


Abstract
Recent years have witnessed the discovery of novel selective agonists of the M1 muscarinic acetylcholine (ACh) receptor (mAChR). One mechanism invoked to account for the selectivity of such agents is that they interact with allosteric sites. We investigated the molecular pharmacology of two such agonists, 1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone (77-LH-28-1) and 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl] piperidine hydrogen chloride (AC-42), at the wild-type M1 mAChR and three mutant M1 mAChRs. Both agonists inhibited the binding of the orthosteric antagonist [3H]N-methyl scopolamine ([3H]NMS) in a manner consistent with orthosteric competition or high negative cooperativity. Functional interaction studies between 77-LH-28-1 and ACh also indicated a competitive mechanism. Dissociation kinetics assays revealed that the agonists could bind allosterically when the orthosteric site was prelabeled with [3H]NMS and that 77-LH-28-1 competed with the prototypical allosteric modulator heptane-1,7-bis-[dimethyl-3′-phthalimidopropyl]-ammonium bromide under these conditions. Mutation of the key orthosteric site residues Y381A (transmembrane helix 6) and W101A (transmembrane helix 3) reduced the affinity of prototypical orthosteric agonists but increased the affinity of the novel agonists. Divergent effects were also noted on agonist signaling efficacies at these mutants. We identified a novel mutation, F77I (transmembrane helix 2), which selectively reduced the efficacy of the novel agonists in mediating intracellular Ca2+ elevation and phosphorylation of extracellular signal regulated kinase 1/2. Molecular modeling suggested a possible “bitopic” binding mode, whereby the agonists extend down into the orthosteric site as well as up toward extracellular receptor regions associated with an allosteric site. It is possible that this bitopic mode may explain the pharmacology of other selective mAChR agonists.
The five mAChRs are prototypical family A members of the G protein-coupled receptor superfamily and are widespread throughout the central nervous system and periphery (Hulme et al., 1990; Christopoulos, 2007; Wess et al., 2007). Based on anatomical, pharmacological, and preclinical animal model studies, the M1 mAChR subtype has long been considered a potential therapeutic target for the treatment of cognitive deficits associated with disorders such as Alzheimer's disease and schizophrenia (Langmead et al., 2008b). It is noteworthy that human clinical data derived from trials using the M1/M4 mAChR-preferring mAChR agonist xanomeline have also provided evidence for cognitive improvement (Bodick et al., 1997; Shekhar et al., 2008). However, the development of truly subtype-selective mAChR agonists has been hampered by the very high degree of sequence homology within the orthosteric (acetylcholine-binding) site across all five mAChRs (Hulme et al., 1990).
It is now well established that mAChRs also possess topographically distinct allosteric binding sites that may offer a novel avenue toward attaining greater receptor subtype selectivity (Conn et al., 2009). Although early studies in this area focused on allosteric modulators, such as gallamine and C7/3-phth, which regulated the actions of orthosteric ligands but lacked any appreciable intrinsic efficacy of their own (Gregory et al., 2007), recent progress has been made in identifying novel selective agonists that also display pharmacological characteristics suggestive of an allosteric mode of action. The best studied agonist in this regard is 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl] piperidine hydrogen chloride (AC-42; Fig. 1), which possesses significant functional selectivity for activating the M1 mAChR relative to other subtypes (Spalding et al., 2002), interacts allosterically with the antagonists [3H]NMS and atropine (Langmead et al., 2006; Spalding et al., 2006), and is relatively insensitive to key mutations in the orthosteric binding pocket, such as Y106A, Y381A, and N382A (residues 3.33, 6.51, and 6.52, respectively, using the Ballesteros and Weinstein, (1995) convention), which have profound inhibitory effects on the binding and/or function of the prototypical nonselective orthosteric agonist, carbachol (Spalding et al., 2002, 2006). However, additional mutational analysis of key residues in transmembrane domain (TM) 3 of the rat M1 mAChR identified L102A (3.29) as a mutation that inhibited the function of both carbachol and AC-42 and W101A (3.28) as a mutation that profoundly increased the function of AC-42, but inhibited that of carbachol (Spalding et al., 2006). Thus, an interesting picture is emerging in which novel agonists, such as AC-42, although clearly seeming to bind in a mode different from those of prototypical orthosteric agonists, can still perhaps display sensitivity to some receptor epitopes commonly associated with the orthosteric binding pocket. One possible explanation for such findings may be that the novel agonists are “bitopic” ligands able to interact with regions in both orthosteric and allosteric sites, as has been demonstrated recently for the partial agonist 4-I-[3-chlorophenyl]carbamoyloxy)-2-butynyltrimethylammnonium chloride (McN-A-343), at the M2 mAChR (Valant et al., 2008).

Structure of the novel agonists used in this study.
More recently, we have described the new agonist 1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone (77-LH-28-1; Fig. 1), which is a structural analog of AC-42 that also displays functional selectivity for the M1 mAChR over other subtypes (May et al., 2007; Langmead et al., 2008a). 77-LH-28-1 is bioavailable, brain-penetrant, and, importantly, displays higher efficacy than AC-42 when tested at native M1 mAChRs (Langmead et al., 2008a). At the M2 mAChR, 77-LH-28-1 can act as an allosteric modulator of [3H]NMS dissociation kinetics (May et al., 2007), and a study at the M1 mAChR (Lebon et al., 2009) suggested a novel binding mode for both 77-LH-28-1 and AC-42 that may involve a conformational isomerization of the side-chain of Trp101 (3.28). Given the growing interest in such novel agonists of the M1 mAChR, the current study aimed to investigate the pharmacological properties of 77-LH-28-1 at the M1 mAChR in detail and to compare them with those of AC-42 and the prototypic orthosteric agonists ACh and pilocarpine. We also included a comparison of the effects on agonist affinity and efficacy of two key orthosteric site mutations, W101A (3.28) and Y381A (6.51), as well as a novel TM2 mutant, F77I (2.56), that had been suggested to affect the activity of AC-42 in preliminary studies (Jacobson et al., 2004). We reveal that 77-LH-28-1 can interact with part of the “common” allosteric binding site used by the prototypical modulator, C7/3-phth, on a [3H]NMS-occupied M1 mAChR and suggest a new role for Phe77 (2.56) at the top of TM2 of the M1 mAChR in selectively controlling the efficacy of novel agonists such as 77-LH-28-1 and AC-42.
Materials and Methods
Materials.
Chinese hamster ovary (CHO) Flp-In cells and Hygromycin B were purchased from Invitrogen (Carlsbad, CA). U2OS osteosarcoma cells were obtained from the American Type Culture Collection (Manassas, VA). Dulbecco's modified Eagle's medium and fetal bovine serum (FBS) were from Invitrogen and JRH Biosciences (Lenexa, KS), respectively. The AlphaScreen SureFire phospho-ERK1/2 reagents were kindly donated by Dr. Michael Crouch (TGR Biosciences, Adelaide, Australia), whereas the AlphaScreen streptavidin donor beads and anti-IgG (Protein A) acceptor beads used for pERK1/2 detection, [3H]quinuclidinyl benzilate ([3H]QNB; specific activity 52 Ci/mmol) and [3H]NMS (specific activity, 72 Ci/mmol), were purchased from PerkinElmer Life and Analytical Sciences (Waltham, MA). C7/3-phth was synthesized in-house at Monash University by Dr. Celine Valant. AC-42 and 77-LH-28-1 were synthesized in-house at GlaxoSmithKline as described previously (Skjaerbaek et al., 2003; Langmead et al., 2008a). All other chemicals were from Sigma Chemical Co. (St. Louis, MO).
cDNA Constructs and Generation of Stable Cell Lines.
cDNA encoding the human M1 mAChR was obtained from Missouri University of Science and Technology cDNA Resource Center (Rolla, MO), and was used to generate CHO Flp-In cells stably expressing the receptor as described previously (Avlani et al., 2007; May et al., 2007). For experiments performed on U2OS cells, transient expression of wild-type and mutant M1 mAChR constructs was achieved by infection with BacMam virus for 24 h at varying transduction concentrations. To achieve this, the virus was mixed at the desired dilution (plaque-forming units per milliliter) with U2OS cell suspension immediately before plating/passaging of the cells. Modified baculovirus containing the mammalian cytomegalovirus promoter was produced using the Bac-to-Bac Baculovirus expression system (Invitrogen). Mutant receptor constructs were made using either the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) or by the method of polymerase chain reaction gene splicing by overlap extension (Horton et al., 1990). The accuracy of all polymerase chain reaction-derived sequences was confirmed by dideoxy sequencing of the mutant plasmids.
