


Abstract
Ligands of the benzodiazepine binding site allosterically modulate γ-aminobutyric acidA receptors. Their binding pocket is made up of amino acid residues located on both α and γ subunits. We transiently expressed wild-type α1β2γ2 and mutant GABAA receptors in human embryonic kidney 293 cells and determined their binding properties. Receptors containing the mutant αY209A showed ∼40-fold decrease in affinity for [3H]Ro 15–1788 and diazepam, whereas zolpidem displayed no measurable affinity. Receptors containing the mutant αY209F showed a small-to-moderate decrease in affinity for [3H]Ro 15–1788, diazepam, zolpidem, methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate, and Cl 218872, amounting to 2–8-fold. Receptors containing the mutant αY209Q appeared in the surface membrane of transfected cells, bound [3H]muscimol with wild-type affinity, but failed to bind [3H]Ro 15–1788 or [3H]flunitrazepam with detectable affinity. If these mutant receptors were expressed inXenopus laevis oocytes, the apparent affinity for GABA was only slightly decreased, whereas the ability of the currents to be stimulated by low concentrations of flunitrazepam was abolished. Receptors containing a point mutant of another amino acid residue, αT206A, surprisingly showed an increase in affinity of 5- and 16-fold, for the negative allosteric modulator methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate and the partial positive allosteric modulator Cl 218872, respectively, whereas there was only a small decrease in affinity for Ro 15–1788, diazepam, and zolpidem, amounting to 2-, 4-, and 5-fold. Both α206 and α209 are thus both important in determining the binding affinities for ligands of the benzodiazepine binding site. The residues are spaced at an interval of three amino acids and may be part of an α helix.
The GABAA receptor is one of the major inhibitory neuronal ion channels in mammalian brain. Two subunits were initially purified (1), and their coding DNA was cloned (2). A total of 14 mammalian subunits were subsequently cloned (3) (see Refs. 4-8 for reviews). The subunits show homology to subunits of the nicotinic acetylcholine receptor, glycine receptor and serotonin3 receptor.
The GABAA receptor is also the site of binding of benzodiazepines and related compounds (see Ref. 7 for review). Both the β and α subunits are important for interaction with the channel agonist GABA (9-11). In addition to the well established importance of an α subunit, the presence of a γ subunit is indispensable for benzodiazepine stimulation of GABA-induced currents (12, 13). Three amino acid residues in the γ2 subunit affect benzodiazepine pharmacology (14-17). Both α and γ subunits are thought to contribute to the benzodiazepine binding site, but its localization remains unknown.
We recently described three point mutations in the α and γ subunits that individually affect benzodiazepine effects in GABAA receptors on expression inXenopus laevis oocytes (15). All of the mutated channels respond ∼3-fold to diazepam compared with the wild-type. Two point mutations also result in channels with an enhanced response to zolpidem, whereas the mutation in the γ subunit results in a loss of the zolpidem effect (15). The loss in sensitivity toward zolpidem is accompanied by a loss in sensitivity toward alpidem, Cl 218872, and zopiclone (16). Although the benzodiazepine antagonist Ro 15–1788 can counteract diazepam effects, zolpidem cannot, indicating that zolpidem has lost its ability to interact with the receptor; direct binding studies on GABAA receptors transfected in HEK 293 cells confirmed this (16). Interestingly, some of the amino acid residues important for allosteric modulation by benzodiazepines are directly homologous to amino acid residues on other subunits implicated in the formation of the receptor agonist binding site.
In a previous functional study, the mutated amino acid Y209A of the α subunit gave varying results with different ligands of the benzodiazepine binding site. Although there was a significant functional difference between wild-type channels and channels containing this mutated subunit toward 1 μm zolpidem, this divergent behavior was not significant for 1 μmdiazepam. To decide whether this amino acid residue is important for the binding of ligands of the benzodiazepine binding site, we investigated the binding properties of the respective receptors after transient expression in HEK 293 cells. We analyzed two additional point mutants of this amino acid residue and the T206A mutant of the α subunit. Results obtained suggest involvement of both amino acid residues α206 and α209 in the formation of the binding pocket for ligands of the benzodiazepine binding site. The positional difference of the two residues is suggestive of an α helix.
Materials and Methods
Transfection of recombinant GABAA receptors in cultured cells.
The cDNAs coding for the α1, β2, and γ2 subunits of the rat GABAA receptor channel and the construction of point mutants have been previously described (18-20). HEK 293 cells were maintained in minimum essential medium (GIBCO BRL, Gaithersburg, MD) supplemented with 10% fetal calf serum, 2 mm glutamine, 50 units/ml penicillin, and 50 μg/ml streptomycin through standard cell culture techniques. Equal amounts (total of 20 μg of DNA/90-mm dish) of GABA receptor subunits were transfected into HEK 293 cells (American Type Culture Collection, Rockville, MD) through the calcium phosphate precipitation method (21). After overnight incubation, the cells were washed twice with serum-free medium and fed again with medium.
