


Abstract
The three α2-adrenergic receptor subtypes (α2a, α2b, and α2c) are highly homologous G protein-coupled receptors. These receptors all couple to pertussis toxin-sensitive G proteins and have relatively similar pharmacological properties. To further explore functional differences between these receptors, we used immunocytochemical techniques to compare the ability of the three α2-receptor subtypes to undergo agonist-mediated internalization. The α2a-receptor does not internalize after agonist treatment. In contrast, we observed that the α2b-receptor is able to undergo agonist-induced internalization and seems to follow the same endosomal pathway used by the β2-adrenergic receptor. Attempts to examine internalization of the α2c-receptor were complicated by the fact that the majority of the α2c-receptor resides in the endoplasmic reticulum and cis/medial Golgi and there is relatively little cell surface localization. Nevertheless, we were able to detect some internalization of the α2c-receptor after prolonged agonist treatment. However, we observed no significant movement of α2c-receptor from the intracellular pool to the plasma membrane during a 4-hr treatment of cells with cycloheximide, suggesting that these cells are unable to process α2c-receptors in the same way they process the α2a or α2b subtypes.
The α2-AR family consists of three highly homologous subtypes: α2a, α2b, and α2c (1, 2). They belong to the superfamily of G protein-coupled receptors and mediate the physiological actions of the endogenous catecholamines, epinephrine and norepinephrine. These receptors are targets for therapeutic agents for hypertension, anesthesia, and analgesia. Although the three α2-ARs are highly homologous (50–60% identity), several differences in their coupling to G proteins α subunits and their desensitization have been reported. In reconstitution experiments. Kurose et al. (3) have shown that the human α2a- and α2c-ARs couple to the same G proteins, with only slight differences in efficiency. In contrast, using transfected cell lines, others (4) have shown that the α2c subtype (the rat RG10 receptor) preferentially couples to Go, whereas α2a- and α2b-ARs couple to Gi (5). Initial studies indicated that α2-ARs transduce their signal through a pertussis toxin-sensitive G protein (Gi or Go); however, several more recent studies have demonstrated that all three α2-AR subtypes can stimulate adenylyl cyclase in pertussis toxin-treated cells (6). The EC50 value of agonists for stimulation of adenylyl cyclase by α2-ARs, presumably through Gs, is higher than the EC50value for inhibition of adenylyl cyclase through Gi (6). The three α2-AR subtypes seem to differ in their efficiency of coupling to Gs (6). Moreover, the capacity and efficacy of Gs coupling by the three α2-ARs are dependent on the agonist (7) and the cell line used to express the α2-AR subtype (6, 8). Similarly, others have shown that depending on the cell line studied, α2-ARs can inhibit or increase cellular cAMP levels, suggesting that these differences are due to coupling to different G proteins or isoforms of adenylate cyclase (9). Cotecchia et al. (10) demonstrated that α2a- and α2c-ARs can stimulate phospholipase C activity. The α2a-AR has also been shown to inhibit voltage-dependent calcium currents and increase inwardly rectifying potassium currents (11); however, the ability of α2b and α2cto couple to these ion channels has not been reported. Finally, differences in receptor regulation have been described for the three α2-ARs. Agonist-promoted desensitization has been observed for all α2-AR subtypes; however, the extent of desensitization is greater for the α2a subtype (12, 13). It has also been shown that phosphorylation of the α2a-AR is required for desensitization (14, 15).
In addition to these functional differences in signal transduction and regulation, recent studies have identified differences in the intracellular trafficking of this receptor family. The α2a- and α2b-ARs have been observed to reside primarily in the plasma membrane, whereas a large portion of the α2c-AR is found in an intracellular compartment (16, 17). We previously reported that there is no detectable agonist-induced internalization of the α2a-AR in cells in which rapid agonist-induced internalization of the β2-AR is seen (16). Previously, agonist-induced internalization of the α2b and α2c-AR subtypes have not been thoroughly characterized using immunocytochemical techniques; however, these receptors are structurally and functionally more similar to the α2a-AR than to the β2-AR and might be predicted to have similar trafficking properties. In this study, we examined the steady state distribution and agonist-induced subcellular sorting of each of the three α2-AR subtypes in the same cell line. We observed that each subtype has distinctive trafficking properties. The α2a- and α2b-ARs are located on the plasma membrane at steady state, whereas most of the α2c-AR is found in the endoplasmic reticulum. The α2b- and α2c-AR subtypes are internalized into endosomes after exposure to agonists, whereas the α2a subtype is not.
Experimental Procedures
Materials.
[3H]Atipamezole (50 Ci/mmol) and dexmedetomidine were kindly provided by Orion-Farmos (Turku, Finland). All other ligands and chemicals were purchased from Sigma Chemical (St. Louis, MO) unless indicated.
Plasmid construction and receptor expression in tissue culture cells.
All cells were grown in DMEM (University of California, San Francisco Cell Culture Facility) supplemented with 10% fetal bovine serum (Gemini Bio-Products, Calabasas, CA) and 25 μg/ml gentamicin (Boehringer-Mannheim Biochemicals, Indianapolis, IN). The three mouse α2-ARs were cloned into pBC 12BI, pREP4, or pcDNA3 expression vectors (InVitrogen, San Diego, CA) and epitope-tagged as previously described (16). Briefly, the 12CA5 epitope (sequence MGYPYDVPDYA) was inserted at the amino terminus of the three mouse α2-ARs, using oligonucleotide linker-adapters into theNcoI site located at the 5′-end of the receptor coding sequence. The sequence of the recombinant α2-ARs was confirmed by dideoxy sequencing. To monitor the trafficking pathway of α2-ARs between intracellular organelles and the cell surface, a double epitope tag with a thrombin cleavage site between the two epitopes was added to the amino termini of the α2a- and α2c-ARs (see Fig. 6). For this purpose, part of the extracellular amino-terminal sequence of a flag epitope-tagged thrombin receptor (18, 19) was combined with the coding sequences of the murine α2a- and α2c-ARs. The thrombin epitope tag consists of the following domains: prolactin signal sequence, flag epitope (italic), amino acids 53–64 of the human thrombin receptor containing a thrombin cleavage site (dash) and the hirudin domain epitope (bold) (20) (amino acid sequence: MDSKGSSLLCQGVVSDYKDDDDVDATLDPR–SFLLRNPNDKYEPFWEDEEKNESGLTEYRLVSINDS). The sequence of the recombinant α2-ARs was confirmed by dideoxy sequencing and subcloned into the pREP4 plasmid for expression in eukaryotic cells. The thrombin epitope tag can be detected by immunostaining using the monoclonal M1 antibody (Kodak IBI, Rochester, NY) recognizing the flag epitope and a polyclonal antiserum that was raised against a peptide containing the hirudin epitope domain of the human thrombin receptor (19). Epitope-tagged receptors were transfected by either DEAE-Dextran, calcium-phosphate precipitation, or electroporation methods. Stable cell lines were selected under 250–500 μg/ml G418 (GIBCO, Grand Island, NY) or 200 IU/ml hygromycin (Boehringer-Mannheim) in the media.

