


Abstract
Genistein (5,7,4′-trihydroxyisoflavone), an isoflavinoid found in soy beans, has been identified as potentially causal for the low incidence of metastatic prostate cancer (PCa) in certain countries. Although genistein-induced PCa cell adhesion has been identified as a possible causative mechanism, direct growth inhibition by genistein has been reported and also could be causal. If in vivo growth inhibition was significant, then growth inhibition should occur at concentrations attained with dietary consumption, the mechanism of growth inhibition should be relevant to PCa, and genistein (a broad-spectrum in vitro protein-tyrosine kinase inhibitor) should have relatively specific kinase inhibitory effectsin vivo. These considerations were investigated by measuring growth inhibitory activity in a variety of PCa cell lines. Growth inhibitory effects were shown not to occur with concentrations below the low micromolar range (i.e., 3 logs above that attained in serum). In-depth mechanistic studies with the PC3-M metastatic variant cell line demonstrated that growth inhibition was independent of genistein’s estrogenic effects. Genistein was shown to decrease the viability of nonadherent cells, suggesting a lack of dependence on cell adhesion for growth inhibition. However, important molecular and kinetic differences between genistein’s effects on growth in adherent versus nonadherent cells were identified. Specific suppression of focal adhesion kinase activity (without global decreases in phosphotyrosine) was shown to precede induction of apoptosis, which was responsible for growth inhibition in adherent cells. These findings do not support anin vivo growth inhibitory role by genistein consumed in quantities associated with a soy-based diet. They do, however, identify genistein as a potential therapeutic agent for PCa and as a tool with which to study the control of apoptosis in PCa.
PCa will be diagnosed in approximately 317,100 men in the United States in 1996, accounting for 41% of all male cancers; it will cause death in approximately 41,400 individuals, accounting for 14% of all male cancer deaths (1). Disease progression is inevitable when patients no longer respond to hormone therapy (see Ref. 2 for review). Cytotoxic agents and other forms therapy are associated with low response rates and have no significant effect on survival.
Ineffective therapy, high prevalence, and unique biology have spurred the search for novel acting agents. One approach is the rational targeting of processes unique to PCa. PCa is uniquely recognized as a slow-growing malignancy, the growth of which is controlled by specific growth factors. Rates of PCa cellular proliferation of 3.0% and below are typical (3), making this one of the slowest growing malignancies known. Indeed, this low growth fraction may be responsible, at least in part, for the ineffectiveness of cytotoxic agents, the activity of which is diminished in slower growing tumors. Specific growth factors have been identified that affect PCa cell growth and seem to be involved in neoplastic transformation (4, 5). Slow growth rates and unique growth-factor dependence suggest that pharmacologic agents that induce apoptosis and/or modulate signal transduction processes may be particularly effective against PCa (4, 6-9).
There is insufficient knowledge regarding both the control of apoptosis and of signal processing in PCa to allow easy identification of intracellular targets or of agents that can modulate those targets. The demographics of PCa do, however, provide some insight into the identification of potential therapeutic agents. Clinical PCa rates are higher in Western countries than in countries in Southeast Asia, where soy beans are a dietary staple; when Southeast Asians migrate to the West, PCa incidence increases over the course of two generations to the level seen in the West (10, 11). Genistein (5,7,4′-trihydroxyisoflavone), an isoflavinoid derived from soy beans, has been identified as a putative PCa-preventive agent that may account for these demographics (10-13). This is significant in that genistein is a recognized PTK inhibitor (see Ref. 14 for review), and PTKs are known to play key roles in both growth factor-related signal modulation (see Ref. 15 for reviews) and in apoptosis (16).
Previous studies from this laboratory have shown that genistein increases the adhesion of PCa cells (8). Such a mechanism, if operatingin vivo, would block the initial step of the metastatic cascade (i.e., detachment of cells from supporting matrix) and would be consistent with current demographic findings. However, Peterson and Barnes (17) have shown that genistein can directly inhibit PCa cell growth in vitro, raising the possibility that it may do soin vivo; such a mechanism also would be consistent with the demographic findings.
If genistein has significant in vivo PCa growth inhibitory effects, then the following conditions should apply: genistein (a “nonspecific” in vitro PTK inhibitor) should have relatively specific in vivo effects on PTK activity; growth inhibition should be occurring through a mechanism relevant to PCa; finally, growth inhibitory effects should be evident at genistein concentrations that are attained with dietary consumption.
A series of studies were undertaken to investigate these considerations. Taken together, they indicate that genistein has unique effects on PCa cells of potential therapeutic and definite investigative significance.
Materials and Methods
Cell culture.
