induced FN mRNA and, to a lesser extent, TSP1 and PAI-1 mRNA. This demonstrates that p38 MAPK activation plays a role in TGF-β1 regulation of FN, TSP-1, and possibly PAI-1 synthesis.
Activation of p38 MAPK leads to activation of transcription factors, including ATF-2, CREB, and AP-1 (Xing et al., 1998; Zhu and Lobie, 2000; Wilmer et al., 2001), which in turn can bind the respective response elements found in regulatory sequences of the fibronectin gene (Srebrow et al., 1993; Nahman et al., 1996; Tamura et al., 1998). Therefore, TGF-β1–induced increase of FN mRNA may be mediated indirectly via p38 MAPK activation of ATF-2, CREB, and AP-1. It should be noted that whereas TGF-β1 increases collagen Iα1 and PAI-1 mRNA rapidly within a few hours, FN mRNA increases are not significant until more than 16 h. This further suggests that FN mRNA regulation by TGF-β1 is indirect.
In summary, this study shows that SB-431542 is a potent inhibitor of the TGF-β type I receptor ALK5. Although SB-431542 has significant activity against the closest related kinase (ALK4), it is selective against 25 other kinases and an excellent tool to evaluate the role of TGF-β and ALK5 in cellular mechanisms. Finally, use of SB-431542 and the selective p38 MAPK inhibitor SB-242235 also showed that TGF-β1 can transcriptionally regulate extracellular matrix genes by several mechanisms, including the Smad pathway, the p38 MAPK kinase pathway, and possibly a combination of the two, as well as other kinase pathways. Thus, TGF-β1 seems to regulate collagen Iα1 mRNA levels by ALK5 independent of p38 MAPK activation. In contrast, TGF-β1 induction of FN mRNA requires p38 MAPK activity. TGF-β1 induction of PAI-1 and TSP-1 mRNA uses at least ALK5 and possibly the p38 MAPK pathway. It is certain that the Smad proteins interact with other transcription factors, thereby providing a mechanism for modulation of TGF-β1–responsive genes by other factors and signaling pathways.
Acknowledgments
We thank Drs. David Brooks and Caroline Hill for critical discussions.
Footnotes
- Received November 28, 2001.
- Accepted March 21, 2002.
Abbreviations
- TGF-β1
- transforming growth factor β1
- GS
- glycine-serine
- ALK
- activin receptor-like kinase
- MAPK
- mitogen-activated protein kinase
- SB-203580
- 4-[4′-fluorophenyl]-2-[4′-methylsulfinylphenyl]-5-[4′-pyridyl] imidazole
- SB-431542
- 4-(5-benzo[1,3]dioxol-5-yl-4-pyridin-2-yl-1H-imidazol-2-yl)-benzamide
- SB-242235
- 4-[4-(4-flurophenyl)-1-(4-piperidinyl)-1H-imidazol-5-yl]-2-methoxypyrimidine
- GST
- glutathione S-transferase
- RPTEC
- renal proximal tubule epithelial cells
- XTT
- 2,3-bis[2-methoxy-4-nitro-5-sulphophenyl]-2H-tetrazolium-5-carboxyaniline
- BMP
- bone morphogenetic protein
- PBS
- phosphate-buffered saline
- PAI-1
- plasminogen activator inhibitor-1
- TSP-1
- thrombospondin-1
- FN
- fibronectin
- rpL32
- ribosomal protein L32
- RT-PCR
- reverse transcription-polymerase chain reaction
- SB-202190
- 4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)-1H-imidazole
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}