Amino-terminal thrombin/M1 Flag epitope-tagged α2c-AR. A recombinant α2c-AR was constructed with an additional amino-terminal sequence containing the M1 Flag epitope (first box), preceding a thrombin cleavage site (arrow) and the hirudin epitope (second box). When this epitope-tagged receptor is present in the plasma membrane of intact cells, thrombin treatment cleaves off the flag epitope, resulting in a loss of staining for flag, but retains staining for hirudin. Therefore, this construct allowed the differentiation between intracellular α2c-AR, which had not yet been delivered to the plasma membrane (stains positive for both epitopes), and receptor presented to the plasma membrane, which had its M1 flag epitope irreversibly cleaved by thrombin (stains positive for hirudin only). This thrombin epitope-tagged receptor binds the antagonist atipamezole and agonists epinephrine and norepinephrine similar to wild-type α2c-AR (Table 1) and shows subcellular distribution typical of the wild-type α2c-AR (see Fig. 7, E and F).
Radioligand binding assay.
For binding studies, COS-7 cells were transiently transfected using the DEAE-Dextran method, and cell membranes were harvested 3 days after transfection as previously described (21). Binding experiments were performed in 500-μl volumes of binding buffer (75 mm Tris, 12.5 mmMgCl2, and 1 mm EDTA, pH 7.4) for 90 min at room temperature. The bound radioactivity was separated from free by filtration through GF/C filters and washed three times with 5 ml of ice-cold binding buffer using a Brandel cell harvester. Saturation isotherms were performed by incubating the membranes with varying concentrations of [3H]atipamezole. Nonspecific binding was determined by the addition of 100 μm yohimbine. Competition binding experiments were carried out by incubating membranes with varying concentrations of competing ligand and 2 nm [3H]atipamezole. Nonspecific binding was determined by the addition of 100 μm yohimbine. Equilibrium dissociation constants were determined from saturation isotherms and competition curves using GraphPAD software (San Diego, CA). All binding experiments were done in duplicate and repeated at least three times.
Production of antisera.
The antigenic epitopes selected for the polyclonal antibodies were the carboxyl termini of each α2-AR. The antigenic peptides were synthesized and MBS-coupled to thyroglobulin, purified, and injected into New Zealand White rabbits. Polyclonal sera directed against the α2band α2c epitopes were affinity-purified over peptide coupled to epoxy-activated Sepharose columns and eluted with either 3m KSCN, pH 7.4, or 20 mm glycine, pH 3.0. The three polyclonal antisera are specific for their respective α2-AR subtype and do not cross-react with other α2-AR subtypes at the dilutions used (α2a, 1:500; α2b and α2c affinity-purified antisera, 1:100; data not shown). The previously described β2 antibody was kindly provided by Dr. M. Von Zastrow (University of California, San Francisco).
Immunocytochemical studies and techniques.
Receptor trafficking and subcellular distribution were examined using indirect immunocytochemical staining. Nonspecific binding was blocked with blotto (5% nonfat dry milk in 50 mm Tris, pH 7.6) for 45 min. For staining in permeabilized cells, nonionic detergent (Nonidet P-40, Sigma) was added to blotto incubations to a final concentration of 0.2%. Cells were stained using 12CA5 hemagglutinin antibody (Berkeley Antibody Co., Berkeley, CA) monoclonal antibody (at 1:500 in blotto). For nonpermeabilized studies, receptors present on the plasma membrane were stained by incubation of live cells grown on coverslips at 37° with 6% CO2 for 60 min with 12CA5 monoclonal antibody at 1:500 in DMEM supplemented with 10% fetal calf serum. After a 60-min incubation, cells were washed with cold PBS and fixed with 4% formaldehyde for 30 min and then rinsed three times in room-temperature PBS. Nonpermeabilized immunocytochemistry was also performed by growing cells on coverslips for 2 or 3 days and then fixing them for 30 min in 4% formaldehyde and deleting detergent from the blotto. Secondary antibodies were either fluorescein-conjugated (fluorescein isothiocyanate) or Texas red-conjugated directed against the species of primary antibody used (Jackson ImmunoResearch, West Grove, PA).
One day after transient transfections, cells were trypsinized off tissue culture dishes and replated onto sterilized cover slips. For steady state studies, cells were grown on coverslips for 2 days and then fixed in 4% formaldehyde or 4% paraformaldehyde for 45 min, followed by three washes with room-temperature PBS. For adrenergic agonist treatment studies, after 2 days’ growth on coverslips, cells were incubated for 30 min at 37° with 6% CO2 in either serum-free DMEM (control) or agonist in serum-free DMEM. After incubations, the medium was aspirated, and cells were fixed with 4% formaldehyde for 45 min at room temperature and then washed three times with room-temperature PBS. Studies involving basal trafficking of α2c thrombin epitope-tagged receptors were carried out in the absence of agonist after a 30-min treatment with 5 nmthrombin to cleave off the M1 epitope from receptors present on the plasma membrane. The effect of agonist stimulation on α2cthrombin epitope-tagged receptor trafficking was examined by exposing cells to 10 μm epinephrine in DMEM with 40 mmHEPES, pH 7.4, and 10 μm cycloheximide to inhibitde novo protein synthesis for 30 min at 37°. Steady state trafficking of the cytosolic α2c thrombin epitope-tagged receptor pool was also studied in the absence of de novoprotein synthesis by treated cells with 10 μmcycloheximide for 4 hr. During inhibition of protein synthesis, cells were concomitantly treated with 5 nm thrombin to remove the M1 epitope from all receptors that were translocated to the plasma membrane.
For studies using thrombin epitope-tagged α2-ARs, after 2 days of growth on coverslips, cells were washed three times with PBS and incubated at 37° with 6% CO2 in DMEM with 40 mm HEPES, pH 7.4, for 30–45 min. After various treatments as outlined above, cells were rinsed three time with PBS and fixed using cold methanol (−20°) for 5 min. After fixation, cells were washed three times with PBS supplemented with Ca2+/Mg2+ over a 5-min period. To block nonspecific binding, 5% nonfat dry milk in PBS with Ca2+/Mg2+ supplemented with 50 mmHEPES, pH 7.4, was applied to cells for a period of 30–45 min. Selective dual antibody labeling of cells was performed in the blocking agent applied at room temperature for 1 hr with either the monoclonal M1 antibody (Kodak-IBI) at 1:500 or a polyclonal antibody to the hirudin epitope (Dr. S. Coughlin, University of California, San Francisco, CA) at 1:1000.