PC3 and DU-145 prostate carcinoma cells were obtained from American Type Culture Collection (Rockville, MD); PC3-M prostate carcinoma cells were obtained from L. Whitesell (National Cancer Institute, Bethesda, MD) and are a metastatic variant of PC3 cells as described by Kozlowski et al. (18). PC3 and PC3-M cells were grown in Roswell Park Memorial Institute 1640 medium (GIBCO/BRL, Gaithersburg, MD) supplemented with 10% FBS (Whittaker, Walkersville, MD); DU-145 cells were grown in Dulbecco’s modified Eagle’s medium (Biofluids, Rockville, MD) supplemented with 5% FBS. All cells were maintained at 37° in a humidified atmosphere of 5% carbon dioxide, with biweekly media changes.
Growth inhibition assays.
Three-day growth inhibition assays were performed in Falcon TC microtiter plates (Becton Dickinson, San Jose, CA) unless otherwise stated. Initially, 800-2000 PCa cells per 100 μl of cell culture media were plated into each well. Twenty-four hours later, genistein (LC Laboratories, Woburn, MA) suspended in cell culture media was added to give a final volume of 200 μl per well. Before use, genistein was suspended in DMSO (Sigma Chemical, St. Louis, MO) stock solution and stored at −70°. Genistein was thawed just before use. The final DMSO concentration did not exceed 0.5% in any experiment.
Cell viability, as determined by trypan-blue exclusion, was reflected accurately by MTT reduction for PC3 and PC3-M cells; for DU-145 cells, it was reflected most accurately by thymidine uptake.1 MTT reduction was measured as described (19), with modifications: 20 μl of a solution containing 5 mg/ml MTT in PBS were added to each well of a microtiter plate, which was then incubated for 4 hr at 37°. Cells were then lysed in 150 μl DMSO, and optical density was measured at 540 nm on a Bio Tek microplate reader (model EL312E; Bio Tek Instruments, Winooski, VT). Thymidine uptake was measured as described (4), with modifications: 0.5 μCi of [3H]thymidine in 20 μl of cell culture media was added to each well of a microtiter plate and incubated for 6 hr at 37°. Cells were then harvested onto glass fiber filters (Packard, Meriden, CT) with a Packard cell harvester and counted in a Matrix 9600 microplate counter (Packard). All microtiter assays in this study were run in replicates of 3–5; all cell growth assays in this study (microtiter and clonogenic) were performed at least twice.
Falcon TC microtiter plates, which were coated with 1 μg/cm2 fibronectin according to the manufacturer (Collaborative Research, Bedford, MA), and Primeria Falcon (Becton Dickinson) microtiter plates were used in some growth assays as indicated. Estradiol (Sigma Chemical), used in some experiments, was suspended in ethanol and stored as stock solutions at −70°; final ethanol concentrations in growth assays did not exceed 0.25%. Cytochalasin D (Sigma Chemical) was added in some experiments as indicated.
Eleven-day growth assays were based on colony formation; 500 cells in 4 ml of growth media were allowed to adhere to 6-cm Falcon tissue culture dishes for 24 hr. For continuous drug exposure experiments, media and genistein were replaced every 4 days. For pulse treatment experiments, genistein-containing media were removed after 4 days and replaced with fresh, genistein-free, media. After 11 days, cells were washed with PBS, stained for 1 hr with 0.025% methylene blue in PBS, and rinsed with water. Colonies ≥ 0.5 mm were then counted on a model 880 Artek Counter (Dynatech Labs, Chantilly, VA). All clonogenic assays were run in replicates of at least three.
Flow cytometric analysis.
Cell preparation, DNA staining, and flow cytometric analysis were performed as described (20), on a FACStar Plus (Becton Dickinson).
Hoechst staining and photomicroscopy.
PC3-M cells for Hoechst staining were grown on sterilized coverslips and processed as described (8), with modifications. Briefly, after washing one time with PBS, cells were fixed with 3.7% formaldehyde in PBS for 10 min, washed one time with PBS, stained with 0.4 μg/ml Hoechst (Molecular Probes, Eugene, OR) in PBS for 15 min, washed two times with PBS, and then one time with water. Coverslips were then air-dried and mounted with SlowFade (Molecular Probes) mounting media. Immunofluorescent micrographs were taken on a Zeiss Axiovert (Carl Zeiss, Oberkochen, Germany) microscope.
DNA ladder formation.
Exponentially growing PC3-M cells were treated with genistein for the indicated time periods; both adherent and floating cells were then washed with PBS, combined, and lysed in DNA lysis buffer (0.1% NP40, 10 mm Tris, pH 7.4, 25 mm EDTA). Lysates were digested with 0.5 mg/ml proteinase K (Pharmacia, Uppsala, Sweden) for 16 hr at 55°, centrifuged at 14,000 × g for 3 hr at 4°, and then digested with 100 μg/ml DNase-free RNase A (Pharmacia) for 10 hr at 37°. DNA was ethanol precipitated, resuspended in water, and DNA content measured by optical density. Equal amounts of DNA were loaded onto each lane of a 2.3% agarose (GIBCO/BRL) gel, which was run over 4 hr. DNA bands were visualized by UV illumination after ethidium bromide staining.