Membrane preparation.
At ∼60 hr after transfection, the cells were harvested by washing with ice-cold phosphate-buffered saline, pH 7.0, and centrifuged at 150 × g. Cells were washed with buffer containing 10 mm K phosphate, 100 mm KCl, and 0.1 mm K-EDTA, pH 7.4. Cells were homogenized through sonication in the presence of 10 μmphenylmethylsulfonyl fluoride and 1 mm EDTA. Membranes were collected in three centrifugation-resuspension cycles (100,000 ×g for 20 min), and then used for ligand binding or stored at −20°.
Binding assays.
Resuspended cell membranes (0.5 ml) were incubated for 90 min on ice in the presence of [3H]muscimol (26 Ci/mmol), [3H]Ro 15–1788 (87 Ci/mmol), or [3H]flunitrazepam (86 Ci/mmol) (Dupont-New England Nuclear, Boston, MA) and various concentrations of competing ligands. Nonspecific binding was determined in the presence of 25 μm unlabeled muscimol, Ro 15–1788, or flunitrazepam, respectively. Membranes (20–50 μg of protein/filter) were collected through rapid filtration on GF/C filters presoaked in 0.3% polyethyleneimine. After three washing steps with 4 ml of buffer, the filter-retained radioactivity was determined by liquid scintillation counting. On the basis of IC50 determinations, the Ki value was estimated according to the Cheng-Prusoff equation (22).
Detection of GABAA receptors on the surface of living, transfected HEK 293 cells.
Culture medium was removed from transfected cells. Rabbit polyclonal antibodies raised against a polypeptide representing amino acid residues 1–9 of the rat GABAA receptor α1 subunit (5 μg/ml) (23, 24) and dialyzed, tetramethylrhodamine isothiocyanate-coupled swine anti-rabbit IgG antibody (R 0156, 1:20; DAKO, Carpinteria, CA) were added in succession, each diluted in buffered saline and incubated for 45 min at room temperature. The cells were visualized using a Zeiss Axiovert 35 fluorescence microscope equipped with a 63× objective or on a confocal microscope (Zeiss LSM 100). Where indicated, cells were fixed using 4% paraformaldehyde before immunostaining.
Detection of α1 GABAA receptor subunits on Western blots.
Protein (15 μg) from transfected cells was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and blotted onto nitrocellulose membrane. After blocking for 1 hr with 5% nonfat dry milk/0.05% Tween-80, lanes were successively subjected for 3 hr to 5 μg/ml concentrations of the above-mentioned rabbit primary antibody (25) and peroxidase-coupled anti-rabbit IgG antibody (R-14745, 1:200, 1 hr; Transduction Laboratories, Lexington, KY). Peroxidase activity was visualized by the ECL reaction (Amersham, Arlington Heights, IL).
Expression and functional characterization.
X. laevis oocytes were prepared, injected, and defolliculated, and currents were recorded as previously described (13, 26). Briefly, oocytes were injected with 50 nl of cRNA dissolved in 5 mmK-HEPES, pH 6.8. This solution contained the transcripts coding for the different subunits at a concentration of 10 nm for α1, 10 nm for β2, and 100 nm for γ2 (calculated from the UV absorption). Electrophysiological experiments were performed by the two-electrode voltage-clamp method at a holding potential of −80 mV. GABA dose-response curves were fitted using a least-squares method (Gauss-Newton-Marquardt). The equation used was I(c) = Imaxcn/(cn +K a n), where c is the GABA concentration, I is the current elicited,Ka is the GABA concentration eliciting half-maximal current amplitudes (50% Imax), and n is the Hill coefficient. Each of the curves was then standardized to Imax= 100%, and the averaged data were fitted again. Allosteric potentiation via the benzodiazepine site was measured at a GABA concentration eliciting 5–15% of the maximal GABA current amplitude by application of GABA alone and coapplication of GABA with increasing concentrations of flunitrazepam. GABA was applied for 20 sec, and a washout period of 4 min was allowed to ensure full recovery from desensitization. Stimulation of GABA currents was expressed in percentage of the respective control current amplitudes and then standardized, taking the stimulation by 1 μmflunitrazepam of wild-type receptors (328%) as 100%. The perfusion system was cleaned by washing with dimethylsulfoxide to avoid contamination.
Results
Expression of wild-type and mutant receptors in HEK 293 cells.