Membrane Preparation.
CHO Flp-In M1 cells were grown until approximately 90% confluence and harvested using 2 mM EDTA in phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, and 1.5 mM KH2PO4). Cells were pelleted by centrifugation for 10 min at 1200g, and the pellets were resuspended in 30 ml of buffer containing 20 mM HEPES and 10 mM EDTA at pH 7.4. All subsequent steps were performed at 4°C. The cell suspension was homogenized using a Polytron PT 1200CL homogenizer (Kinematica, Littau-Lucerne, Switzerland) with two 10-s bursts separated by cooling on ice. The cell homogenate was centrifuged for 5 min at 1700g, and the supernatant was transferred to new tubes and further centrifuged (90 min, 38,000g) in a Sorvall centrifuge (Thermo Fisher Scientific). The pellet was resuspended in 10 ml of buffer (20 mM HEPES and 0.1 mM EDTA, pH 7.4) and briefly homogenized to ensure uniform consistency. Membranes were aliquoted and stored at −80°C. The protein concentration was determined by the method of Bradford using bovine serum albumin as a standard (Bradford, 1976).
Radioligand Equilibrium Binding Assays.
Saturation and competition binding assays were performed both in membranes derived from CHO FlpIn M1 cells and in intact U2OS cells transduced with either the M1-WT, M1-Y381A, M1-W101A, or M1-F77I. For membrane-based saturation binding assays, 15 μg of CHO FlpIn M1 membranes were incubated with the orthosteric antagonist [3H]NMS in 1 ml of HEPES buffer (20 mM HEPES, 100 mM NaCl, and 10 mM MgCl2, pH 7.4) at 37°C for 1 h before termination of the assay by rapid filtration onto GF/B grade filter paper by using a Brandel harvester, followed by three 2-ml washes with ice-cold NaCl (0.9%). Nonspecific binding was defined in the presence of 10 μM atropine, and radioactivity was determined by liquid scintillation counting. For inhibition binding assays, membranes were incubated in HEPES buffer containing increasing concentrations of the allosteric modulator C7/3-phth or either of the novel agonists (i.e., 77-LH-28-1 and AC-42) for 1 h at 37°C in the presence of a [3H]NMS concentration approximately equal to its equilibrium dissociation constant, unless specified otherwise under Results. Nonspecific binding, reaction termination and radioactivity determination were as described above.
For binding assays performed on intact U2OS cells, cells were first transduced with various numbers of plaque forming units (0.5 or 1.25 pfu/ml as indicated in Tables 1 to 3) and, 24 h later, were harvested by trypsinization and resuspended in Ca2+ assay buffer (150 mM NaCl, 2.6 mM KCl, 1.18 mM MgCl2, 10 mM d-glucose, 10 mM HEPES, 2.2 mM CaCl2, 0.5% BSA, and 2.5 mM probenecid). For saturation binding experiments, the U2OS cells were incubated in a 1-ml total volume of Ca2+ buffer containing 105 cells/ml and concentrations of [3H]QNB ranging from 0.002 nM to 0.5 nM for 60 min at 37°C. For competition binding experiments, the cells were incubated in a 1-ml total volume of Ca2+ buffer containing 0.1 nM [3H]QNB and a range of concentrations of the ACh pilocarpine, AC-42, or 77-LH-28-1 for 30 min at 37°C. Nonspecific binding was defined using 10 μM atropine. Incubation was terminated by rapid filtration through GF/C filters (Whatman, Maidstone, UK) using a cell harvester (Brandel Inc., Gaithersburg, MD). All other details were as described above.
Ligand binding parameters for M1 mAChR constructs derived from U2OS whole-cell binding assays
Data represent the mean ± S.E.M. of three experiments performed in duplicate. Values in parentheses are normalized expression levels relative to the wild type.
Agonist coupling efficiency parameters for M1 mAChR-mediated Ca2+ elevation in U2OS cells
Data represent the mean ± S.E.M. of four to eight experiments performed in duplicate. The logarithm of the relative coupling efficiency parameter, τ, was determined via nonlinear regression of the data to an operational model of agonism and corrected for receptor expression levels to yield a corrected τc parameter (Logτc). Antilogarithm shown in parentheses.
Agonist coupling efficiency parameters for M1 mAChR-mediated ERK1/2 phosphorylation in U2OS cells
Data represent the mean ± S.E.M. of three experiments performed in duplicate. The logarithm of the relative coupling efficiency parameter, τ, was determined via nonlinear regression of the data to an operational model of agonism and corrected for receptor expression levels to yield a corrected τc parameter (Logτc). Antilogarithm shown in parentheses.
[3H]NMS Dissociation Kinetics Assays.
CHO-FlpIn cell membranes (15 μg) were equilibrated with [3H]NMS (0.5 nM) in a 1-ml total volume of HEPES buffer [also containing 100 μM Gpp(NH)p] for 60 min at 37°C. Atropine (10 μM) alone or in the presence of test ligand was then added at various time points to prevent the re-association of [3H]NMS with the receptor. In subsequent experiments designed to investigate the effect of a range of modulator concentrations on [3H]NMS dissociation rate, a “two-point kinetics” experimental paradigm was used, where the effect of increasing concentrations of allosteric modulator on [3H]NMS dissociation was determined at 0 and 10 min. This approach is valid to determine [3H]NMS dissociation rate constants if the full-time course of radioligand dissociation is monophasic in both the absence and the presence of modulator (Lazareno and Birdsall, 1995; Kostenis and Mohr, 1996); this was the case in the current study. Termination of the reaction and determination of radioactivity were performed as described above.
U2OS Cell Intracellular Calcium Elevation Assays.
U2OS cells were transduced as described above and plated at a density of 5 × 104 cells/well in 96-well clear flat- bottomed plate. After 22 h in a 37°C, 5% CO2 atmosphere, the cells were washed twice with Ca2+ assay buffer, loaded with Ca2+ assay buffer containing 10 μM Fluo-4, and incubated in dark. The cells were then washed twice with Ca2+ buffer to remove excess Fluo-4. The calcium elevation in response to addition increasing concentrations of ligand was measured for 3 min at 1.5-s intervals using a Flexstation (Molecular Devices, Sunnyvale, CA). For functional interaction studies, cells were incubated at 37°C with varying concentrations of ACh in the absence and presence of different concentrations of a second compound (see Results) that was added 60 s before the addition of ACh for a further 120 s. Calcium elevation was measured using the Flexstation at an interval of 1.5 s for a total of 195 s.
U2OS Cell Extracellular Signal-Regulated Kinase 1/2 Phosphorylation Assays.
Initial ERK1/2 phosphorylation time course experiments were performed to determine the time at which ERK1/2 phosphorylation was maximal after stimulation by each agonist. Cells were seeded into transparent 96-well plates at 4 × 104 cells per well and grown overnight or until confluent. Cells were then washed twice with phosphate-buffered saline and incubated in serum-free Dulbecco's modified Eagle's medium at 37°C for at least 4 h to allow FBS-stimulated phosphorylated ERK1/2 (pERK1/2) levels to subside. Cells were stimulated with agonist using a staggered addition approach. For subsequent agonist-stimulated concentration-response experiments, cells were incubated at 37°C with each agonist for the 8 min required to achieve peak response. For all experiments, 10% FBS was used as a positive control, and vehicle controls were also performed. The reaction was terminated by removal of drugs and lysis of cells with 100-μl SureFire lysis buffer (provided by TGR Biosciences). The lysates were agitated for 1 to 2 min and diluted at a ratio of 4:1 (v/v) lysate/Surefire activation buffer in a total volume of 50 μl. Under low-light conditions, a 1:240 (v/v) dilution of AlphaScreen beads/Surefire reaction buffer was prepared, and this was mixed with the activated lysate mixture in a ratio of 6:5 (v/v), respectively, in a 384-well opaque Optiplate (PerkinElmer Life and Analytical Sciences). Plates were incubated in the dark at 37°C for 1.5 h before the fluorescence signal was measured using a Fusion-α™ plate reader (PerkinElmer Life and Analytical Sciences) using standard AlphaScreen settings.