Phosphotyrosine and FAK Westerns.
Cell lysis and Western blotting for FAK (8) and phosphotyrosine (21) were performed as described previously, with modifications. Briefly, PC3-M cells were lysed in TNESV lysis buffer (50 mm Tris, pH 7.5, 1% Nonidet P40, 2 mm EDTA, 0.1% SDS, 100 mm NaCl, and 1 mm vanadate) (22), and the resultant clarified lysates, normalized for protein, were run on an 8% sodium dodecyl sulfate polyacrylamide gel. Proteins were then transferred onto 0.45 μm nitrocellulose (Schleicher & Schuell, Keene, NH) in either a semidry transfer apparatus (Pharmacia, for FAK) or in a wet transfer cell (BioRad, Hercules, CA, for phosphotyrosine). For FAK, blots were blocked with nonfat dry milk and probed with an anti-FAK monoclonal antibody (Transduction Laboratories, Lexington, KY) at 1 μg/ml. For phosphotyrosine, blots were blocked with 5% bovine serum albumin (fraction V; Sigma) in Tris-buffered saline/Tween 20 (21) for 1 hr at 45° and probed with a 1:750 dilution of antiphosphotyrosine antibody (clone 4G10; Upstate Biotechnology, Lake Placid, NY) in blocking solution for 1 hr at room temperature. Proteins were then detected using an anti-mouse-HRP conjugated secondary antibody (Amersham, Arlington Heights, IL) diluted 1:13, 000 in Tris-buffered saline/Tween 20, and visualized with the ECL detection system (Amersham) according to the manufacturer.
Focal adhesion kinase immunoprecipitation kinase assay.
FAK immunoprecipitation kinase assays were performed as described (8). Reactions were run in 40 μl of kinase assay buffer (10 mmTris, pH 7.5, 5 mm MnCl2) and initiated by the addition of 10 mCi [gamma-32P] ATP (3000 Ci/mmol; Amersham Corp.). After 4 min, the reaction was quenched by the addition of gel-loading buffer and heating to 95°; products were then run on an 8% sodium dodecyl sulfate polyacrylamide gel and visualized by autoradiography with intensifying screens using Kodak X-O-MAT AR film at −70°.
Results
Genistein inhibits PCa cell growth.
Short-term (3-day) growth assays were performed in microtiter plates with the PC3, PC3-M, and DU-145 PCa cell lines. After allowing the cells to adhere for 24 hr, cells were treated with various concentrations of genistein for 3 days. For PC3 and PC3-M cells, the ability to reduce MTT was measured; for DU-145 cells, thymidine uptake was measured. Separate studies have demonstrated that these methods of analysis, in the respective cell lines, correlate most closely with cell viability as measured by trypan-blue exclusion.1 Long-term (11-day) growth assays were performed by first allowing 500 cells to adhere in 6-cm culture dishes and then treating with various concentrations of genistein. After 11 days, cells were stained with methylene blue, and colonies ≥ 0.5 mm were counted. A 3-day growth inhibition curve for the PC3-M cell line is depicted in Fig. 1A and is typical of all cell lines tested. IC50 values for each cell line are shown in Fig. 1B and range from 18 to 43 μm(mean; n ≥ 3 for each cell line) for 3-day growth assays and from 10 to 17 μm (mean; n ≥ 2 for each cell line) for 11-day growth assays.