Wild-type α1β2γ2 (hereafter referred to as αβγ) were mutated in the tyrosine residue α209 to alanine (αY209Aβγ), phenylalanine (αY209Fβγ), or glutamine (αY209Qβγ) or the threonine residue α206 to alanine (αT206Aβγ). Wild-type and point mutant αY209Aβγ, αY209Fβγ, αY209Qβγ, and αT206Aβγ receptors were expressed by transient transfection in HEK 293 cells. Expression was verified by measuring binding of [3H]Ro 15–1788 to membrane fractions. On coexpression with wild-type subunits to yield αβγ combinations, the mutants αT206A and αY209F each resulted in maximal [3H]Ro 15–1788 binding comparable with wild-type receptors, whereas αY209A resulted in ∼10% of the binding of wild-type receptors.
In αY209Qβγ receptors, no binding was detectable of [3H]Ro 15–1788 or [3H]flunitrazepam, and the ligand for the agonist site of the receptor [3H]muscimol was used. On coexpression with βγ, αY209Q resulted in maximal [3H]muscimol binding approximately half that of wild-type receptors (Fig. 1). Specific binding of [3H]muscimol to αY209Qβγ and wild-type receptors showed a KD value of 14 ± 6 and 14 ± 5 nm, and a maximal binding capacity of 0.74 ± 0.14 and 1.6 ± 0.6 pmol/mg of protein, respectively, showing that at least this mutation, which destroyed the binding of two different radiolabeled ligands of the benzodiazepine binding site, did not significantly affect agonist binding affinity. In nontransfected cells, a very small endogenous component of specific [3H]muscimol binding was seen similar to that previously observed (27), but because of its small size, it could not be properly quantified.

Binding of [3H]muscimol to wild-type and αY209Qβγ receptors. HEK 293 cells were transiently transfected with the wild-type or the mutated α subunit, each combined with wild-type β and γ subunits. After harvesting and washing of the membranes, binding assays with [3H]muscimol were carried out as described in Materials and Methods. Wild-type (•) and mutant αY209Qβγ receptors (○) bound the agonist with a K d value of 6 and 13 nm and a B maxvalue of 1.1 and 0.6 pmol/mg of protein, respectively, in this experiment. Values are mean ± standard deviation of two determinations performed in triplicate.
Expression of the α1 subunit was also verified on Western blot analysis (25) for these subunit combinations. Although nontransfected cells did not result in a signal, wild-type and mutant αY209Qβγ receptors resulted in a signal of comparable intensity (not shown).
Surface expression in living HEK 293 cells was followed for wild-type receptors and αY209Aβγ and αY209Qβγ mutant receptors. This was achieved using fluorescent staining of GABAAreceptors on living cells with an α1-specific polyclonal antibody (23, 24) recognizing an extracellular epitope followed by fluorescence microscopy. Although cells transfected with βγ resulted only in very little autofluorescence (Fig. 2b), cells transfected with wild-type receptors showed an intense fluorescence organized in clusters at the surface membrane with little background fluorescence in the cell interior (Fig. 2d). Cells transfected with mutant αY209Qβγ (Fig. 2f) displayed a surface staining similar to wild-type receptors, and αY209Aβγ receptors seemed to show slightly less fluorescence (not shown). The organization of the fluorescence into clusters is induced by the polyclonal antibody and lateral diffusion of the receptors during the incubation; a random distribution was found in transfected cells that were first fixed and then immunolabeled (Fig. 2j). Again, cells transfected with βγ only did not show any specific fluorescence (Fig. 2h). The results demonstrate clearly that in these experiments, mutant subunits αY209Q reach the surface membrane to a similar extent as the wild-type subunits. Furthermore, these observations made with the αY209Qβγ mutant parallel our observations in [3H]muscimol binding measurements.

Surface immunofluorescence of GABAAreceptors expressed in HEK 293 cells. GABAA receptors were stained on living and fixed cells, using an antibody recognizing an extracellular epitope as described in Materials and Methods and visualized using a Zeiss Axiovert 35 fluorescence microscope. a, c, e, g, and i, Differential interference contrast (DIC) optics. b, d, f, h, and j, Corresponding immunofluorescence pictures. a, b, g, and h, Cells transfected with βγ receptors. c, d, i, and j, Cells transfected with wild-type αβγ receptors. e and f, Cells transfected with αY209Qβγ receptors. a–f, Living cells. g–j, Fixed cells. Scale bar, 10 μm.
Binding properties of αY209Aβγ, αY209Fβγ, and αY209Qβγ receptors.
In the previous functional study (15), the mutated amino acid Y209A of the α subunit yielded similar but somewhat differing behavior toward diazepam and zolpidem. Although there was a significant difference between wild-type channels and channels containing this mutation toward 1 μm zolpidem, this difference was not significant for 1 μm diazepam; therefore we investigated whether binding of ligands was affected.