Construction of an M1 mAChR Model.
The initial model of the TM domain of the human M1 mAChR was constructed by homology with the published X-ray crystal structure of β2 adrenergic receptor (Protein Data Bank code 2RH1) (Cherezov et al., 2007). Alignment between the M1 mAChR sequence and β2 receptor was based on the characteristic motifs found in each TM region, such as the asparagine in TM1, the aspartate in TM2, the “DRY” motif of TM3, the tryptophan in TM4, and the conserved prolines in TM5, TM6, and TM7. This alignment was used with the standard homology modeling tools in the Quanta program (Accelerys Software, San Diego, CA) to construct the seven helical bundle domain of the M1 mAChR. The extracellular loop regions were subsequently added using a procedure developed in-house at GlaxoSmithKline that uses a combined distance geometry sampling and molecular dynamics simulation (Blaney et al., 2001). The side chains of this model were then refined using the Karplus standard rotamer library (Dunbrack and Karplus, 1993). The final model was optimized fully (500 steps of Steepest Descent followed by 5000 steps of Adopted Basis Newton Raphson) using the CHARMm force field. Helical distance constraints between the ith and i + 4th residues (except proline) within a range of 1.8 to 2.5Å were used to maintain the backbone hydrogen bonds of the helix bundles.
Ligand Docking Studies.
AC-42 and 77-LH-28-1 were docked into the receptor model manually using a variety of low-energy starting conformations. Adjustments of the receptor protein side chains were made where necessary, always ensuring that these side chains were only in allowed rotameric states (Dunbrack and Karplus, 1993). Once again, full optimization of the receptor-ligand complexes was performed using CHARMm, the only constraints used being those that maintained the hydrogen bonding pattern of the helical bundle. This procedure allows full relaxation of both the ligand and the whole protein, something that is not possible with automated docking procedures. Although numerous binding orientations were found to be possible, one docked binding mode that accommodated both novel agonists satisfied much of the site-directed mutagenesis data generated.
Data Analysis.
All data were analyzed using Prism 5.01 (GraphPad Software, San Diego, CA). For radioligand saturation binding data, the following equation was globally fitted to nonspecific and total binding data.
 where Y is radioligand binding, Bmax is the total receptor density, [A] is the radioligand concentration, KA is the equilibrium dissociation constant of the radioligand, and NS is the fraction of nonspecific radioligand binding.
where Y is radioligand binding, Bmax is the total receptor density, [A] is the radioligand concentration, KA is the equilibrium dissociation constant of the radioligand, and NS is the fraction of nonspecific radioligand binding.
For radioligand inhibition binding experiments, a one-site binding equation was fitted to the specific binding of each orthosteric ligand:
 where Top and Bottom are the maximal and minimal asymptotes of the curve, respectively, log[B] is the concentration of inhibitor, and logIC50 is the logarithm of the concentration of inhibitor that reduces half the maximal radioligand binding for each binding site. IC50 values were converted to KA values (inhibitor equilibrium dissociation constant) using the Cheng and Prusoff (1973) equation.
where Top and Bottom are the maximal and minimal asymptotes of the curve, respectively, log[B] is the concentration of inhibitor, and logIC50 is the logarithm of the concentration of inhibitor that reduces half the maximal radioligand binding for each binding site. IC50 values were converted to KA values (inhibitor equilibrium dissociation constant) using the Cheng and Prusoff (1973) equation.
For some experiments, as indicated under Results, the following version of a simple allosteric ternary complex model (Lazareno and Birdsall, 1995) was also fitted to inhibition binding data:
 where Y/Ymax denotes fractional specific binding, [A] is the radioligand concentration, KA is the equilibrium dissociation constant of the radioligand, KB denotes the allosteric modulator dissociation constant, and α denotes the cooperativity factor. Values of α > 1 denote positive cooperativity, values < 1 (but greater than 0) denote negative cooperativity, values = 1 denote neutral cooperativity, and values approaching zero denote inhibition that is indistinguishable from competitive (orthosteric) antagonism.
where Y/Ymax denotes fractional specific binding, [A] is the radioligand concentration, KA is the equilibrium dissociation constant of the radioligand, KB denotes the allosteric modulator dissociation constant, and α denotes the cooperativity factor. Values of α > 1 denote positive cooperativity, values < 1 (but greater than 0) denote negative cooperativity, values = 1 denote neutral cooperativity, and values approaching zero denote inhibition that is indistinguishable from competitive (orthosteric) antagonism.
Dissociation kinetics data all followed a monoexponential decay. Thus, the following equation was fitted to these data.
 where t denotes incubation time, Bt denotes specific radioligand binding at time t, B0 denotes the specific radioligand binding at time at equilibrium (time = 0) and koff represents the observed radioligand dissociation rate constant. For the two-point dissociation experiments, where the effects of a range of concentrations of allosteric modulators were investigated, individual koff values determined in the presence of modulator were normalized to the control koff value (absence of modulator) and then plotted as a function of modulator concentration.
where t denotes incubation time, Bt denotes specific radioligand binding at time t, B0 denotes the specific radioligand binding at time at equilibrium (time = 0) and koff represents the observed radioligand dissociation rate constant. For the two-point dissociation experiments, where the effects of a range of concentrations of allosteric modulators were investigated, individual koff values determined in the presence of modulator were normalized to the control koff value (absence of modulator) and then plotted as a function of modulator concentration.
The following three-parameter logistic equation was fitted to concentration-response data generated from the functional U2OS cell-based assays:
 where E is response, Emax and Bottom are the top and bottom asymptotes of the curve, respectively, [A] is the agonist concentration, and pEC50 is the negative logarithm of the agonist concentration that gives a response halfway between Emax and Bottom. Where appropriate, the following form of an operational model of agonism (Black and Leff, 1983) was also fitted to agonist data.
where E is response, Emax and Bottom are the top and bottom asymptotes of the curve, respectively, [A] is the agonist concentration, and pEC50 is the negative logarithm of the agonist concentration that gives a response halfway between Emax and Bottom. Where appropriate, the following form of an operational model of agonism (Black and Leff, 1983) was also fitted to agonist data.
 where Em is the maximal possible response of the system (not the agonist), Basal is the basal level of response in the absence of agonist, KA denotes the functional equilibrium dissociation constant of the agonist (A), τ is an index of the coupling efficiency (efficacy) of the agonist and is defined as RT/KE where RT is the total concentration (Bmax) of receptors and KE is the concentration of agonist-receptor complex that yields half the maximum system response (Em). Throughout this study, all references to the term “efficacy” specifically relate to the property quantified by τ. To define the Em and τ for each mutant and assay, the KA for each agonist was constrained to equal the KA value derived from radioligand binding assays (see Results) in the nonlinear regression procedure. In addition, because the τ parameter can be influenced by variations in the expression level of various receptor constructs, the values reported in this study have been normalized according to the following ratio: τcorrected (τc) = τestimated × (Bmax-mutant /Bmax-wildtype) (see Gregory et al., 2010).
where Em is the maximal possible response of the system (not the agonist), Basal is the basal level of response in the absence of agonist, KA denotes the functional equilibrium dissociation constant of the agonist (A), τ is an index of the coupling efficiency (efficacy) of the agonist and is defined as RT/KE where RT is the total concentration (Bmax) of receptors and KE is the concentration of agonist-receptor complex that yields half the maximum system response (Em). Throughout this study, all references to the term “efficacy” specifically relate to the property quantified by τ. To define the Em and τ for each mutant and assay, the KA for each agonist was constrained to equal the KA value derived from radioligand binding assays (see Results) in the nonlinear regression procedure. In addition, because the τ parameter can be influenced by variations in the expression level of various receptor constructs, the values reported in this study have been normalized according to the following ratio: τcorrected (τc) = τestimated × (Bmax-mutant /Bmax-wildtype) (see Gregory et al., 2010).