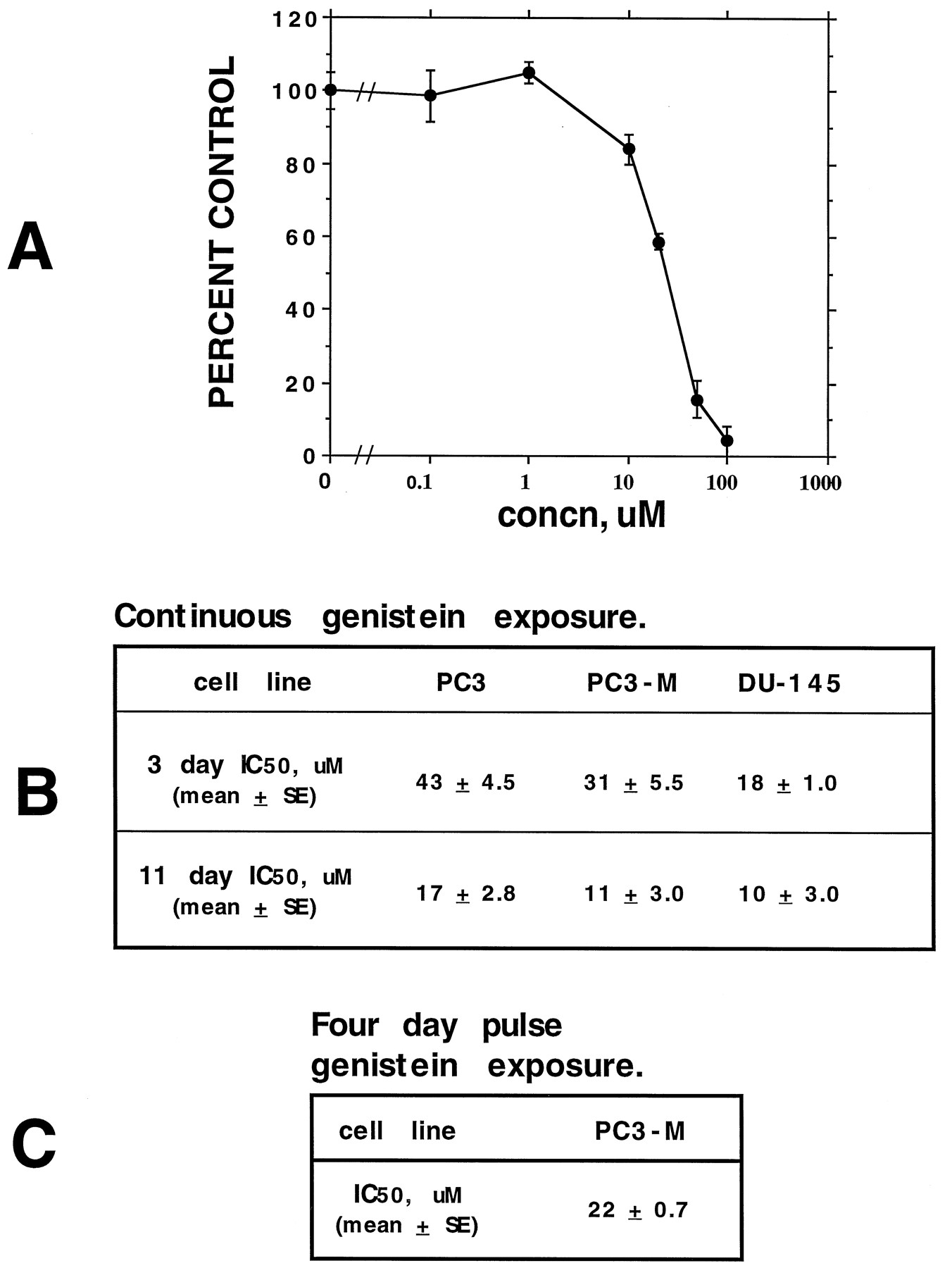
Growth inhibition of PCa cells by genistein. Three- and 11-day growth inhibition assays were performed as described in Materials and Methods. A, Typical growth inhibition curve for PC3-M cells, representative of all cell lines tested. B, Three- and 11-day continuous drug exposure IC50 values for a variety of PCa cell lines. C, IC50 value for pulse-treated PC3-M cells; cells were treated initially with genistein for 4 days and then were grown in the absence of genistein for the remainder of the 11-day study period. Values represent the mean ± standard error of three separate experiments (each run in replicates of 3–5) for the 3-day assays and of two separate experiments (each run in replicates of 3) for the 11-day assays.
A series of studies were then undertaken to examine both the schedule dependence and the mechanism of genistein-mediated growth inhibition. These studies were conducted with the PC3-M cell line, which is a metastatic variant of the parental androgen-independent PC3 cell line; the PC3-M cell line has a relatively short doubling time and is highly metastatic in athymic mice (8, 18). As such, it is representative of clinical hormone refractory metastatic PCa.
The effects of exposure time were first determined by treating PC3-M cells with genistein for various time periods. No detectable MTT reduction was observed for exposure times of less than 3 days, as measured in 3-day growth assays in which cells initially were exposed to genistein (up to 50 μm) for 1 or 2 days, followed by a genistein-free washout period for the remainder of the study period.1 Eleven-day clonogenic assays were then performed in which cells were treated initially for 4 days, followed by a genistein-free washout period for the remainder of the study. A 4-day treatment time was chosen because initial growth inhibitory effects were detectable after 3 days; therefore, 4 days ensured adequate time for growth inhibition. When performed in this manner, an IC50 value of 22 ± 0.7 μm (mean ± standard deviation; n = 2) was obtained (Fig. 1C), representing a 100 ± 3.2% increase from the 11 ± 3.0 μm IC50 value obtained when cells were cultured for 11 days with continuous genistein exposure (Fig. 1B).