When the wild-type tyrosine was replaced by alanine, affinities for [3H]Ro 15–1788 and diazepam (Fig.3) were reduced 40–50-fold, whereas zolpidem was unable to displace all specific [3H]Ro 15–1788 binding at concentrations up to 30 μm and had therefore lost the ability to bind to the receptor site (Table 1).

Displacement of [3H]Ro 15–1788 binding from αY209Aβγ receptors by diazepam. HEK 293 cells were transiently transfected with the wild-type or mutated α subunit, each combined with wild-type β and γ subunits. After harvesting and washing of the membranes, displacement of 2 and 20 nm[3H]Ro 15–1788 for wild-type (•) and mutant (○) receptors with varying concentrations of diazepam is shown. Values are mean ± standard deviation of two determinations performed in duplicate.
Binding properties of receptor containing α206 and α209 mutants
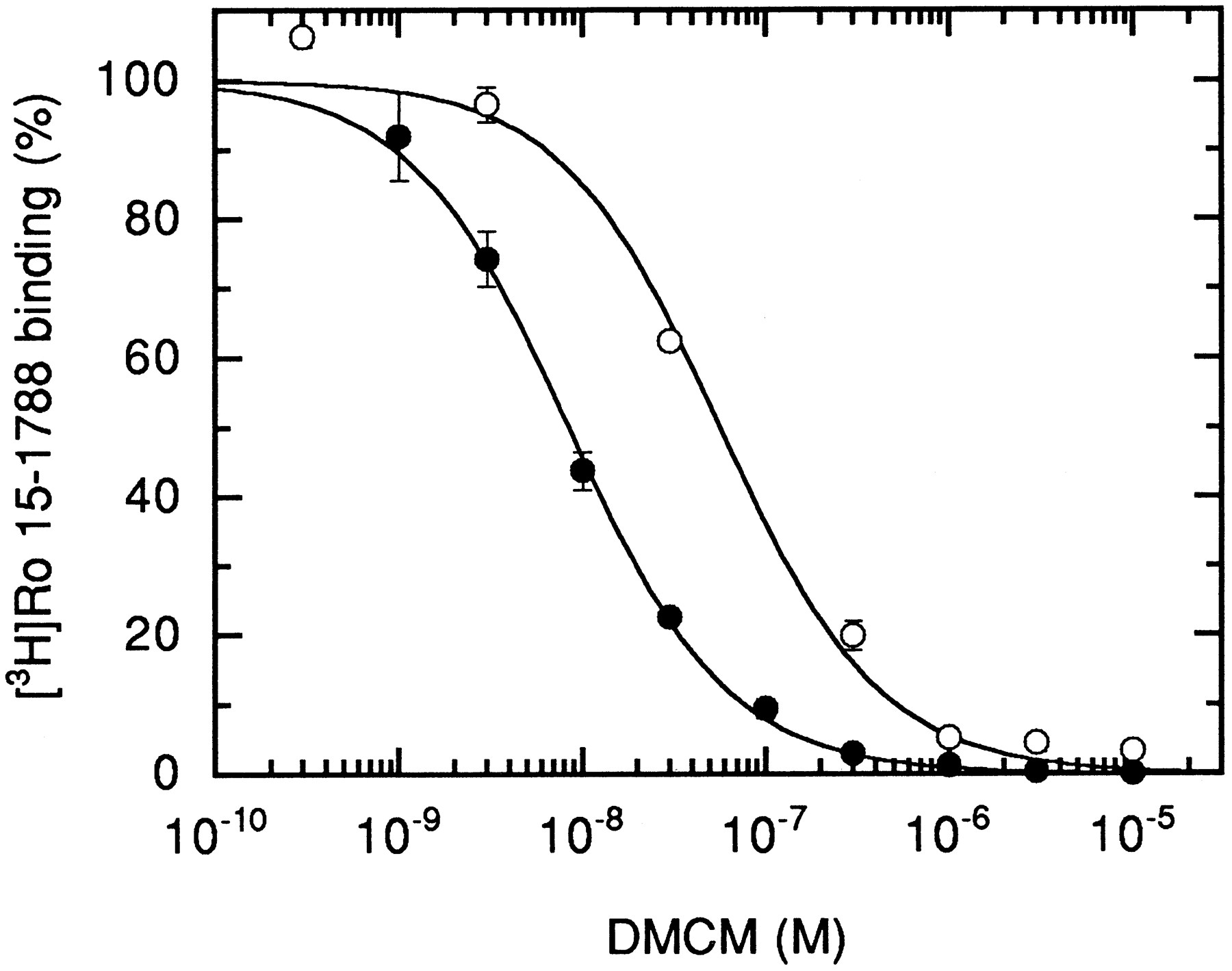
Replacement of tyrosine with phenylalanine (removing a hydroxyl group from residue 209 of the α subunit) led to a small decrease (2–8-fold) for [3H]Ro 15–1788, diazepam, zolpidem, DMCM, and Cl 218872 (Table 1). The largest effect was seen on DMCM (Fig. 4) binding, indicating that this hydroxyl residue is important for interaction of the receptor with this compound.