A logistic equation of competitive agonist-antagonist interaction (Motulsky and Christopoulos, 2004) was globally fitted to data from functional experiments measuring the interaction between ACh and atropine:
 where s represents the Schild slope for the antagonist, and pA2 represents the negative logarithm of the molar concentration of antagonist that makes it necessary to double the concentration of agonist needed to elicit the original submaximal response obtained in the absence of antagonist; all other parameters are as defined above in eq. 5. The following operational model for the competitive interaction between an orthosteric full and partial agonist, as derived previously by Leff et al. (1993), was also fitted to the interaction data between ACh and 77-LH-28-1.
where s represents the Schild slope for the antagonist, and pA2 represents the negative logarithm of the molar concentration of antagonist that makes it necessary to double the concentration of agonist needed to elicit the original submaximal response obtained in the absence of antagonist; all other parameters are as defined above in eq. 5. The following operational model for the competitive interaction between an orthosteric full and partial agonist, as derived previously by Leff et al. (1993), was also fitted to the interaction data between ACh and 77-LH-28-1.
 where the parameters are as described above for eqs. 6 and 7.
where the parameters are as described above for eqs. 6 and 7.
All parametric measures of potency, affinity, operational efficacy and cooperativity were estimated as logarithms (Christopoulos, 1998). In all instances, models were fitted to pooled datasets. Statistical comparisons between parameters were performed using Student's t test or F test, where appropriate, with p < 0.05 taken as indicating significance.
Results
77-LH-28-1 and AC-42 Inhibit the Binding of the Orthosteric Antagonist [3H]NMS at the M1 mAChR.
AC-42 and 77-LH-28-1 were initially studied using an equilibrium binding assay to examine their interaction with the radioligand, [3H]NMS (pKA = 9.7 ± 0.3; Bmax = 4.5 ± 1.3 pmol/mg; n = 3). Both compounds inhibited binding in a monophasic manner almost down to nonspecific levels at [3H]NMS concentrations of 0.2 nM (data for 77-LH-28-1 shown in Fig. 2A). To reveal any potential allosteric mechanism of action, the same experiment was performed using 2 nM [3H]NMS (equivalent to approximately 10 × KA). Global analysis of both datasets according to eq. 3 (Materials and Methods) indicated that the compounds possessed micromolar affinity for the M1 mAChR with high degrees of negative cooperativity with respect to [3H]NMS (77-LH-28-1, pKB = 6.34 ± 0.14; Log α = −1.78 ± 0.17 (α = 0.017; n = 3); AC-42, pKB = 5.87 ± 0.06; Log α = −2.03 ± 0.06 (α = 0.009; n = 3). In contrast, the prototypical mAChR allosteric modulator, C7/3-phth, displayed much weaker negative cooperativity, as evidenced by its inability to fully inhibit specific radioligand binding even at 0.2 nM [Fig. 2B ; C7/3-phth, pKB = 5.53 ± 0.04; Logα = −0.81 ± 0.02 (α = 0.15; n = 3)].

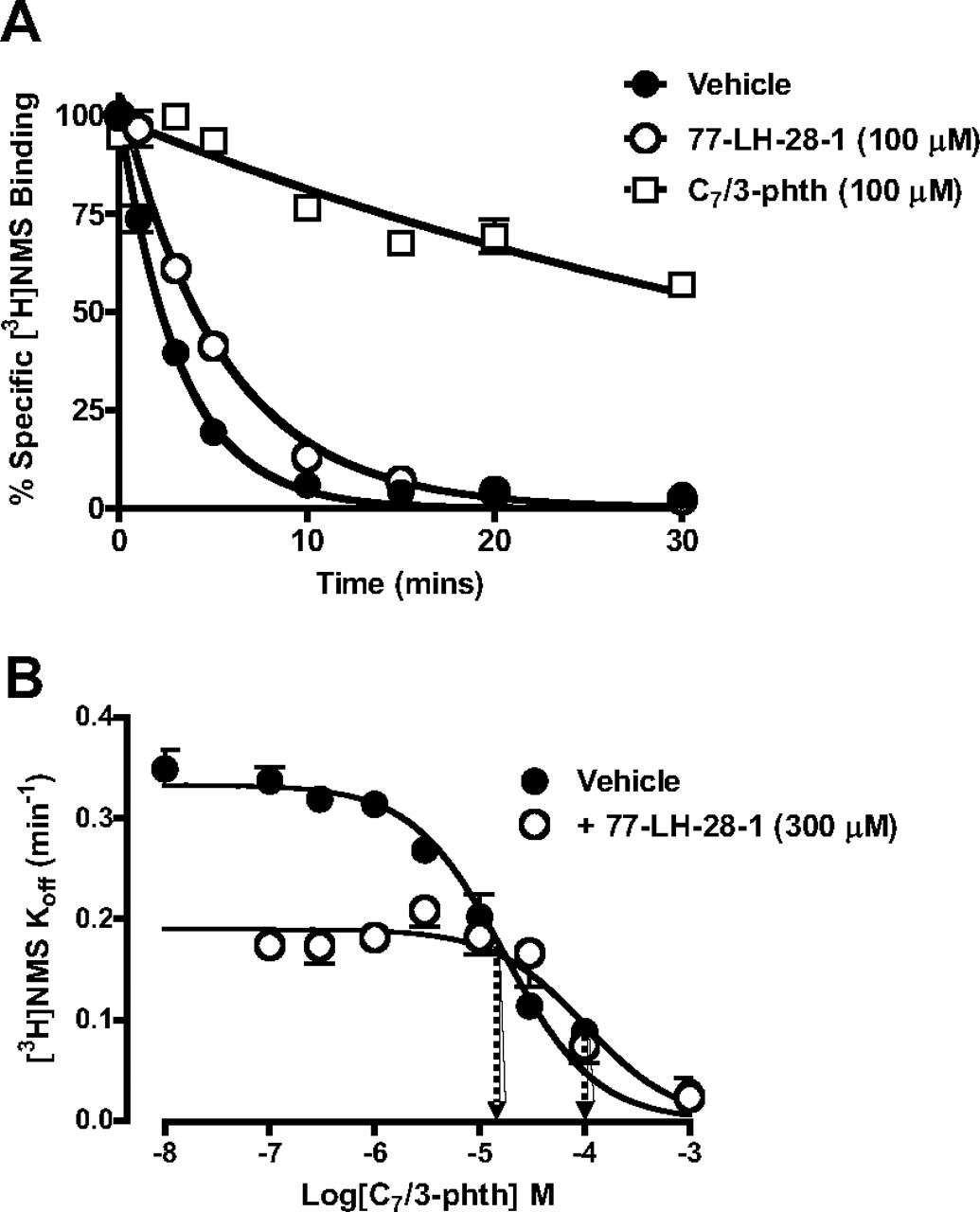
Comparison of equilibrium binding between 77-LH-28-1 and the prototypical allosteric mAChR modulator C7/3-phth. The interaction between each ligand and the orthosteric ligand, [3H]NMS, was assessed at 37°C on membranes from CHO FlpIn cells stably expressing the human M1 mAChR. The curves superimposed on the data points represent the best global nonlinear regression curve fit of an allosteric ternary complex model. Points represent the mean ± S.E.M. of three experiments performed in duplicate. Where error bars are not shown, they lie within the dimensions of the symbol.
77-LH-28-1 Recognizes Part of the “Common” Allosteric Site on the [3H]NMS-Occupied M1 mAChR.
Because of the high degree of negative cooperativity observed between [3H]NMS and AC-42 or 77-LH-28-1, the conclusion that these agonists interact allosterically with respect to NMS at the M1 mAChR was equivocal. To validate their mechanism of action further, their effects on the dissociation of a preformed complex of M1 mAChR and [3H]NMS was examined. In the absence of allosteric modulator, [3H]NMS dissociation was rapid and monophasic, with a koff value of 0.35 ± 0.01 min−1 (n = 3). The presence of C7/3-phth (100 μM) significantly slowed the [3H]NMS dissociation rate (Fig. 3A; koff = 0.04 ± 0.01 min−1), consistent with previous observations of its allosteric mechanism of action. Likewise, 100 μM concentrations of either AC-42 (koff = 0.28 ± 0.01 min−1) or 77-LH-28-1 (koff = 0.22 ± 0.01 min−1) significantly retarded the rate of [3H]NMS dissociation from the M1 mAChR compared with control, although not to the same extent as observed with C7/3-phth (P < 0.05; one-way ANOVA followed by Dunnett's post test; Fig. 3A). These effects clearly show that both AC-42 and 77-LH-28-1 are able to interact allosterically with the [3H]NMS-occupied receptor.