Growth inhibition is not dependent on estrogen.
Genistein is a phytoestrogen (a plant-derived compound with estrogenic properties) and has known estrogenic effects in mammals (23). Estrogen receptors have been detected in the parental PC3 cell line, and high levels of estradiol have been shown to inhibit their growth (24). To examine the effect of estrogen on genistein-mediated growth inhibition, PC3-M-associated estrogen binding sites were measured first. No estrogen-receptor-binding sites were detected using the dextran-coated charcoal radioimmune estrogen binding assay and analysis for specific estrogen binding, as described (24, 25, 26), on whole-cell lysates as described (27).1
To rule out the possibility that low levels of functional estrogen receptors were present and were contributing to genistein-mediated growth inhibition, a functional assay was performed. In this assay, the ability of 0.1 μm estradiol to modulate PC3-M cell growth in the presence or absence of genistein was measured in a 3-day growth assay system. As can be seen in Fig. 2, high concentrations of estradiol did not directly inhibit PC3-M cell growth, nor did they affect genistein-mediated growth inhibition. This assay was performed in the presence of 10% FBS that had not been subjected to either dialysis or charcoal stripping; the final concentration of serum-derived estrogens in this system was <1 pm and was, therefore, >5 logs below the concentration of either genistein or estradiol used in this study. As charcoal stripping and/or dialysis will remove other potentially important growth modulatory factors that may be important in genistein- and/or estrogen-related signaling response pathways, this system was believed to be most accurate.

The effect of estrogen on genistein-mediated growth inhibition. Three-day growth assays were performed in which PC3-M cells were treated with genistein ± 0.1 μm estradiol. Values represent the mean ± standard error of a single experiment run in triplicate; similar results were attained in a separate experiment.
Growth inhibition is not dependent on cell attachment.
Cell adhesion is an important cell growth modulatory parameter (see Ref. 28for review). As we have demonstrated previously that genistein increases cell attachment of PCa cells (including PC3-M cells) (8), the role of cell attachment in genistein-mediated growth inhibition was investigated. Changes in supporting substrata were evaluated first. PC3-M cells were plated onto one of three types of solid matrix: standard microtiter plates, standard microtiter plates coated with 1 μg/cm2 fibronectin, or Primeria microtiter plates (these plates are coated with a highly charged, proprietary, protein-like material that serves to enhance cell adhesion). After allowing cells to attach for 24 hr, cells were treated with genistein; their ability to reduce MTT was measured after 3 days (Fig. 3A). Changes in solid matrix support did not affect genistein-mediated growth inhibition under these experimental conditions.

The effect of cell adhesion on genistein-mediated growth inhibition. A, Effect of various substrata on growth inhibition. PC3-M cells were plated onto each of 3 types of solid matrix support: standard tissue culture microtiter plates (STC), standard tissue culture microtiter plates coated with 1 μg/cm2 fibronectin (STC+FBN), or Primeria microtiter plates (PRIM). Twenty-four hours later, genistein was added and 3-day growth assays were performed as described in Materials and Methods. B, Effect of cell adhesion on growth inhibition. PC3-M cells were prevented from attaching by suspending in culture media while subjecting them to gentle mechanical agitation. After growing for 3 days in the presence or absence of 25 μm genistein, cell viability was measured by trypan-blue exclusion and compared with the viability of adherent cells treated in an identical manner. Values represent the mean ± standard error of a single experiment run in triplicate; similar results were attained in a separate experiment.
The effect of changes in the state of cell attachment was investigated by measuring the ability of genistein to inhibit cell growth in detached cells. Exponentially growing PC3-M cells were detached by exposure to trypsin/EDTA and resuspended in culture media; cells were then maintained in suspension for the 3-day treatment period by gentle mechanical agitation in the presence or absence of genistein. Under these experimental conditions, the number of viable cells (trypan-blue excluding) decreased by 35 ± 9.4% (mean ± standard error;n = 5) after exposure to 25 μm genistein (Fig. 3B); this is not significantly different from the 28 ± 4.8% (mean ± standard error; n = 3; Student’st test p value = 0.26) decrease observed when attached cells are exposed to 25 μm genistein (Fig.3B). Of note, however, untreated detached control PC3-M cells did not increase in number over the course of the 3-day assay period.1
Genistein does not cause cell cycle block in PCa cells.