Displacement of [3H]Ro 15–1788 binding from αY209Fβγ receptors by DMCM. HEK 293 cells were transiently transfected with the wild-type or mutated α subunit, each combined with wild-type β and γ subunits. After harvesting and washing of the membranes, displacement of 2.1 and 7.6 nm[3H]Ro 15–1788 for wild-type (•) and mutant (○) receptors with varying concentrations of DMCM is shown. Values are mean ± standard deviation of two determinations performed in duplicate.
When the same amino acid residue tyrosine in position 209 of the α subunit was replaced by glutamine, the binding properties of the benzodiazepine site were altered dramatically. Indeed, we first analyzed for binding of [3H]Ro 15–1788 and were unable to detect any specific binding. Before immunofluorescence, Western blot experiments, and [3H]muscimol binding, we interpreted this as a failure in the expression of the mutant receptor.
When [3H]flunitrazepam was used for binding, a saturable binding with a KD value of 43 ± 6 nm (two experiments) and a maximal specific binding capacity of ∼400 fmol/mg of protein could be measured (not shown). Displacement studies with Ro 15–1788, zolpidem, DMCM, and diazepam were then carried out. An affinity of <30 μm was not detectable for Ro 15–1788, zolpidem, DMCM, or Cl 218872, indicating that these ligands were unable to displace [3H]flunitrazepam. These binding properties are reminiscent of the [3H]flunitrazepam binding site endogenous to HEK 293 cells (28). Fuchs et al. (28) reported an endogenous binding component for [3H]flunitrazepam. They found a specific binding of 20 ± 1 fmol/mg of protein at a 2 nm concentration. TheKd value was estimated at >100 nm. If the value for specific binding is extrapolated to 20 nm[3H]flunitrazepam, binding amounts to ∼200 fmol/mg of protein. Specific binding also could not be displaced by Ro 15–1788 or a β-carboline. We found that in the presence of 20 nm [3H]flunitrazepam, ∼120 fmol/mg of protein was specifically bound to the membranes, regardless of whether cells were untransfected or transfected with the Y209Q mutant. Flunitrazepam