Colocalization immunocytochemical studies were performed on either the wild-type α2c or 12CA5 epitope-tagged α2c-AR and a variety of antibodies selective to various intracellular compartments. Cells were costained with either the 12CA5 monoclonal antibody or affinity-purified polyclonal antibody directed against the wild-type α2c-AR as described above and one of the following: lgp120 (Dr. S. Green, University of Virginia, Charlottesville, VA) at 1:1000; cation-independent M6PR (Dr. William Brown, Cornell University, Ithaca, NY) antibody at 1:500; mannosidase II (Berkeley Antibody, Berkeley, CA) at 1:500; calnexin (StressGen Biotechnologies, Victoria, BC, Canada) at 1:750; or BiP (GRP-78; Affinity Bioreagents, Nashanic Station, NJ) at 1:500. After primary antibody labeling, the cells were rinsed three times with PBS supplemented with Ca2+/Mg2+. Blocking agent was again applied for 30 min, and the aforementioned conjugated secondary antibodies were applied in blocking agent for 1 hr at room temperature. Cell were subsequently rinsed three times with PBS supplemented with Ca2+/Mg2+, and the specimens were mounted on glass slides for viewing.
Conventional immunofluorescence microscopy was performed using a Zeiss Axiophot microscope with a Zeiss Neofluar 63×/1.25 or Zeiss Neofluar 100×/1.3 objective. Confocal microscopy was performed using a Zeiss Axioskop microscope and a Sarastro Phoibos 1000 instrument with either a Zeiss Apochromat 40×/1.0 or Zeiss Apochromat 63×/NA 1.4 objective, a 50-μm confocal pinhole, and optical filters set as described in the manufacturer’s instructions for fluorescein isothiocyanate/Texas red imaging.
ELISA.
For quantification of receptor internalization, ELISA assays were performed. Briefly, nonclonal stable HEK 293 cell lines were plated onto 24-well tissue culture dishes. To improve adhesion of this cell line to plastic, the wells were treated for 30 min with 0.0005% poly-l-lysine in PBS immediately before plating out ∼10 × 104 cells/well. At ∼48 hr later, when cells were ∼75% confluent, they were incubated for 30 min at 37° with 6% CO2 in serum-free DMEM or 10 μmnorepinephrine in serum-free DMEM. After incubations, the medium was aspirated, and cells were fixed with 4% paraformaldehyde for 5 min at room temperature. Wells were washed three times with room-temperature PBS. Nonspecific binding was blocked with 1% BSA for 45 min with occasional gentle shaking. The 12CA5 antibody was diluted to 1:1000 in 1% BSA for 60 min, followed by three gentle PBS washes. Cells were briefly reblocked with 1% BSA for 15 min and then incubated with anti-mouse conjugated alkaline phosphatase (BioRad, Richmond, CA) diluted 1:1000 in 1% BSA for 60 min with occasional gentle shaking. Wells were then washed three times with PBS, and a colorimetric alkaline phosphate substrate was added. Plates were continuously but gently shaken (40 rotations/min) until an adequate color change had occurred, at which time a 100-μl sample was taken for colorimetric readings. Nontransfected cells were studied concurrently to determine background. All experiments were done in triplicate.
Results
Immunocytochemical techniques were used to examine the distribution of α2-AR subtypes expressed in COS 7, HEK 293, MDCK, NRK and Rat1 fibroblast cell lines. Subtype-specific rabbit polyclonal antibodies were prepared against a carboxyl-terminal peptide as described in Experimental Procedures. The specificity of these antibodies was verified by staining untransfected COS 7 cells or COS 7 cells transfected with each of the α2-AR subtypes (data not shown). In addition, we used receptors modified with an amino-terminal 12CA5 epitope, which is recognized by a commercially available monoclonal antibody. The amino-terminal tag permits examination of cell surface receptor density in nonpermeabilized cells. We previously observed that the use of this epitope does not alter the trafficking of the β2-AR (16). Moreover, the distribution and trafficking of epitope tagged α2-ARs (examined with either 12CA5 monoclonal antibody or polyclonal subtype-specific antibody) are indistinguishable from those of nontagged receptors (examined with polyclonal antibody; data not shown). Binding affinity for the antagonist atipamezole, the endogenous catecholamines epinephrine and norepinephrine, and the specific α2agonist dexmedetomidine are also comparable between wild-type and epitope-tagged α2-ARs (Table 1).
Equilibrium dissociation constants of various adrenergic ligands for wild-type and amino-terminal 12CA5 or thrombin epitope-tagged mouse α2-adrenergic receptors expressed in COS-7 cells
Fig. 1 shows the distribution of the three α2-AR subtypes in HEK 293 cells at steady state and after agonist treatment. It can be seen that α2a and α2b subtypes are predominantly localized in the plasma membrane at steady state, whereas a large portion of α2c-AR is found in an intracellular compartment. After agonist treatment (10 μm norepinephrine) for 30 min at 37°, the α2b-AR is internalized, but the α2a subtype remains in the plasma membrane. This analysis does not allow us to determine whether the α2c-AR is internalized because of the large background of α2-AR already in an intracellular membrane compartment. This intracellular pool of α2c-AR is be further characterized below.

Distribution of the three α2-AR subtypes in permeabilized HEK 293 cells at steady state and after agonist treatment. Agonist treatment, cell fixation, permeabilization, and staining are described in Experimental Procedures. A, At steady state, the α2a-AR was primarily localized to the plasma membrane. B, After a 30-min incubation with 10 μmnorepinephrine, the α2a-AR did not redistribute. C, Similar to the α2a, at steady state the α2b-AR was primarily localized in the plasma membrane. D, However, 10 μm norepinephrine treatment caused the α2b-AR to internalize into intracellular vesicles. E, In contrast to α2a- and α2b-AR sorting at steady state, the α2c-AR was primarily localized in an intracellular compartment; the α2c-AR appears as punctate intracellular staining. In nonpermeabilized cells at steady state, there is a relatively small amount of the α2c-AR present in the plasma membrane (data not shown). F, A 30-min incubation with 10 μm norepinephrine did not cause notable redistribution of the α2c-AR.
To further investigate differences in agonist-mediated internalization, 12CA5 epitope-tagged α2b-AR was coexpressed with a nontagged α2a-AR, allowing us to examine differences in trafficking between these two subtypes in the same cell. The α2a distribution was monitored with the polyclonal α2a subtype-specific antibody, and the epitope-tagged α2b-AR distribution was monitored with a monoclonal antibody to the 12CA5 epitope. At steady state, both receptors are found in the plasma membrane of the same cell (Fig. 2, A and B). After exposure to 10 μm norepinephrine for 30 min, only the α2b-AR is internalized (Fig. 2, C and D). Similar treatment with 10 nm of the nonselective α2 agonist dexmedetomidine also selectively internalized the α2b-AR in cotransfected cells (Fig. 2, E and F). Additional agonist treatment paradigms with the α2a-AR (30-min incubations with 10 μm epinephrine or 100 μm norepinephrine or ≤4-hr incubation with 10 μm norepinephrine) resulted in similar findings (data not shown).

Receptor distribution after agonist treatment in HEK 293 cells coexpressing α2a- and α2b-ARs. The 12CA5 epitope-tagged α2b-AR was transiently transfected into a nonclonal HEK 293 cell line stably expressing the α2a-AR. After treatment, cells were fixed and permeabilized as described in Experimental Procedures. At steady state, receptor was localized to the plasma membrane in cells expressing both (A) α2a- and (B) α2b-AR subtypes. C, After a 30-min incubation with 10 μmnorepinephrine, the α2a-AR did not redistribute. D, In contrast, agonist treatment caused the α2b-AR coexpressed in the same cell to internalize into endosomes. Similar treatment with a 10 nm concentration of the nonselective α2agonist dexmedetomidine also selectively internalized the α2b-AR (F), whereas no α2a-AR internalization could be detected in the same cells (E).