77-LH-28-1, recognizes part of the prototypical allosteric site on the [3H]NMS-occupied M1 mAChR. A, [3H]NMS dissociation determined in the absence or presence of C7/3-phth or 77-LH-28-1 at 37°C on membranes from CHO FlpIn cells stably expressing the human M1 mAChR. B, full concentration-response relationship of the effect of C7/3-phth on the dissociation rate of [3H]NMS at 37°C in the absence or presence of 77-LH-28-1. Data represent the mean ± S.E.M. obtained from three experiments conducted in duplicate. Where error bars are not shown, they lie within the dimensions of the symbol. Arrows indicate midpoint potency values.
We then used a two-point kinetics experimental paradigm (Kostenis and Mohr, 1996) to determine the concentration-dependence of the effect of C7/3-phth on [3H]NMS dissociation rate, which allowed for an estimate of its potency at the [3H]NMS-occupied receptor; pIC50 = 4.77 ± 0.06 (n = 3; Fig. 3B). Unfortunately, it was not possible to establish a similar concentration-response profile for either AC-42 or 77-LH-28-1 because of a combination of their low solubility and the relatively modest effect they have on [3H]NMS dissociation rate. However, it was possible to probe the interaction between C7/3-phth and 77-LH-28-1 using this experimental design. As shown in Fig. 3B , 77-LH-28-1 (300 μM) produced a significant 6-fold rightward shift in the concentration-response curve of C7/3-phth to inhibit the rate of [3H]NMS dissociation (pIC50 in the presence of 77-LH-28-1 = 3.99 ± 0.14; n = 3) as well as exerting an effect to slow [3H]NMS dissociation in its own right. Assuming a competitive interaction, this corresponds to a pA2 value of 4.3 for 77-LH-28-1. According to the allosteric ternary complex model, this pA2 value corresponds to the negative log of the dissociation constant of 77-LH-28-1 for the [3H]NMS-occupied receptor, which we can also calculate separately from the KB and α values derived from eq. 4 above (yielding a predicted pA2 value of 4.6). These data indicate, for the first time, that 77-LH-28-1 probably shares some of its binding site with that of the prototypical mAChR allosteric modulator, C7/3-phth (at the [3H]NMS-occupied receptor).
Determination of Functional Efficacies and Interactive Properties of Novel Agonists at the M1 mAChR.
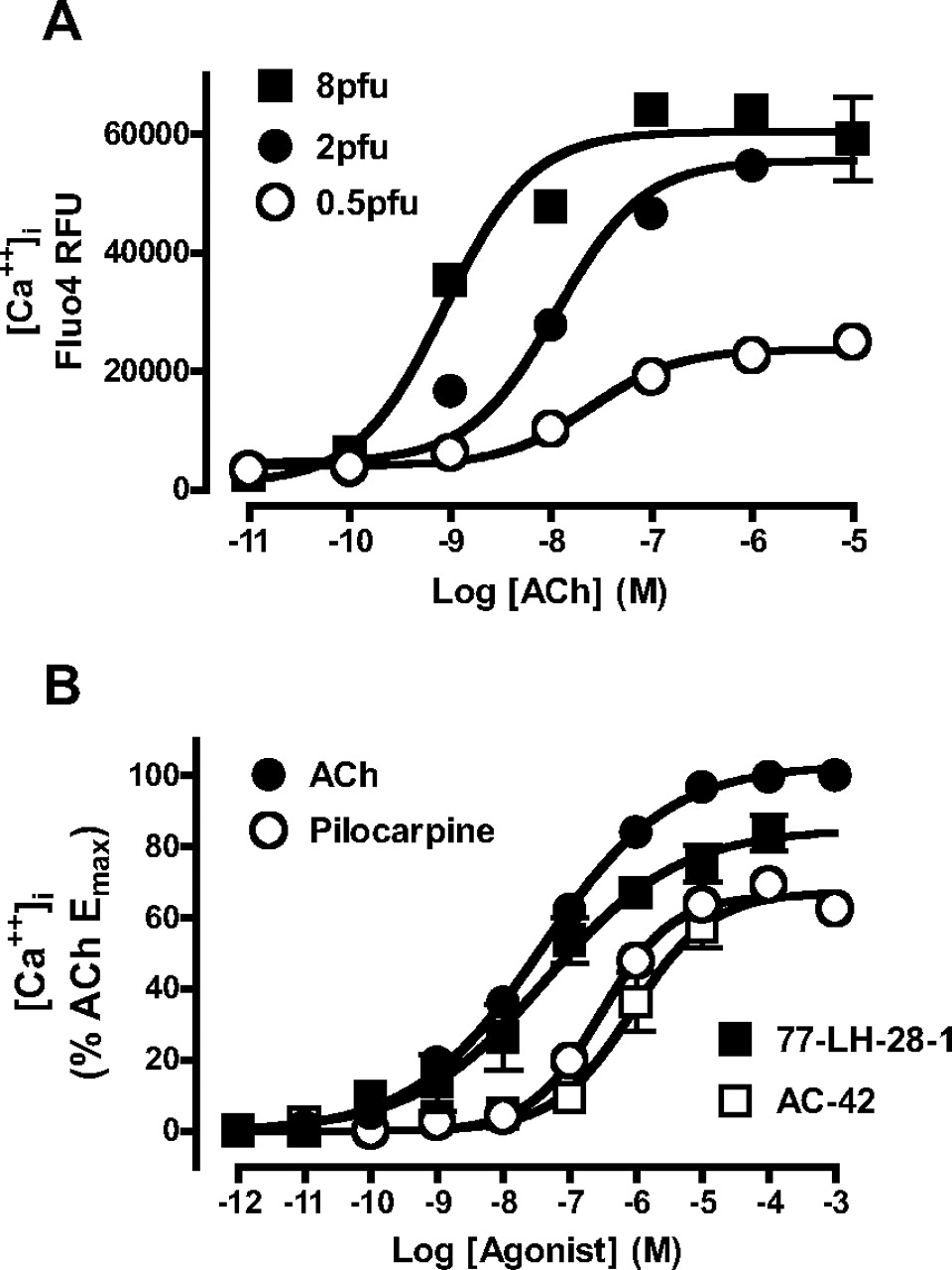
To evaluate the functional pharmacology of AC-42 and 77-LH-28-1 in comparison with prototypical orthosteric agonists, intracellular calcium elevation studies were performed in U2OS cells transiently transduced with the M1 mAChR using BacMam (modified baculovirus) technology. One advantage of BacMam transductions is that transient receptor expression level can be controlled by varying the multiplicity of infection during the transduction. Figure 4A shows the effect of varying transduction concentrations (0.5, 2 and 8 pfu per cell) on the concentration-response curve to ACh, indicating that different receptor expression levels allow for clear separation of agonist efficacies. Two pfu/cell was chosen as an optimal multiplicity of infection for allowing ACh to elicit a close-to-system-maximal Ca2+ response. These conditions clearly revealed that 77-LH-28-1 and AC-42 were lower efficacy agonists than ACh; pilocarpine was included as a prototypical orthosteric partial agonist comparator (Fig. 4B).

Novel agonists have lower efficacy at the M1 mAChR relative to ACh. A, effect of different expression levels of M1 mAChR on ACh potency and maximal effect for mediating intracellular Ca2+ elevation in U2OS cells transiently transduced using BacMam technology. B, comparison of orthosteric agonist (ACh, pilocarpine) and novel agonist (77-LH-28-1, AC-42)-mediated intracellular Ca2+ elevation in U2OS cells transiently transduced with 2 pfu/cell of M1 mAChR. Points represent the mean ± S.E.M. of three experiments performed in duplicate. Where error bars are not shown, they lie within the dimensions of the symbol.