therefore does not bind to expressed receptors but rather to an unidentified endogenous binding site. As shown above, measurement of [3H]muscimol binding showed that the receptor was present and, remarkably, the binding affinity for the receptor agonist muscimol was unaltered. Because [3H]muscimol was bound to αY209Qβγ and the receptor appears on the cell surface, the mutation Y209Q leads to an intact receptor but to the complete loss of binding ability for the radiochemicals [3H]flunitrazepam and [3H]Ro 15–1788.
Functional properties of αY209Qβγ receptors.
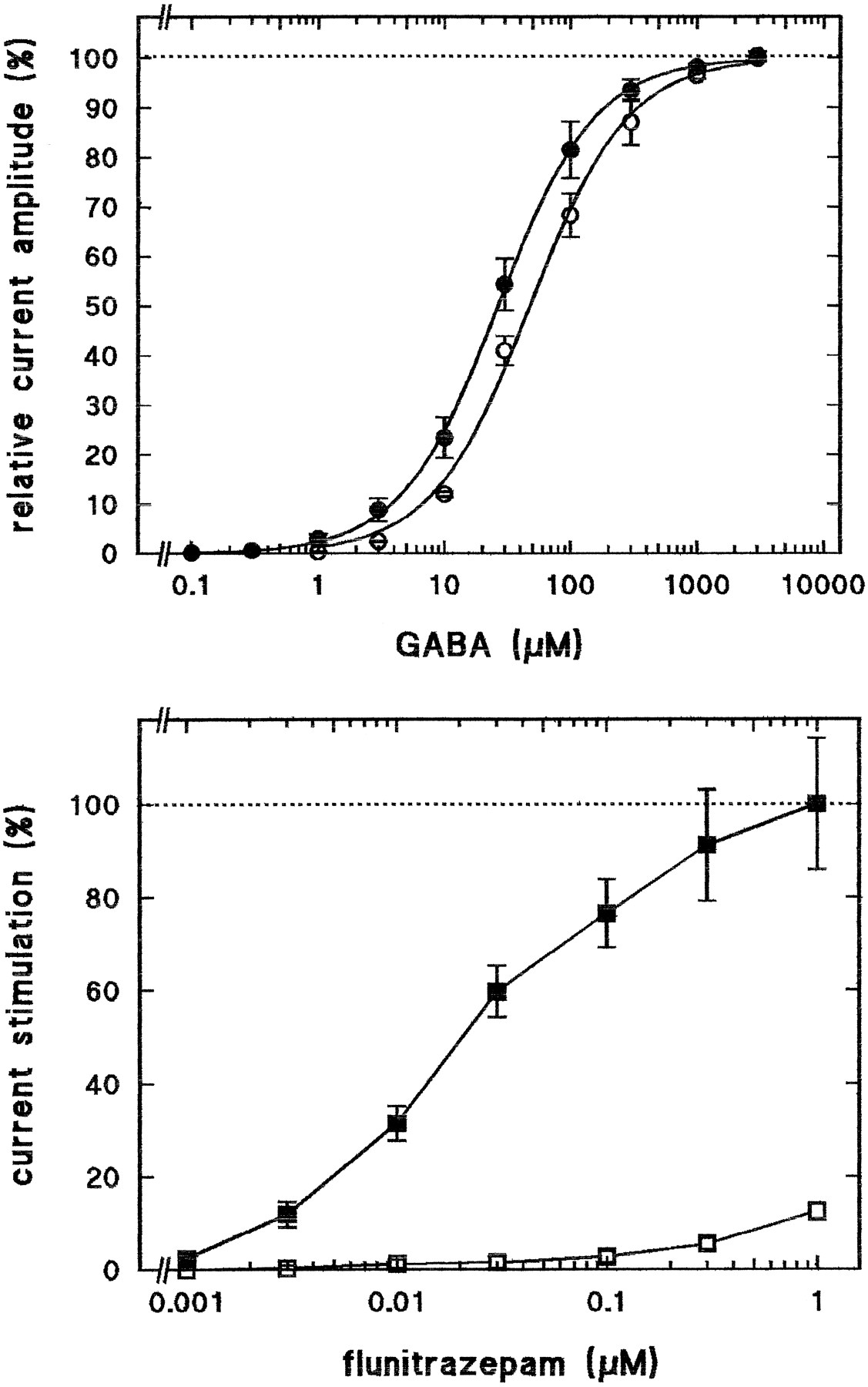
Wild-type αβγ and mutant αY209Qβγ receptors were expressed inX. laevis oocytes. The mutant receptor resulted in maximal GABA current amplitudes approximately three times smaller than those of wild-type receptors. The apparent affinities for GABA to activate ion currents (Ka ) were determined for both types of receptor. Fig. 5,top, shows that there is a small, nearly 2-fold reduction inKa value from 27 ± 4 (three experiments) in wild-type to 48 ± 7 μm(three experiments) in mutant receptors. Concentrations of flunitrazepam of <30 nm failed in contrast to wild-type receptors to significantly stimulate GABA-induced currents (Fig. 5, bottom). Higher concentrations of flunitrazepam resulted in a small current stimulation in mutant receptors, amounting to <15% of the wild-type at 1 μmflunitrazepam..

Loss of stimulation by low concentrations of flunitrazepam of αY209Qβγ receptors. Wild-type αβγ (•, ▪) and mutant αY209Qβγ (○, □) receptors were functionally expressed in X. laevis oocytes. Top, GABA dose-response curves; increasing concentrations of GABA were applied, and data were treated as indicated in Materials and Methods. Values are mean ± standard deviation of three determinations.Bottom, effect of flunitrazepam; increasing concentrations of flunitrazepam were applied together with 5 μm GABA. The relative stimulation of GABA currents in the absence of flunitrazepam is shown. Values are mean ± standard deviation of four determinations.
Binding properties of αT206Aβγ receptors.