The α2b- and β2-ARs internalize by a similar mechanism.
The mechanism of agonist-mediated internalization of the β2-AR has been well characterized. We therefore coexpressed the epitope-tagged α2b-AR along with the β2-AR and examined the distribution of the two receptors after agonist treatment. At steady state, both receptors are on the plasma membrane (Fig. 3, A and B). Only the β2-AR is internalized in cells exposed to the β-AR agonist isoproterenol (Fig. 3, C and D), and only the α2b-AR is internalized in cells exposed to the α2-AR agonist dexmedetomidine (Fig. 3, E and F). Using confocal microscopy, we demonstrated that both receptors are internalized by exposure to the common agonist norepinephrine and seem to reside in the same endocytic vesicles (Fig. 3, G and H).
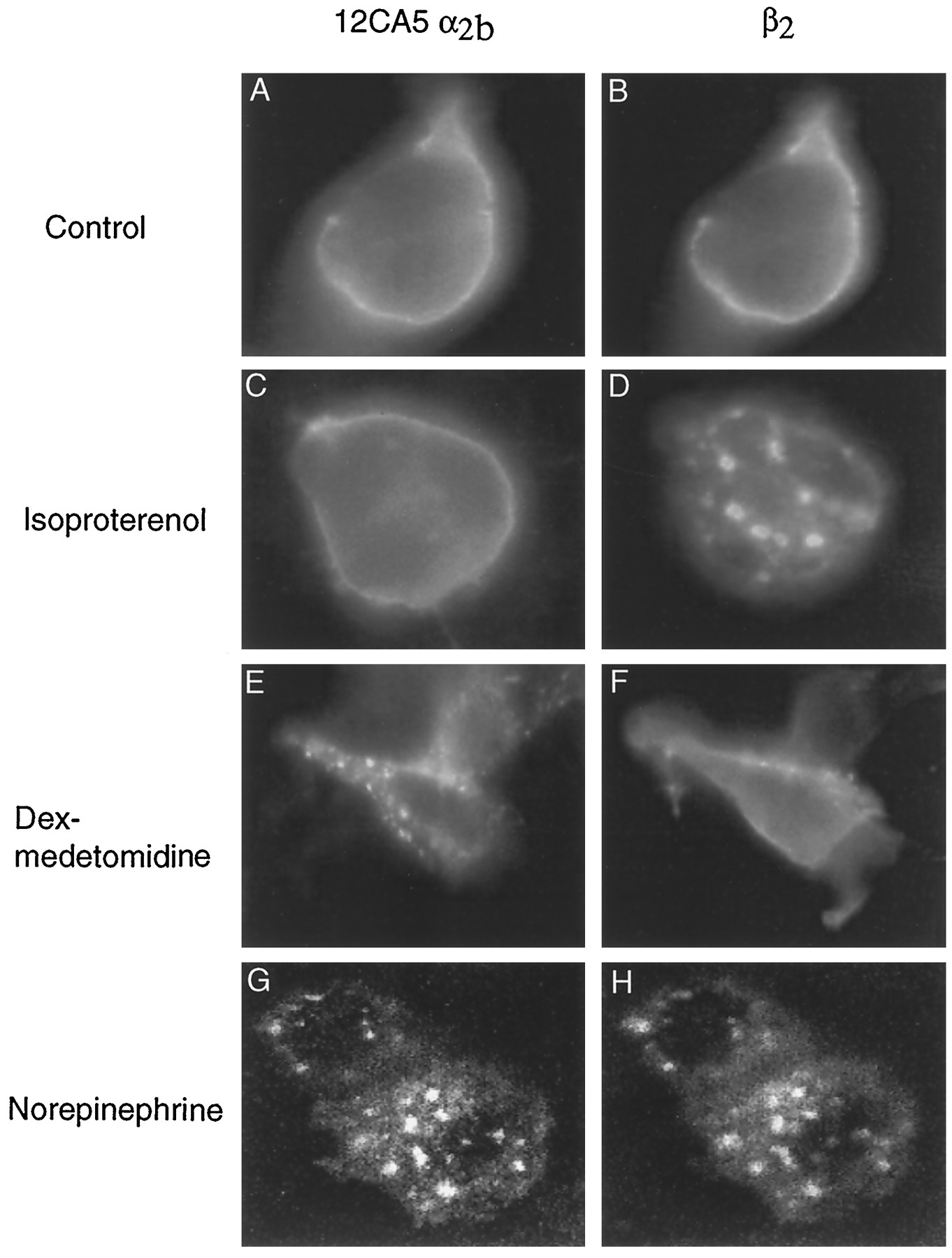
Effect of subtype-selective and -nonselective agonist treatment of HEK 293 cells coexpressing α2b- and β2-ARs. The 12CA5 epitope-tagged mouse α2b-AR was transiently cotransfected with the human β2-AR in HEK 293 cells. At ∼24 hr after transient transfection, cells were plated onto glass coverslips and grown for 2 days. For treatments, coverslips were incubated for 30 min in serum-free DMEM (control) or with the appropriate agonist in serum-free DMEM at 37° and 6% CO2. After fixation, cells were permeabilized before staining. A and B, At steady state, both receptors are localized in the plasma membrane. In cells coexpressing both ARs, 10 μm isoproterenol, a β-selective agonist, induced internalization of β2-ARs (D), whereas α2b-ARs did not redistribute (C). Similarly, 10 nm dexmedetomidine, an α2-selective agonist, promoted internalization of α2b-ARs (E), whereas β2-ARs did not redistribute (F). When cells coexpressing both α2b- and β2-ARs were treated with 10 μm norepinephrine, a common agonist for α2- and β2-ARs, both receptors internalized into the same endocytic vesicles as observed by confocal microscopy (G and H, respectively).
The results of these immunocytochemical studies suggest that there is no internalization of the α2a-AR after agonist treatment; however, it may not be possible to detect internalized receptor by fluorescence microscopy if it is not concentrated in endosomes. To investigate this possibility, we used an ELISA to monitor the loss of cell surface receptor rather than accumulation of intracellular receptor protein after agonist activation. Epitope-tagged α2a-and α2b-ARs were expressed in HEK 293 cells, and the density of receptors was monitored in nonpermeabilized cells with the 12CA5 antibody. At steady state, the cell surface expression of the α2a- and α2b-ARs was comparable. After treatment with norepinephrine for 30 min, there was a loss of 31 ± 2.4% (mean ± standard error) of α2b-ARs and a loss of 7 ± 1.4% of α2a-ARs. These results confirm the difference between α2a- and α2b-ARs; however, they suggest that there is a small but significant agonist-induced internalization of the α2a-AR, possibly by a nonendosomal mechanism.