Subsequently, we transduced U2OS cells with 0.5 pfu/cell to reduce receptor expression, and hence agonist efficacy, to study the functional interaction between 77-LH-28-1 and ACh; the prototypical orthosteric antagonist atropine was included as a comparator. The effects of atropine and 77-LH-28-1 on the ACh concentration-response curve are shown in Fig. 5. Increasing concentrations of both atropine and 77-LH-28-1 produced concentration-dependent, parallel, rightward shifts in the ACh concentration-response curve. In addition, a slightly elevated basal response was observed in the presence of 77-LH-28-1, indicative of the residual partial agonist activity of this compound. Analysis of the ACh/atropine data according to eq. 7 (Materials and Methods) and the ACh/77-LH-28-1 data according to eq. 8 yielded Schild slopes not significantly different from unity (0.97 ± 0.05 for atropine and 0.85 ± 0.11 for 77-LH-28-1); constraining them as such yielded the following pA2 values of 9.30 ± 0.08 (n = 5) for atropine and 6.21 ± 0.12 (n = 4) for 77-LH-28-1, respectively. Therefore, the interaction between 77-LH-28-1 and ACh, at least under the conditions tested, seems indistinguishable from simple competition, suggesting that the binding of 77-LH-28-1 must either overlap with that of the orthosteric site or be linked to the binding of ACh with a very high degree of negative cooperativity that could not be differentiated from orthosteric competition over the concentration range tested in the calcium assay.

Functional interaction between ACh and 77-LH-28-1 is consistent with competition. Effect of increasing concentrations of atropine (A) or 77-LH-28-1 (B) on ACh-mediated intracellular Ca2+ elevation in U2OS cells transiently transduced with 0.5 pfu/cell of M1 mAChR. The curves superimposed on the data points represent the best global nonlinear regression curve fit of a competitive Schild model (A) or operational model (B). Points represent the mean ± S.E.M. of four or five experiments performed in duplicate. Where error bars are not shown, they lie within the dimensions of the symbol.
Effects of Key M1 mAChR Mutations on Agonist Pharmacology.
The preceding studies indicated that the novel agonists display pharmacology that is consistent with both orthosteric and allosteric modes of action, depending on the experimental design. It is possible that such compounds recognize epitopes in both these regions of the receptor and adopt more than one binding pose depending on whether the orthosteric site is occupied by another ligand or not. To further probe these interactions at the molecular level, we examined the pharmacology of the novel agonists at M1 mAChRs containing key point mutations. Specifically, we determined the affinity and efficacy of AC-42 and 77-LH-28-1 at Y381A, W101A and F77I mutant M1 mAChRs. Tyr381 in TM6 is a known key component of the ACh orthosteric binding site that, when mutated to alanine, reduces the potency of prototypical orthosteric agonists such as ACh and carbachol but has relatively little effect on the response to novel agonists such as AC-42 and N-desmethylclozapine (Ward et al., 1999; Spalding et al., 2002; Sur et al., 2003). Trp101 in TM3 of the M1 mAChR has previously been shown to be part of a “second shell” of residues that surround the orthosteric binding site (Hulme et al., 2003), and the W101A mutation markedly increases the potency of structurally related, selective agonists such as AC-260584 (Spalding et al., 2006) and 77-LH-28-1 (Lebon et al., 2009) at the receptor. Finally, Phe77 was identified in preliminary work as a residue that, when mutated to isoleucine, caused a reduction in the potency of AC-42 (Jacobson et al., 2004).
U2OS cells were transduced with different numbers of plague-forming units per cell to attain roughly similar levels of receptor expression for mutant versus wild-type receptors, as determined by whole-cell [3H]QNB binding (Table 1); we used this radioligand because its affinity is substantially less affected by mutation of key residues, such as W101A and Y381A, compared with the binding of [3H]NMS (Ward et al., 1999; Lebon et al., 2009). Although variations in expression were still noted between mutants, the estimated Bmax values were used to correct for these variations after application of an operational model of agonism (eq. 6) to obtain agonist efficacy estimates (Logτc values; Table 2) for Ca2+ elevation. [3H]QNB and the agonists tested each displayed affinities at the wild-type M1 mAChR consistent with their previously described pharmacology at this receptor subtype. The W101A mutation resulted in a small, but significant, reduction in affinity of ACh, pilocarpine, and [3H]QNB (Table 1), consistent with a role for Trp101 as part of the second shell of residues forming the orthosteric binding site. Conversely, this mutation resulted in significant increases in the affinity of AC-42 and 77-LH-28-1 (Table 1), in agreement with previous binding studies (Lebon et al., 2009). It is noteworthy that this mutation did not alter the efficacies of ACh and pilocarpine (Table 2; Fig. 6), but enhanced the efficacy of AC-42 (though not 77-LH-28-1).

Novel agonists display divergent sensitivity to key M1 mAChR mutations relative to prototypical orthosteric agonists. Effect of increasing concentrations of agonist on M1 on mAChR-mediated intracellular Ca2+ elevation in U2OS cells transiently transduced with the indicated mutant M1 mAChR. Points represent the mean ± S.E.M. of three experiments performed in duplicate.
Mutation of Y381A in TM6 did not result in a change of [3H]QNB affinity (Table 1), consistent with previous observations that this residue is able to discriminate between the binding of [3H]QNB and [3H]NMS (Ward et al., 1999). However, mutation of Y381A markedly reduced the affinity of orthosteric agonists ACh and pilocarpine, consistent with its role as a key binding partner in the orthosteric agonist binding site. This mutation also reduced ACh and pilocarpine efficacy, suggesting that Tyr381 is also involved in receptor activation by orthosteric agonists (Table 2; Fig. 6). Likewise, Y381A reduced the efficacies of AC-42 and 77-LH-28-1 but moderately increased their affinity compared with the wild-type receptor, suggesting that although the role of Tyr381 in receptor activation may be common to all the agonists tested, only orthosteric agonists rely on this residue for binding to the receptor.
Phe77 is located in TM2, and had been reported in a preliminary study to selectively diminish the potency of AC-42 when mutated to isoleucine (Jacobson et al., 2004). We have now found that this mutation did not alter the affinity of any of the ligands tested (except for a small reduction in 77-LH-28-1 affinity; Table 1). However, the efficacies (Log τc values) of both AC-42 and 77-LH-28-1 were substantially reduced by F77I, whereas those of ACh and pilocarpine were unaltered (Table 2; Fig. 6). This indicates that mutation of F77I selectively affects the ability of AC-42 and 77-LH-28-1 to signal, unlike the Y381A mutation, which reduced the efficacy of all the agonists tested.
To further verify this novel observation, the activity of all four agonists was examined at both the wild-type and F77I constructs in a second signaling assay, that of ERK1/2 phosphorylation. All agonists displayed Logτc values somewhat lower than those observed in the Ca2+ assay (Table 3; Fig. 7), suggesting that the receptor coupling efficiency in the pERK1/2 assay is lower. In agreement with the previous dataset, mutation of Phe77 did not alter the efficacy of ACh or pilocarpine but reduced the efficacy of 77-LH-28-1 and abolished the agonist activity of AC-42 (Table 3; Fig. 7). Thus the selective modulation of the efficacy of the novel agonists by F77I was not restricted to a single pathway but observed for both Ca2+ elevation and ERK1/2 phosphorylation.

Differential effect of M1 mAChR F77I mutation on novel agonist efficacy is independent of signaling pathway. Effect of increasing concentrations of agonist on M1 on mAChR-mediated phosphorylation of ERK1/2 in U2OS cells transiently transduced with the indicated mutant M1 mAChR. Points represent the mean ± S.E.M. of three experiments performed in duplicate and are normalized to the response mediated by 10% FBS. Where error bars are not shown, they lie within the dimensions of the symbol.
Modeling and Ligand Docking.