Genistein induces a G2/M phase cell cycle block in HGC-27 colon cancer cells (29); therefore, similar effects were sought in PCa cells. When viable (trypan-blue excluding) PC3-M cells were measured over a 3-day genistein treatment period, cell numbers remained constant (Fig. 4A), consistent with growth arrest due to cellular cytostasis. Flow cytometric analysis, however, demonstrated only minor effects on the cell cycle. The percentage of cells in S/G2/M phases rose from a baseline value of 32 ± 2.9% (mean ± standard error; n = 3) to 42 ± 0.2% (Student’s t test p value = 0.05) after PC3-M cells were exposed to 50 μm genistein for 3 days. No detectable effects were evident with shorter exposure times.1

Kinetics of PCa growth inhibition by genistein. A, Viability of PC3-M cells exposed to genistein. The viability (trypan-blue excluding) of adherent PC3-M cells exposed to 50 μm genistein was measured daily over the course of a 3-day treatment period. Values represent mean of a single experiment run in triplicate (mean ± standard error ≤ 5% for all values); similar results were observed in a separate experiment. B, Comparison of thymidine uptake and MTT reduction in PC3-M cells exposed to genistein for 3 days. MTT reduction and thymidine uptake assays were performed as described in Materials and Methods on PC3-M cells that had been exposed to genistein for 3 days. Values represent the mean ± standard error of a single experiment run in triplicate (MTT) or replicates of five (thymidine); similar results were observed in a separate experiment.
If this small degree of cell cycle block is enough to account for the near-total growth stasis, a disproportionate decrease in thymidine uptake (a measure of actively cycling cells) relative to MTT reduction (a measure of viable cells) should be evident. The results of such a comparison are shown in Fig. 4B; no significant decrease in thymidine uptake relative to MTT reduction was detected.
Genistein inhibits cell growth by inducing apoptosis.
A failure of cell numbers to increase with time, in the absence of cell cycle blockade, suggests that there is an increase in the rate of cell death. This is consistent with the notion that genistein is inducing apoptosis without significantly affecting cell-cycle transition. Induction of apoptosis was sought by first looking for the formation of nuclear apoptotic bodies. PC3-M cells were grown in the presence of 50 μm genistein for various time periods and analyzed by light microscopy for the formation of apoptotic bodies (Fig.5A). Although nuclear segmentation was evident after 48 hr (data not shown), it was more pronounced at 72 hr. To confirm these findings, low-molecular-weight DNA ladder formation was measured by agarose gel electrophoresis. Under the same experimental conditions used for detecting apoptotic body formation, DNA ladder formation was evident by 48 hr (Fig. 5B).

Induction of apoptosis by genistein in PC3-M cells. Exponentially growing PC3-M cells were treated with 50 μmgenistein for up to 3 days. A, Photomicrographs of Hoechst-stained cells that had been exposed to genistein (GE) for 3 days. B, Photomicrographs of Hoechst-stained cells that had not (CO) been exposed to genistein (magnification 1000×). C, a photograph of an UV light-illuminated agarose gel containing total cellular DNA; lanes were loaded with DNA preparations made 1, 2, or 3 days after cells were treated with genistein, control cells (C) were not treated.