The α2c-AR is found in the endoplasmic reticulum in Rat1 fibroblast cells.
Although there is some plasma staining, the α2c-AR seems to reside primarily in an intracellular compartment in HEK 293 cells (Fig. 1C). To further study this, we expressed the α2a- and α2c-ARs in a variety of cell lines. Similar to studies reported by Wozniak et al., (17) when expressed in a MDCK II cell line, the abundance of α2c-AR was localized in an intracellular compartment, whereas the α2a-AR was targeted to the basolateral plasma membrane (Fig. 4, A and B). When the 12CA5 epitope-tagged α2c-AR was transiently cotransfected in an HEK-293 cell line stably expressing wild-type α2a-AR, the α2c subtype was also localized to an intracellular compartment (Fig. 4, C and D). Coexpression of the α2cdid not alter the trafficking of the α2a-AR subtype. We attempted to quantify the amount of 12CA5 epitope-tagged α2c-AR in the plasma membrane using the ELISA method discussed above. In contrast to the α2a- and α2b-AR subtypes, the signal generated from α2c-AR transfected cells was not significantly different from that of untransfected cells. Thus, although we were able to detect a small amount of 12CA5 epitope-tagged α2c-AR in the plasma membrane of nonpermeabilized cells by the sensitive technique of immunofluorescence microscopy, the amount was too small to quantify by ELISA.

Distinct subcellular localization of α2a- and α2c-ARs at steady state. Immunolocalization of 12CA5 epitope-tagged (A) α2a- and (B) α2c-ARs in permeabilized MDCK cells is shown. A, At steady state in stably transfected MDCK cells, the α2a-AR was localized to the basolateral plasma membrane. B, The α2c-AR subtype was predominantly localized in an intracellular compartment when stably transfected in MDCK cells. Consistent with our findings of subcellular distribution in other cell lines, a relatively small amount of this receptor is localized in the basolateral plasma membrane (arrows). The 12CA5 epitope-tagged α2c-AR was also transiently cotransfected in HEK 293 cells stably expressing the wild-type α2a-AR. C, Field of permeabilized cells viewed by confocal microscopy at 40× magnification. D, Confocal image at higher magnification (63×). At steady state, the α2a-AR is predominantly sorted to the plasma membrane (stained red). Although some α2c-AR is localized in the plasma membrane (seen asyellow from overlap of red andgreen), the abundance of this receptor is localized in an intracellular compartment (stained green).
The identity of the intracellular compartment containing the α2c-ARs was difficult to assess in HEK 293 cells because of their relatively small size. To further investigate the subcellular distribution of α2c-ARs, we studied Rat1 fibroblast and NRK cells, which have relatively large cytoplasms. The distribution of the receptor in Rat1 cells colocalizes with both BiP and mannosidase II (Fig. 5, A–D), suggesting that the α2c-AR is found primarily in the endoplasmic reticulum and cis/medial Golgi. No overlap was observed with markers for trans-Golgi network (M6PR), endosomes (M6PR), or lysosomes (lgp120) (Fig. 5, E–H). We observed a similar distribution of the α2c-AR in NRK cells (data not shown).
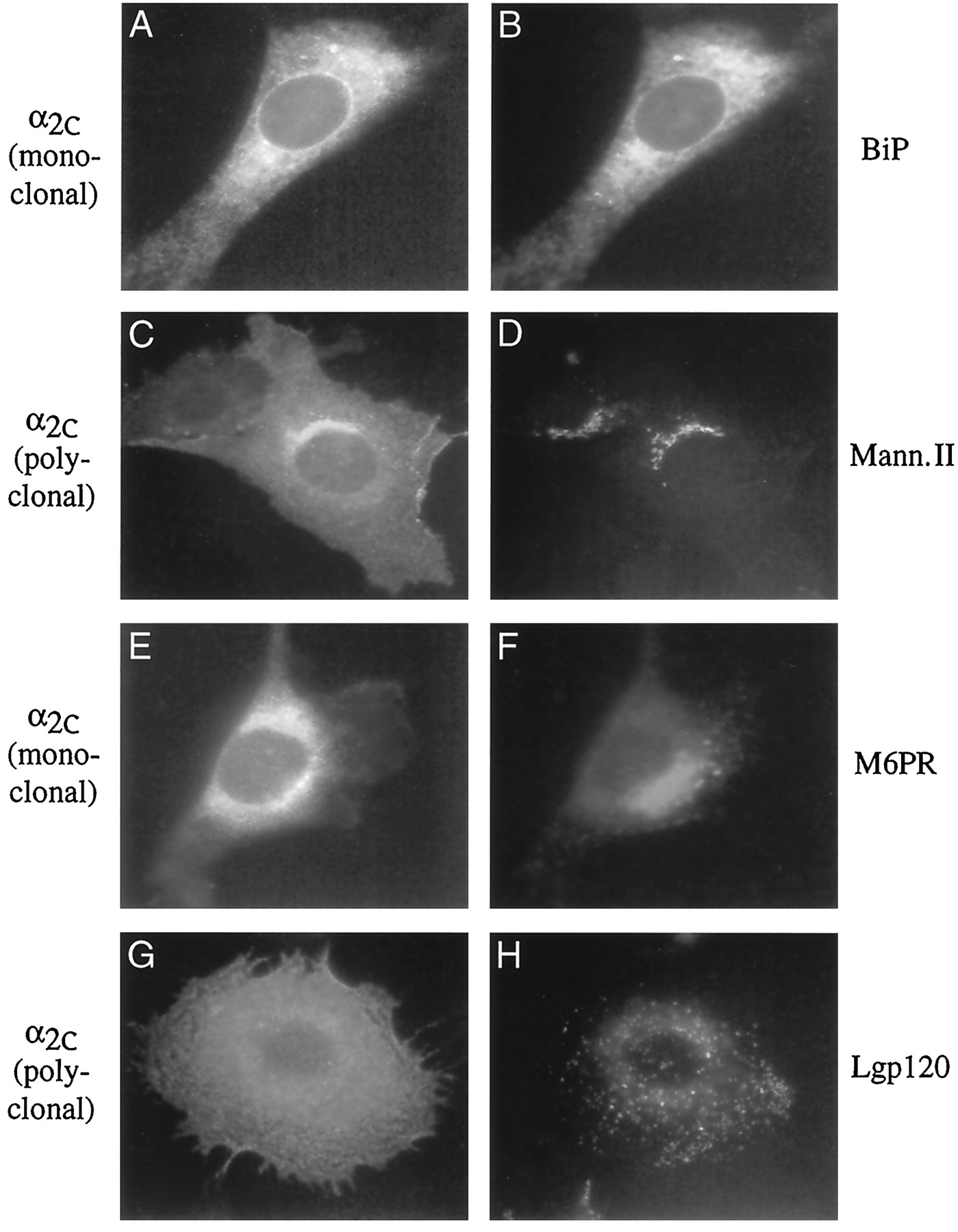
Immunocytochemical identification of the subcellular compartment containing α2c-ARs. Colocalization studies using antibodies that specifically stain various subcellular compartments and antibodies specific for the wild-type (polyclonal) or 12CA5 (monoclonal) epitope-tagged α2c-AR have allowed identification of the distribution of this receptor in permeabilized Rat1 fibroblast cells. A, Cell is stained for the 12CA5 epitope-tagged α2c-AR. B, Same cell is costained for BiP, which specifically labels the endoplasmic reticulum. There is considerable overlap between these two antibodies, indicating that a significant proportion of the α2c-AR is localized to this organelle. Similarly, the wild-type α2c-AR (C) colocalizes with mannosidase II (Mann. II), a marker for cis/medial Golgi (D). Conversely, the 12CA5 epitope-tagged α2c-AR (E) does not overlap with M6PR, which labels the trans-Golgi network and endosomes (F). There also is no significant colocalization between the wild-type α2c-AR (G) and lgp120, a lysosomal marker (H).