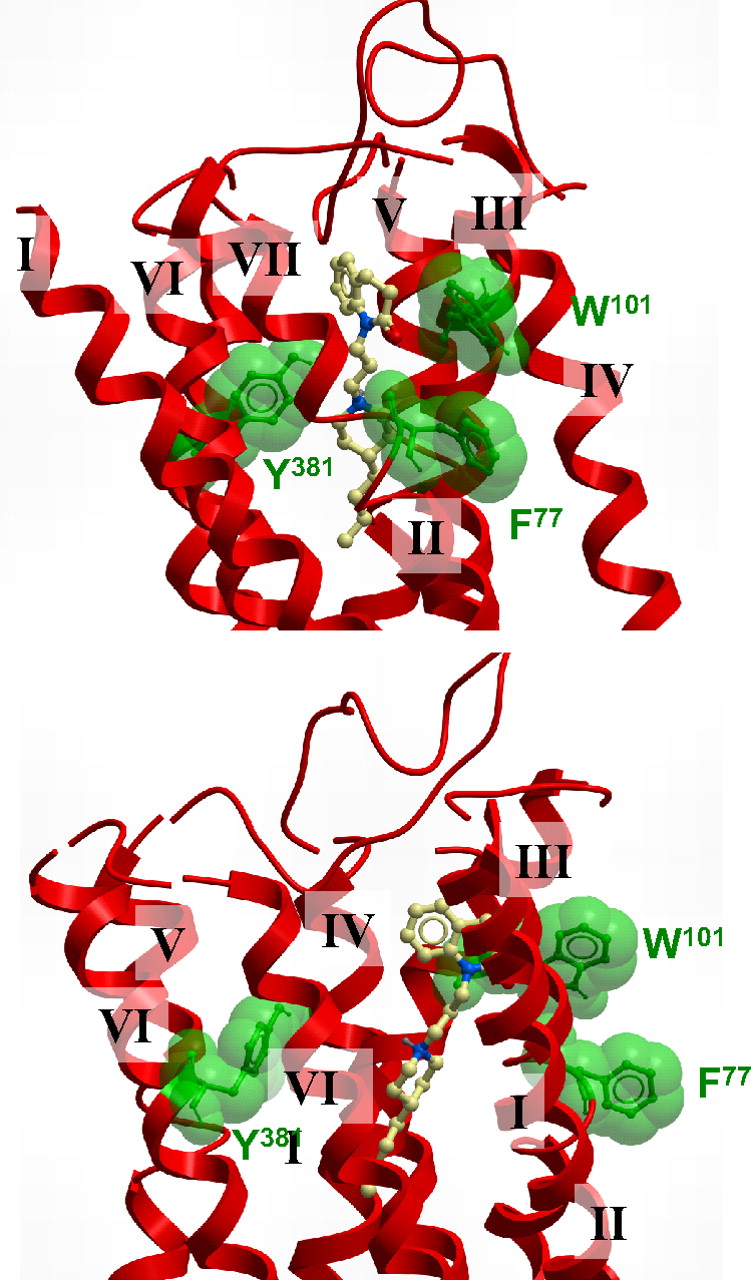
In addition to the radioligand binding and functional studies, molecular modeling and ligand docking were performed to rationalize the results seen with AC-42 and 77-LH-28-1. All reasonable docking solutions suggested that the aspartate residue (Asp105) in TM3 was forming a charge-charge interaction with the protonated nitrogen of 77-LH-28-1. This placed the aromatic benzoyl portion of 77-LH-28-1 between helices 2, 3, and 7, with the phenyl ring adjacent to the flipped-out tryptophan (Trp101) and encircled by a number of other aliphatic and aromatic residues: Trp91, Leu102, Tyr82, and Tyr85. The butyl linker between the aromatic ring and the piperidine ring transverses a hydrophobic region defined by Tyr82, Leu83, Leu102, and Ile180, and the piperidine ring is interacting with Ser78 and Tyr404. The aliphatic butyl tail of AC-42 is located deep within the receptor and is forming hydrophobic interactions with Ile74, Trp378, and Cys407. It is noteworthy that the phenylalanine residue Phe77, located on the external side of TM2, does not seem to be interacting with the ligand; it does, however, seem to be playing a role in the stabilization of the flipping of Trp101 by forming a π-π stacking interaction when Trp101 is in its flipped-out gauche negative state. 77-LH-28-1 sits some distance from Tyr381 in TM6, consistent with previous reports of a lack of effect of mutation of this residue on AC-42 function (Spalding et al., 2002). The tyrosine residue Tyr381 is behind the Tyr404 residue on TM7 that is only partly in contact with the piperidine portion of 77-LH-28-1.
Discussion
The past decade has witnessed a virtual renaissance in the pharmacology of mAChR agonists, spearheaded by the discovery of compounds such as AC-42, which preferentially activate the muscarinic M1 mAChR subtype (Spalding et al., 2002; Langmead et al., 2006). It is noteworthy that a common mechanism invoked to explain the functional selectivity of these agonists has been one involving the possibility that they are allosteric (Sur et al., 2003; Langmead et al., 2006; Spalding et al., 2006; Jones et al., 2008). Our current findings suggest that these novel agonists indeed adopt different poses within the M1 mAChR relative to prototypical orthosteric agonists, such as ACh, but are unlikely to result in a purely allosteric mode of action.
It is noteworthy that in previous studies (Langmead et al., 2006; Lebon et al., 2009) and in our current study, the interactions between 77-LH-28-1 or AC-42 with orthosteric antagonists were consistently characterized by very high degrees of negative cooperativity (Log α values ≥ −2), as determined by application of an allosteric ternary complex model to the data. Such highly negative allosteric interactions are virtually indistinguishable from a competitive interaction at equilibrium. This is in contrast to prototypical allosteric modulators, such as C7/3-phth, which can be shown to interact via a purely allosteric mode in both equilibrium (Fig. 2B) and kinetics (Fig. 3) studies. The ability of ligands such as C7/3-phth and 77-LH-28-1 to alter the dissociation rate of an orthosteric ligand is a key indicator of an allosteric interaction, but it should be noted that this type of assay monitors interactions on a receptor that has been prelabeled with an orthosteric radioligand; dissociation kinetics experiments can reveal whether a ligand is able to adopt an allosteric binding pose but cannot be used to conclude that this pose is relevant to a receptor that does not have an orthosteric ligand present. Indeed, functional interaction studies using 77-LH-28-1 were in agreement with the equilibrium binding studies in that the interaction between this agent and the orthosteric agonist ACh were consistent with a simple competitive mechanism (Fig. 5) or very high negative cooperativity. This finding also complements previous data that showed the functional interaction between 77-LH-28-1 and scopolamine or pirenzepine to be indistinguishable from simple competition (Langmead et al., 2008a). Although our finding of apparent competition between 77-LH-28-1 and C7/3-phth on the [3H]NMS-occupied receptor indicates that the novel agonist has the capacity to recognize epitopes that constitute the prototypical allosteric binding site on the M1 mAChR, the log affinity of the agonist for the occupied receptor (∼4.3) is markedly lower than its log affinity for the free receptor (∼6.3). Given that the novel agonists have a much lower affinity for the allosteric site on the M1 mAChR when the receptor is occupied by orthosteric ligand, it is unlikely that their purely allosteric properties will play a prominent role in their pharmacological effects at concentrations that are physiologically relevant.
Overall, the profile of behaviors exhibited by the novel agonists are qualitatively and quantitatively similar to that displayed by the mAChR partial agonist McN-A-343, the mechanism of action of which was the subject of debate in the literature (Birdsall et al., 1983; Christopoulos and Mitchelson, 1997; May et al., 2007). More recently, radioligand binding and functional and mutagenesis studies using fragments of McN-A-343 revealed it to be a bitopic ligand (i.e., a hybrid molecule capable of interacting concomitantly with the receptor via both orthosteric and allosteric sites) (Valant et al., 2008). This is a mode of interaction that is distinct from a “pure” allosteric mode. Because of its recognition of epitopes within the orthosteric pocket, McN-A-343 seems competitive in equilibrium binding or functional assays but uses regions of the allosteric site to derive functional selectivity; when the orthosteric site is prebound with radioligand, it adopts a second purely allosteric binding mode with much lower affinity. Given the parallels in the pharmacology, this may be a likely mechanism by which 77-LH-28-1 interacts with the unoccupied muscarinic M1 mAChR, one that we have proposed for this agonist at the M2 mAChR (Gregory et al., 2010).
Our modeling and ligand docking also support the notion that 77-LH-28-1 and AC-42 are bitopic ligands (Valant et al., 2008, 2009). Both agonists have basic centers that are likely to interact with Asp105 in the orthosteric binding site; previous mutational data support the requirement of this residue for receptor activation by these agonists (Lebon et al., 2009). However, the benzoyl aromatic group of 77-LH-28-1 is predicted to occupy space between TM domains 2, 3, and 7, near the extracellular loops. It is noteworthy that Trp101 is thought to form the base of the binding site for the prototypical allosteric modulators such as C7/3-phth (Matsui et al., 1995). This binding mode would explain the observation that 77-LH-28-1 seems to compete (at least in part) for the same binding site as C7/3-phth (Fig. 3B). It is likely that interactions in this region are responsible for the observed allosteric effects of 77-LH-28-1 when the receptor is prebound with [3H]NMS. When the receptor is simultaneously exposed to 77-LH-28-1 and either [3H]NMS (equilibrium binding) or ACh (functional Ca2+ studies), the interaction of 77-LH-28-1 seems competitive, primarily because of the interaction with Asp105.