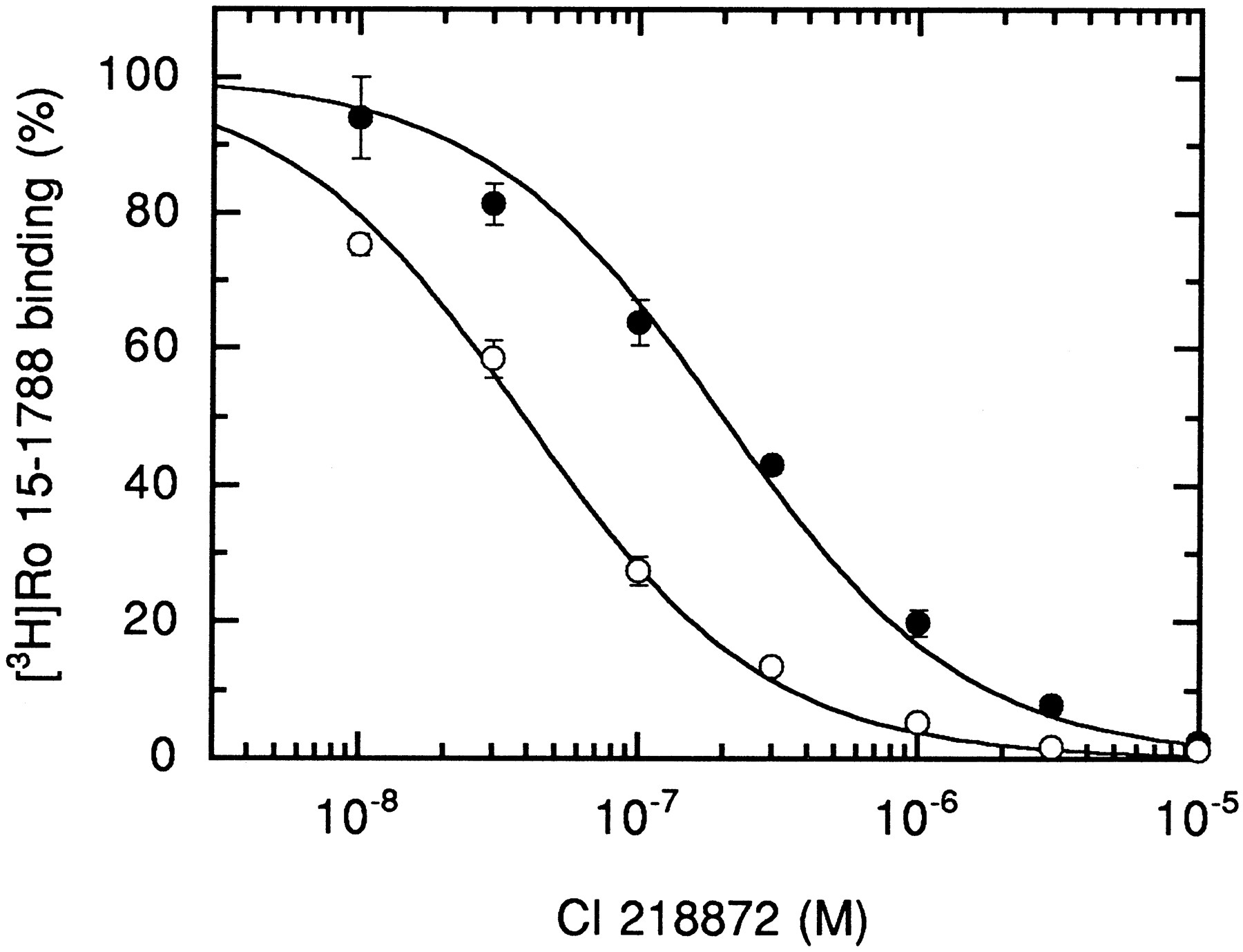
In a previous functional study using receptors expressed in X. laevis oocytes and electrophysiological techniques (15), this mutation resulted in channels displaying an enhanced response to diazepam and zolpidem. It was therefore interesting to see how the binding of ligands of the benzodiazepine binding site was affected. This point mutation led to the expression of receptors displaying a slightly reduced affinity for Ro 15–1788, diazepam, and zolpidem (2-, 4-, and 5-fold) (Table 1). Interestingly, the affinities for DMCM and Cl 218872 were increased strongly, by 5- and 16-fold (Fig.6), respectively, indicating that these latter ligands can be better accommodated after the replacement of threonine by alanine.

Displacement of [3H]Ro 15–1788 binding from αT206Aβγ receptors by diazepam. HEK 293 cells were transiently transfected with the wild-type or mutated α subunit combined with wild-type β and γ subunits. After harvesting and washing of the membranes, displacement of 2.1 and 3.0 nm[3H]Ro 15–1788 for wild-type (•) and mutant (○) receptors with varying concentrations of Cl 218872 is shown. The mutant displays clearly higher affinity for Cl 218872 than wild-type receptors. Values are mean ± standard deviation of two determinations performed in duplicate.
Discussion
We attempted to further characterize the structural and functional properties of the binding site for benzodiazepine-site ligands. For this purpose, wild-type and mutant GABAAreceptors were expressed in HEK 293 cells and X. laevisoocytes. Expression was verified by Western blot analysis, immunocytochemistry, radioligand binding, and electrophysiology.
Binding properties of wild-type and mutant αY209Aβγ, αY209Fβγ, αY209Qβγ, and αT206Aβγ receptors.