The finding that a large proportion of cellular α2c-AR resides in the endoplasmic reticulum suggests several possibilities. This receptor may be improperly processed, or the cell may lack an important chaperone protein and therefore the receptor is retained in the endoplasmic reticulum. It is also possible that the receptor is processed slowly and undergoes a relatively rapid turnover at the plasma membrane. However, similar half-lives have been reported for all three α2-AR subtypes in MDCK cells (17). Finally, it is possible that the receptor is cycling between the plasma membrane and cytosolic pool. To examine these possibilities, we constructed a modified form of the α2c-AR that would allow us to identify receptor that has been delivered to the plasma membrane. Fig.6 shows the amino-terminal sequence of the modified α2c-AR. An M1 flag epitope and a thrombin cleavage site were added to the amino terminus. Cleavage of the receptor by thrombin present in the media results in loss of the flag epitope (which can be recognized by the commercially available M1 monoclonal antibody), whereas the hirudin binding domain of the thrombin cleavage site remains and can be recognized by a polyclonal antibody. Therefore, receptors that are on the plasma membrane are susceptible to thrombin cleavage. These thrombin-cleaved receptors will stain only with the polyclonal antibody to the hirudin epitope, not with the M1 flag antibody.
Fig. 7 demonstrates that the amino-terminal thrombin epitope does not effect the distribution of the α2c-AR. In permeabilized cells, most of the α2c-AR is found in an intracellular compartment (Fig. 7, E–H); however, cell surface expression can be demonstrated in nonpermeabilized cells (Fig. 7, A–D). Exposure of the cell to thrombin results in loss of cell surface receptor staining by the M1 flag antibody (Fig. 7, C and G); however, the receptor is still recognized by the antibody to the hirudin epitope (Fig. 7, D and H). To investigate the turnover of α2c-AR, cells were exposed to cycloheximide to block protein synthesis and incubated in the presence of epinephrine and thrombin for 4 hr (Fig.8, A–F). Movement of receptor from the intracellular pool to the cell surface would result in cleavage by thrombin and a loss of staining by M1 flag antibody. If these receptors were then internalized and recycled through the endoplasmic reticulum or distributed to lysosomes, they would be recognized by staining with the polyclonal antibody to the hirudin epitope but not the M1 flag antibody. As seen in Fig. 8, there is no significant loss of intracellular staining by the M1 antibody, indicating that the intracellular pool of receptor is stable. Moreover, using the cleavable thrombin/flag epitope, we were able to determine that a small amount of α2c-AR was internalized from the plasma membrane with agonist treatment (Fig. 8, E and F). This internalized receptor stains with the hirudin antibody but not the M1 antibody (Fig. 8, E and F,arrows).
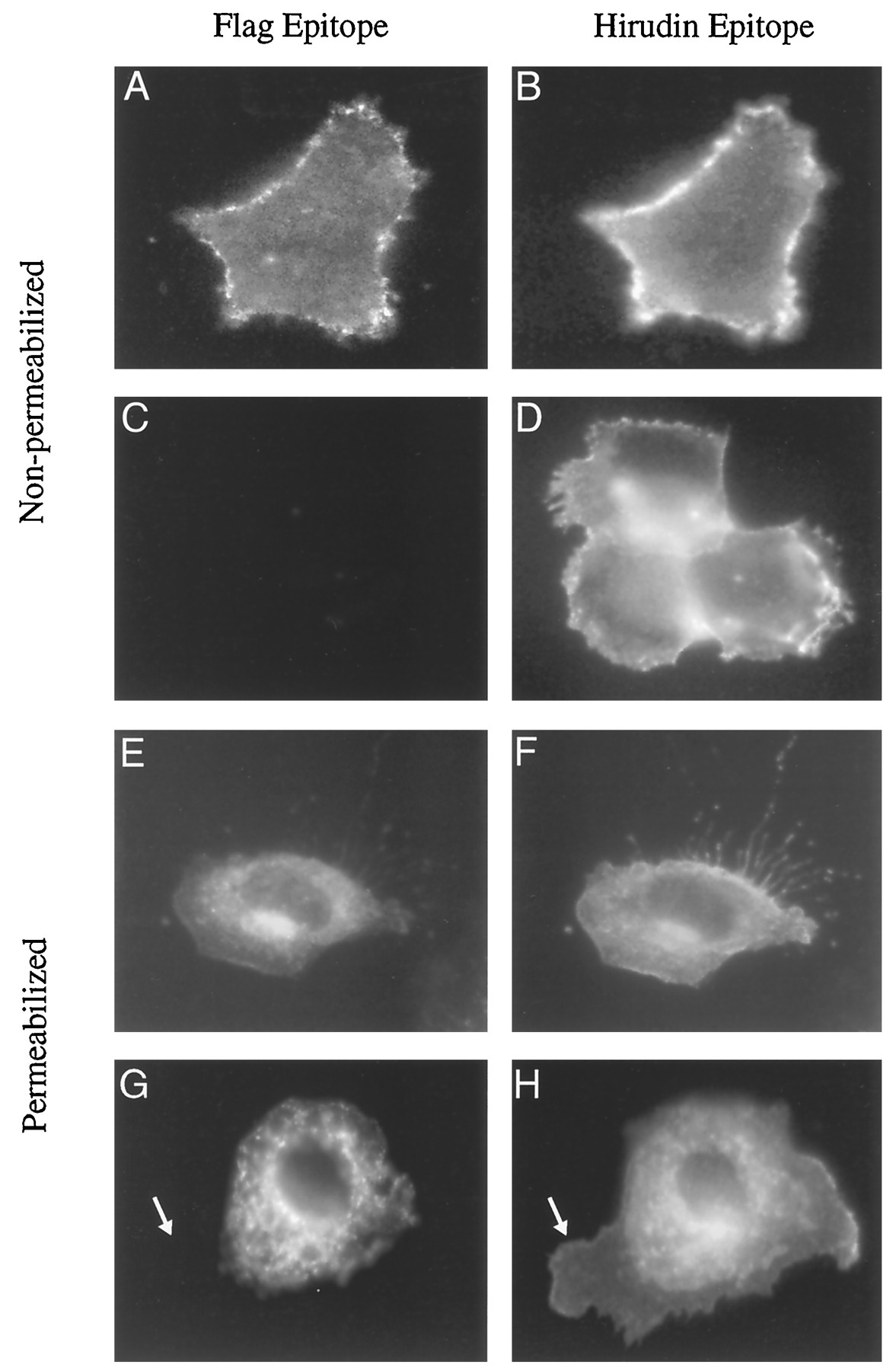
Thrombin cleavage of the amino-terminal flag epitope distinguishes intracellular α2c-AR from that delivered to the plasma membrane. For these experiments, NRK cells were studied because they are large and have a large ratio of cytoplasm to nucleus, which enabled better visualization of intracellular receptor compared with similar studies in HEK-293 cells. Intact NRK cells transfected with the thrombin/flag epitope-tagged α2c-AR were incubated in DMEM (Control) or 5 nm thrombin in DMEM for 30 min. After fixation, immunocytochemical staining was performed using both nonpermeabilizing conditions (A–D) or after cellular permeabilization with 0.2% nonionic detergent (E–H). In nonpermeabilized stained control cells, (A) flag epitope and (B) hirudin epitope colocalized to the plasma membrane. Thrombin treatment caused a complete loss of flag staining in the plasma membrane (C), whereas hirudin staining was retained (D). In control cells that were permeabilized for immunocytochemical staining after fixation, (E) flag epitope and (F) hirudin epitope were colocalized. Thrombin treatment cleaved off the flag epitope of receptor present in the plasma membrane (G, arrow), whereas the intracellular flag epitope remained intact (G). Costaining with hirudin in thrombin-treated cells identifies flag-cleaved receptor in the plasma membrane (H, arrow).