Genistein has unique effects on cellular PTK activity.
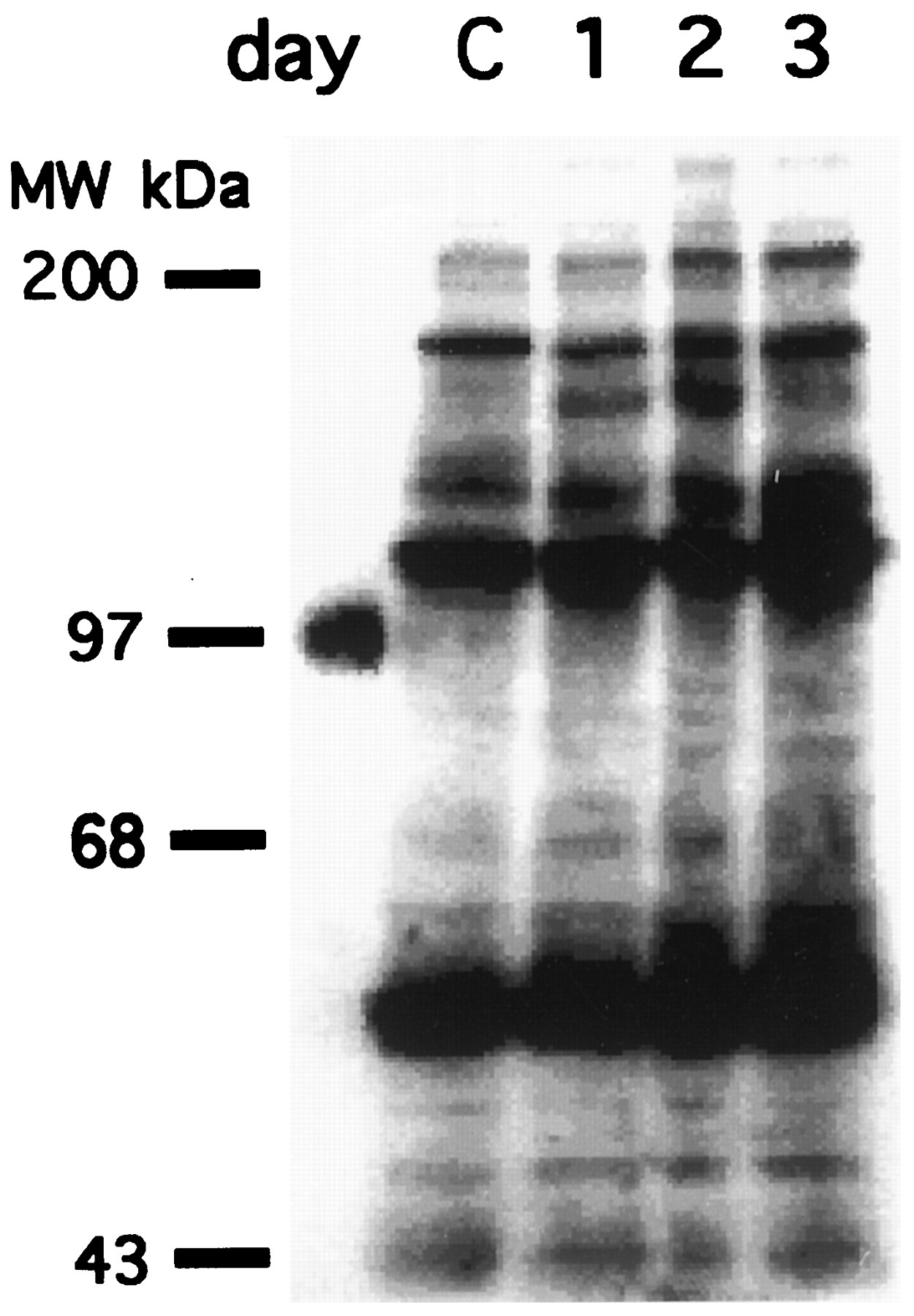
To screen for genistein-mediated inhibition of cellular PTK activity, changes in the level of tyrosine phosphorylation after genistein treatment were sought. After PC3-M cells were exposed to genistein for periods ranging from 1 to 3 days, total cell lysates were probed for phosphotyrosine content by Western blot (Fig. 6A). Although there were selective increases in some band intensities, no significant decrease in phosphotyrosine content was detected.

Effect of genistein on cellular PTK activity. Exponentially growing PC3-M cells were exposed to 50 μmgenistein for either 1, 2, or 3 days; control cells (C) were not exposed to genistein. Equal amounts of total cell lysates were loaded onto each lane and were probed for phosphotyrosine by Western blot as described in Materials and Methods.
Emerging evidence suggests that up-regulation of the PTK, FAK may contribute to the neoplastic phenotype of PCa (8, 30), although another study reports that apoptosis can be induced by decreases in FAK (31). Therefore, the effect of genistein on FAK protein levels and activity was measured. PC3-M cells were exposed to genistein as in Fig. 6A and, at the indicated time points, FAK immunoprecipitation kinase assays were performed (Fig. 7A). FAK activity was decreased at 24 hr and remained low for the remainder of the 3-day study period. FAK protein levels remain constant during this period (Fig. 7A), indicating that the decrease in in vitro kinase activity is an accurate measure of in vivo kinase activity.

The effect of genistein on FAK activity. A, Adherent exponentially growing PC3-M cells were treated with genistein for 3 days as in Fig. 6. B, Detached PC3-M cells were maintained in suspension for 24 hr by mechanical agitation in the presence or absence of 1 μg/ml fibronectin (FBN) and treated with 50 μm genistein (GENI) or not for control cells. FAK immunoprecipitation-kinase assays and FAK Western blots were performed as described in Materials and Methods. Equal amounts of total cellular protein were used for each sample immunoprecipitation or loaded onto each lane.
Changes in FAK kinase activity have been associated with alterations in cell adhesion (8). The role of cell adhesion therefore was examined by treating PC3-M cells, maintained in suspension in the presence or absence of 1 μg/ml fibronectin, with genistein for 24 hr. FAK kinase activity, and protein levels, were then measured (Fig.7B). No alterations in FAK activity nor protein level was detected.
Discussion
Genistein’s ability to inhibit PCa cell growth has been confirmed (17) and expanded to include the PC3 (derived from bone) and PC3-M [metastatic variant of PC3 line (18)] PCa cell lines. Schedule dependency studies indicate that continuous exposure to drug is necessary for ongoing growth inhibition. It is unlikely that IC50 values below the low micromolar range can be attained with the current in vitro system, because there was only a 1.8- to 2.8-fold decrease in IC50 as drug exposure time increased nearly 3-fold.