The residues chosen for mutagenesis, Trp101, Tyr381, and Phe77, also proved useful in identifying patterns of behavior that can be used to differentiate prototypical orthosteric agonists from novel selective agonists (including putative bitopic agonists). The first two residues were selected as their mutation has been shown to have divergent effects on orthosteric and putatively allosteric agonists (Spalding et al., 2002, 2006; Lebon et al., 2009), although their precise in role in ligand binding and receptor activation have not been fully established. Phe77 was highlighted as a residue that, when mutated to isoleucine, caused a reduction in the potency of AC-42 to activate the muscarinic M1 mAChR (Jacobson et al., 2004). Trp101, when in the gauche negative state, is predicted to be adjacent to the phenyl ring of 77-LH-28-1. Mutation of this residue to alanine has been shown to increase the potency of AC-42 and related compounds (Spalding et al., 2006). The same mutation significantly reduced ACh and pilocarpine affinity but significantly enhanced AC-42 and 77-LH-28-1 affinity (Table 1); there was no change in agonist efficacy with the exception of a reduction in the Logτc value for AC-42 (Table 2). These data suggest that Trp101 primarily plays a role in the binding of 77-LH-28-1, probably by “flipping out” to accommodate the ligand; therefore the absence of the side chain reduces the free energy required for ligand binding.
Mutation of Y381A significantly reduced the affinity of pilocarpine and ACh for the M1 receptor (Table 1), consistent with a role in the orthosteric binding site (Ward et al., 1999). It is noteworthy that this mutation moderately increased the affinity of AC-42 and 77-LH-28-1 (Table 1) but significantly reduced the efficacy of all agonists tested (Table 2). Thus, Tyr381 seems key to receptor activation but is only directly involved in the binding of ACh and pilocarpine. This is supported by the ligand docking, where 77-LH-28-1 is positioned some distance from Tyr381 (Fig. 8), unlike dockings of ACh (Hulme et al., 2003; Goodwin et al., 2007).
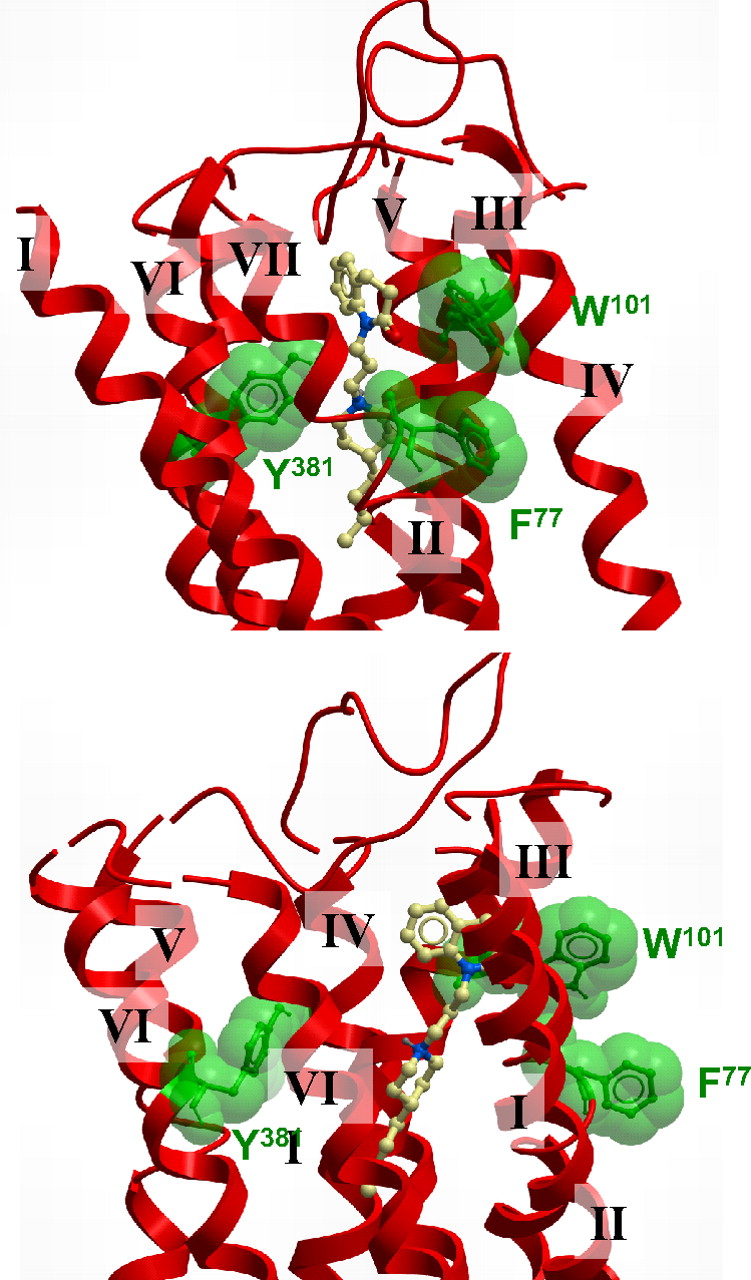
A proposed docking pose of 77-LH-28-1 is consistent with a bitopic mode at the M1 mAChR. A molecular model showing two different views of the top of the TM bundle of the M1 mAChR indicating an extended pose for 77-LH-28-1. The key residues mutated in the current study are also indicated.
At first sight, the role of Phe77 seems to be difficult to resolve based on the receptor model, because the residue faces away from the proposed ligand binding site (Fig. 8). However, mutation of this residue has a clear effect in selectively reducing the efficacy of AC-42 and 77-LH-28-1 (Tables 2 and 3). On this basis, we postulate that the aromatic side chain of Phe77 plays a role to stabilize the flipped-out gauche state of Trp101 via a π-π stacking interaction between the aromatic rings; this selectively enhances the function of AC-42 and 77-LH-28-1 but leaves pilocarpine and ACh unaffected. Therefore, removal of the aromatic ring prevents this stabilization and reduces the ability of 77-LH-28-1 to mediate receptor activation once bound.
Based on the data presented herein, AC-42 and 77-LH-28-1 may be better classed as bitopic, rather than allosteric, agonists, but definitive demonstration that such molecules contain both pure orthosteric and allosteric fragments is still required. Nonetheless, our mutational data have identified Tyr381 and Trp101 as selective differentiators of orthosteric versus novel agonist binding affinity and have revealed Phe77 as a novel and selective regulator of novel agonist efficacy. It will be interesting to determine whether additional functionally selective agonists that have been previously classed as “allosteric agonists” exhibit similar patterns of behavior.
Acknowledgments
We are grateful to Drs. Michael Crouch and Ron Osmond of TGR Biosciences for the generous gift of SureFire ERK1/2 Kit reagents.
Footnotes
-
This work was funded by the National Health and Medical Research Council (NHMRC) of Australia [Program Grant 519461]. A.C. is a Senior Research Fellow of the NHMRC, and P.S. is a Principal Research Fellow of the NHMRC.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
doi:10.1124/mol.110.064345.
-
ABBREVIATIONS:
- mAChR
- muscarinic acetylcholine receptor
- C7/3-phth
- heptane-1,7-bis-[dimethyl-3′-phthalimidopropyl]-ammonium bromide
- AC-42
- 4-n-butyl-1-[4-(2-methylphenyl)-4-oxo-1-butyl] piperidine hydrogen chloride
- NMS
- N-methylscopolamine
- TM
- transmembrane domain
- McN-A-343
- 4-I-[3-chlorophenyl]carbamoyloxy)-2-butynyltrimethylammnonium chloride
- 77-LH-28-1
- 1-[3-(4-butyl-1-piperidinyl)propyl]-3,4-dihydro-2(1H)-quinolinone
- CHO
- Chinese hamster ovary
- FBS
- fetal bovine serum
- ERK
- extracellular signal-regulated kinase
- QNB
- quinuclidinyl benzilate
- WT
- wild type
- pERK1/2
- phosphorylated ERK1/2.
- Received February 23, 2010.
- Accepted April 21, 2010.
- Copyright © 2010 The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}