Agonist treatment does not induce redistribution of the intracellular pool of thrombin/flag epitope-tagged α2c-ARs. To investigate whether agonist treatment could induce a shift in the subcellular localization of α2c-ARs, intact NRK cells transfected with amino terminal thrombin/flag epitope-tagged α2c-ARs were studied. Incubation with 10 μm cycloheximide did not result in an observable loss of α2c-AR from the plasma membrane or shift in the intracellular receptor pool to the plasma membrane over a 4-hr treatment (A and B). Simultaneous 4-hr incubations with 10 μm cycloheximide and 5 nm thrombin showed a loss of receptor flag staining in the plasma membrane (C,arrow), whereas hirudin staining remained intact (D,arrow). There was no reduction of the intracellular pool of α2c (staining with both antibodies). Finally, the effect of agonist on the subcellular trafficking of thrombin/flag epitope-tagged α2c-AR was studied. Concurrent 4-hr incubations with 10 μm cycloheximide, 5 nmthrombin, and 10 μm epinephrine did not produce any observable shift in the intracellular pool of α2c-AR (E and F), indicating that the intracellular pool of α2c-AR is relatively stable. Moreover, the cleavable thrombin/flag epitope on the amino terminus of the α2c-AR allowed us to identify that some receptor is internalized with agonist treatment. In the presence of thrombin, agonist-promoted receptor internalization from the plasma membrane resulted in a relatively small population of intracellular vesicles that stained only for the hirudin epitope (F,arrow), not the flag epitope (E,arrow).
In a comparable experiment, the α2a-AR was similarly tagged with the same thrombin-cleavable epitope (Fig.9). At steady state, most of the receptor is susceptible to cleavage with thrombin, indicating that the majority of α2a-AR is localized in the plasma membrane (Fig. 9, C and D). A small amount of intracellular receptor (M1 flag antibody staining) is observed before but not after exposure to cycloheximide and thrombin for 4 hr (Fig. 9, E and F). This indicates that within 4 hr, all of the α2a-AR in the biosynthetic pathway has been processed and inserted in the plasma membrane.

Trafficking studies of the amino-terminal thrombin/flag epitope-tagged α2a-AR indicates that within 4-hr, all receptor has been processed and inserted into the plasma membrane. Intact NRK cells transfected with the thrombin/flag epitope-tagged α2a-AR were grown on coverslips and incubated in (A and B, Control) DMEM, (C and D) 5 nm thrombin in DMEM for 5 min, or (E and F) 5 nm thrombin, 10 μm cycloheximide, and 10 μm norepinephrine for 4 hr. After fixation, immunocytochemical staining was performed after cellular permeabilization as described in Experimental Procedures. Similar to wild-type or 12CA5 epitope-tagged α2a-ARs, at steady state, thrombin/flag epitope-tagged receptor is primarily localized to the plasma membrane (A and B). Thrombin treatment caused a complete loss of flag staining in the plasma membrane (D), whereas hirudin staining was retained (C). In cells that have been treated with thrombin, cycloheximide, and norepinephrine for 4 hr, there is a complete loss of flag staining (F), whereas hirudin staining in the plasma membrane is retained (E).
Agonist-promoted accumulation of α2c-AR in endosomes may not be as pronounced as that observed for the α2b-AR because of the large amount of α2c-AR in the intracellular compartment at steady state and the comparatively small amount of α2c-AR in the plasma membrane (Fig. 8F). A more sensitive technique for identifying agonist-induced receptor internalization is shown in Fig. 10. Living, nonpermeabilized cells expressing M1 flag-tagged α2c-AR are first labeled with M1 flag monoclonal antibody and then washed and incubated in the presence or absence of agonist for 30 min. Cells were then fixed, permeabilized, and stained with secondary antibody. This approach allowed us to selectively examine only α2c-ARs that are in the plasma membrane. No internalization was detected in control cells (Fig. 10A); however, after agonist treatment, receptor was observed in an intracellular compartment (Fig. 10C, arrow). The intracellular receptor surrounds the nucleus, creating a nuclear shadow in agonist-treated cells (Fig. 10C), whereas no nuclear shadow is observed in control cells (Fig. 10A). The internalized α2c-AR colocalized with the M6PR that is present in the trans-Golgi network and endosomes (Fig. 10D).

Immunocytochemical identification of agonist-induced redistribution of α2c-ARs into endosomes. Thrombin/flag epitope-tagged α2c-ARs were stably expressed in NRK cells and grown on coverslips as described in Experimental Procedures. Coverslips were incubated with monoclonal flag antibody at 4° for 60 min, labeling epitope-tagged α2c-ARs present on the cell surface. After gentle washing, coverslips were returned to 37° with 6% CO2 and incubated in serum-free DMEM in the presence or absence of agonist (10 μm epinephrine) for 30 min. After this 30-min incubation, cells were fixed in 4% PFA, permeabilized, and stained as described. A and C, α2c-AR distribution in control and agonist-treated cells, respectively. After fixation and permeabilization, the polyclonal antibody to M6PR was used to label the trans-Golgi network and endosomes in (B) control and (D) agonist-treated cells. C, Some α2c-AR has internalized with agonist treatment and is localized in the perinuclear region (arrow). This area overlaps with that labeled in D with the M6PR antibody (arrow).