It is, therefore, unlikely that genistein is directly inhibiting PCa growth in vivo. With dietary consumption, serum levels of free genistein do not exceed the low nanomolar range (12) and, therefore, are far below the low micromolar IC50 values attained in the current study. As Peterson and Barnes (17) have shown that genistein-mediated growth inhibition of PCa cells seems to be dependent on the presence of other growth factors, the meaning ofin vitro-generated IC50 values can rightly be questioned, however. It is possible that in vivo, genistein (a known PTK inhibitor) has a more pronounced effect upon growth factor-dependent proliferation (a process known to involve PTK activity).
Genistein may be acting to prevent PCa by mechanisms other than that of growth inhibition (8, 32, 33). Previous studies from this laboratory have characterized the ability of genistein to increase PCa cell adhesion to solid matrix supports at low micromolar levels (8). Ongoing studies are detecting changes in the low nanomolar range.1 With nanomolar levels of free genistein being associated with a low incidence of PCa (10, 11, 13), increased cell adhesion (which would counteract cellular detachment, the first step in metastasis formation) is implicated as a relevantin vivo mechanism.
Genistein is identified as a potential therapeutic agent that inhibits the growth of PCa cells by inducing apoptosis, without affecting cell-cycle transition. PCa cells reside primarily in the resting phase of the cell cycle (3). Therefore, agents that can induce apoptosis without obligatory effects on the cell cycle represent a potentially important class of therapeutic agents for PCa. Pharmacokinetic studies in rodents indicate that micromolar levels of genistein can be attained through the intravenous route (34), suggesting that in vivoefficacy studies should be conducted with high doses of genistein (greater than those taken in through the diet).
Genistein’s specific effects on FAK activity also may be particularly relevant to PCa. Up-regulation of FAK expression has been associated with PCa cell oncogenesis (8, 30) and induced decreases in FAK have been associated with induction of apoptosis (31). Therefore, FAK seems to be emerging as an important pharmacologic target site, at least for PCa. Although FAK is a broad-spectrum PTK inhibitor in vitro, no global decreases in in vivo PTK activity were detected. The decrease in FAK activity by genistein therefore represents a relatively specific effect of genistein; this effect seems to be indirect, however, as genistein does not inhibit FAK kinase activity in vitro (8).
Growth inhibition by genistein was shown to occur in detached cells. However, the lack of cell growth in untreated control detached cells limits the conclusions that can be drawn from these findings. The fact that the growth of epithelial cells in a variety of systems is affected severely when they are forced to exist in a detached state is well recognized (28). The failure of genistein to alter FAK activity in detached cells, even in the presence of the ligand for β-1-integrin (i.e., fibronectin), serves to highlight the difference between the detached and adherent cell systems.
Although a clear mechanistic independence between cell adhesion and cell growth could not be distinguished within the confines of the current study, some findings do suggest that this is indeed the case. The inability of alterations in solid matrix support to affect genistein-mediated decreases in cell growth are consistent with this notion, as is the large difference in genistein concentration associated with the two different biological effects: cell-adhesion-related changes are observed in the low nanomolar range, with growth inhibition only observed at genistein concentrations exceeding 10 μm. This later phenomenon highlights a characteristic of signal modulatory agents wherein differential effects may be observed with a single agent. A differential effect of genistein on PCa cells, as compared with colon cancer cells, also may explain why cell cycle effects were not detected in the former cell type but were detected in the latter type (29).
Growth inhibition also was shown to be independent of estrogenic effects. This is significant in that genistein is a phytoestrogen.In vivo estrogenic effects in humans cannot be ruled out by these studies, however. Although high doses of genistein (i.e., from cattle feed) can lead to a clinical hyperestrogenic state in other mammals (35), no such effects have been identified in humans who subsist on a soy-based diet.
The study of apoptosis in PCa is an emerging area of study and one with important therapeutic implications. By demonstrating that genistein induces apoptosis in PCa cells through an apparently novel mechanism, genistein has been identified as a potentially important anticancer agent, especially in regard to PCa, and as an investigative tool with which to study the control of apoptosis in PCa.
Footnotes
- Received June 6, 1996.
- Accepted October 10, 1996.
-
Send reprint requests to: Dr. Raymond Bergan, NIH/NCI, Building 10, Room 12N226, 9000 Rockville Pike, Bethesda, MD 20892.
-
↵1 E. Kyle and R. Bergan, unpublished observations.
Abbreviations
- PCa
- prostate cancer
- PTK
- protein-tyrosine kinase
- PBS
- phosphate buffered saline
- FBS
- fetal bovine serum
- MTT
- dimethylthiazol-diphenyltetrazolium bromide
- FAK
- focal adhesion kinase
- DMSO
- dimethylsulfoxide
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}