Discussion
In this study, we report differences in steady state targeting and agonist-induced internalization of the three α2-AR subtypes. The α2b-AR behaves more like the β2-AR than the α2a-AR. This is particularly interesting in light of the high degree of identity shared by α2a and α2b (55%, α2a versus α2b; 21%, β2 versus α2b). Moreover, we extend previous observations regarding the intracellular distribution of the α2c-AR subtype. The roles of intracellular targeting and trafficking in signal transduction are not well understood; however, agonist-induced internalization has been shown to play a role in receptor regulation. Studies have implicated agonist-induced internalization in the process of resensitization. Blocking internalization by selective receptor mutations or treatment of cells with concanavalin A or sucrose prevents dephosphorylation of the receptor (22). Recent studies have suggested that binding of β-arrestin to the β2-AR after G protein-coupled receptor kinase phosphorylation is necessary for internalization (23). Of interest, the α2a-AR has been shown to undergo phosphorylation by G protein-coupled receptor kinase (14, 15). The inefficient agonist-induced internalization in the α2a-AR may suggest that this receptor does not bind to β-arrestin. These results may also suggest that the α2a-AR is tethered to the plasma membrane and is prevented from undergoing internalization even when phosphorylated and bound to β-arrestin. This may lead to a difference in the rate of resensitization and therefore account for the more extensive desensitization observed for the α2a-AR.
We observed a small amount of agonist-induced internalization of α2a using a sensitive ELISA technique for quantitative changes in cell surface density of receptor antigen. However, there was no significant accumulation of receptor in endosomes that could be detected by immunocytochemistry. This suggests that the small amount of α2a internalization may be occurring by a different mechanism than that used for internalization of α2b- and β2-ARs. The inefficient agonist-induced internalization of α2a relative to α2b is in contrast to previous reports (12, 24); Eason et al. found that a 30-min exposure to agonist resulted in 35% sequestration of the α2b-AR and 26% sequestration in the α2a-AR. In those studies, CHO cells were used, and internalization was assayed by ligand binding techniques that used hydrophilic agonists to distinguish between cell surface and intracellular receptor. Several possibilities might account for these different results. First, CHO cells may express other proteins that are critical for agonist-induced sequestration in the α2a-AR. Although α2b- and β2-ARs undergo agonist-promoted receptor internalization in HEK 293 cells, it is possible that this cell line lacks a component necessary for sequestration of α2a-ARs that is present in CHO cells. Another possible explanation is that different methods were used to assess sequestration in the two studies. In the current study, we used immunocytochemical methods to examine receptor internalization. These techniques are not influenced by the state of receptor/G protein coupling. This may not be the case when α2-AR internalization is examined by using a hydrophilic agonist to quantify cell surface receptor density. It is possible that agonist treatment could reduce agonist binding of desensitized plasma membrane α2a-AR even in the absence of receptor internalization.
The physiological or functional significance of the large intracellular pool of the α2c-AR subtype is not known at this time. Both we and others have reported this unique intracellular distribution in a variety of cell lines, including COS-7, HEK 293, MDCK II, Rat1 fibroblasts, and NRK (16, 17). Moreover, this subcellular distribution is not a species-specific idiosyncrasy to the mouse α2c-AR because the wild-type human receptor shows similar intracellular localization when transfected in COS-7 or HEK-293 cells (data not shown). Wozniak and Limbird (17) have shown that in MDCK II cells, this distribution is independent of receptor expression levels; consistent with our findings that a relatively small proportion of the α2c-AR is targeted to the plasma membrane (Fig. 4, B–D), the authors showed that this receptor is targeted directly to the basolateral membrane of MDCK II cells. It is also noteworthy that when α2c- and α2a-ARs are coexpressed in the same cell, these differences in receptor sorting are maintained (Fig.4, C and D). The α2c-AR may require a specific chaperone protein for efficient targeting to the plasma membrane, or it may require a specific protein that anchors the receptor to the cytoskeleton. In the latter case, one would expect the receptor to traffic normally to the plasma membrane but not be retained. However, this mechanism is not consistent with the results of experiments using the thrombin/flag epitope-tagged receptor (Fig. 8) that demonstrate no detectable cycling of receptor between intracellular compartment and the plasma membrane.
We noted that plasma membrane α2c-AR was more easily visualized in NRK cells (Figs. 7, 8, and 10) than in HEK 293 cells. It may be that NRK cells are somewhat better in translocating the α2c-AR to the plasma membrane than are HEK 293 cells; however, this may also be due to the fact that the NRK cells are larger and very flat, permitting better visualization of a larger amount of plasma membrane in a single plane of focus.
We were unable to detect agonist-induced internalization of the α2c-AR using conventional immunocytochemical techniques (Fig. 1). This is in part due to the large preexisting pool of intracellular α2c-AR. However, using a cleavable thrombin/flag epitope on the amino terminus of the α2c-AR allowed us to identify that some receptor is internalized with prolonged agonist treatment (Fig. 8, E and F). This was confirmed by prelabeling the amino-terminal M1 flag epitope of plasma membrane α2c-AR with antibody before agonist treatment (Fig. 10).
The differences in intracellular trafficking observed for the α2-AR subtypes is somewhat unexpected considering the high degree of amino acid identity and the functional similarity with respect to ligand-binding properties and G protein coupling. The functional significance of differences in receptor trafficking is unknown but may be more important in vivo in highly differentiated cells. It has been proposed that receptor subtypes may be targeted to specific plasma membrane microdomains in which receptors are found in close proximity with specific G proteins and effector enzymes (25). Thus, unique trafficking behavior of receptors may enable receptors to couple to specific effector systems.
Summary.
We reported that in several cell lines, the three α2-ARs display unique patterns of subcellular distribution and sorting. At steady state, the α2a- and α2b-AR subtypes are localized in the plasma membrane. Agonist treatment induces internalization of the α2b-AR into endosomes, presumably using the same cellular machinery as the β2-AR, whereas no accumulation of α2a-AR in endosomes could be detected after agonist stimulation. In contrast, at steady state the α2c-AR is targeted to the plasma membrane; however, a significant proportion of this receptor is localized in an intracellular pool that has not yet been delivered to the plasma membrane. Through colocalization studies, we determined that the intracellular pool of α2c-ARs is predominantly localized in the endoplasmic reticulum and cis/medial Golgi. Agonist treatment of the α2c-AR results in some receptor internalization from the plasma membrane. The functional significance of the observed differences in subcellular sorting of the three relatively highly homologous α2-ARs remains to be established.
Footnotes
- Received October 28, 1996.
- Accepted February 10, 1997.
-
Send reprint requests to: Dr. Brian K. Kobilka, B159 Beckman Center, Stanford University, Stanford, CA 94305. E-mail:kobilka{at}cmgm.stanford.edu
Abbreviations
- AR
- adrenergic receptor
- HEK
- human embryonic kidney
- DMEM
- Dulbecco’s modified Eagle’s medium
- PBS
- phosphate-buffered saline
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- M6PR
- mannose-6-phosphate receptor
- BSA
- bovine serum albumin
- ELISA
- enzyme-linked immunosorbent assay
- CHO
- Chinese hamster ovary
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}