Wild-type and mutant GABAA receptors were expressed by transient transfection in HEK 293 cells. The agonist binding properties and those for allosteric modulators acting at the benzodiazepine binding site were determined using [3H]muscimol and [3H]Ro 15–1788, respectively. Although the first type of binding site was unaltered, at least in the Y209Q mutant, the second type was profoundly affected in its properties. Because the endogenous [3H]flunitrazepam binding site to HEK 293 cells is unable to bind the antagonist Ro 15–1788, in all cases in which experiments were based on [3H]Ro-15 1788 binding (i.e., all reported mutants mentioned in Table 1), this endogenous site can be safely ignored. Both mutant receptors αT206Aβγ and αY209Fβγ were affected up to 16-fold in their binding properties. Interestingly, replacement of the threonine residue in position 206 by alanine led to an increase in affinity for the negative allosteric modulator DMCM and the partial positive allosteric modulator Cl 218872, amounting to 5- and 16-fold, respectively. It might be speculated that reduction in residue size leads to better accommodation of these ligands in the binding pocket. The mutant receptor αY209Aβγ showed reduced affinities of >40-fold for several ligands. Reduction in size and loss of aromaticity of this amino acid residue lead to the retention of some affinity (40–50-fold reduction) for diazepam and Ro 15–1788, whereas any detectable affinity for zolpidem, DMCM, and Cl 218872 is lost.
Replacement of tyrosine in position 209 in the α subunit by the similar-sized, nonaromatic residue glutamine leads to the complete loss of any detectable affinity for the two benzodiazepine ligands tested, [3H]Ro 15–1788 and [3H]flunitrazepam (Table 1). The binding properties of the αY209Qβγ receptor (i.e., normal presence of agonist binding and absence of the benzodiazepine ligand binding site) are reminiscent of dual subunit receptor αβ, lacking the γ subunit. Because benzodiazepine binding is still present in alanine and phenylalanine mutants of the same residue but each expresses an altered pharmacology, we believe this is an unlikely interpretation of our results. Rather, we believe the binding site for these ligands is compromised by the presence of the glutamine residue. Also, after expression in X. laevis oocytes, the alanine mutant still results in diazepam stimulation (15), which indicates the presence here of a γ subunit. In addition, we have shown using immunocytochemical methods that the altered α subunit reaches the surface membrane in living cells. Because the exclusive expression of α subunits results in the very inefficient formation of homomeric channels (8, 29, 30), the mutant α subunit is probably assembled together with other subunits.
Relation between binding studies and receptor function.
Functional studies on receptors expressed in X. laevisoocytes were previously been performed with the alanine mutants αT206Aβγ and αY209Aβγ (15). The first mutant channel was stimulated by diazepam and zolpidem to a significantly higher degree than the wild-type channel. For the second mutant, diazepam resulted in a nonsignificantly reduced stimulation, and zolpidem resulted in a loss of stimulation. For functional effects to occur, a substance must first bind. The fact that diazepam and zolpidem result in differential decreases in stimulation of the currents elicited in αY209Aβγ can also be rationalized on the basis of the binding data, showing an affinity of ∼0.5 μm for diazepam and a very strongly reduced affinity for zolpidem. Thus, at the 1 μmconcentration used, these agents are expected to occupy their receptor site to a degree of 67% and <4%, respectively.
The glutamine mutant αY209Qβγ showed a <2-fold reduction in the apparent affinity for channel gating by GABA (Fig. 5, top), whereas any stimulation of the GABA-induced currents by flunitrazepam below a concentration of 30 nm was absent (Fig. 5, bottom). This confirms our findings with the immunocytochemistry and binding studies of this receptor variant showing that the mutant receptor reaches the surface membrane and findings for binding properties in transiently transfected 293 cells, with the binding site for [3H]muscimol unaltered and binding sites for [3H]Ro 15–1788 and [3H]flunitrazepam undetectable.
Spacing of mutations affecting the binding properties of ligands of the benzodiazepine binding site.
The mutations described here affect the two amino acid residues T206 and Y209 of the α subunit. These residues are located three amino acids apart, which suggests that this region of the α subunit might form an α helix. A Chou-Fasman analysis (31) predicts for region 199–209 an α helix followed by a β sheet. Amino acid 206 is located in the center of a hydrophilic region, and 209 is located at the interface of the latter and a hydrophobic region. If the prediction of the secondary structure is correct for this stretch of the α subunit and if amino acids altering the binding properties of ligands of the benzodiazepine binding site do take part in the formation of the actual binding pocket, constraints are imposed on the way in which the pocket is formed by the protein.
Homology of α206 and α209 with β202 and β205.
The two residues described here that extensively affect binding of allosteric modulators, Thr206 and Tyr209 on the α1 subunit, are directly homologous to Thr202 and Tyr205 on the β2 subunit, as implicated in the interaction with the channel agonist GABA (9). This indicates a large degree of homology between the binding sites for channel agonists and for channel modulators of the benzodiazepine type. During revision of the current report, a study by Amin et al. (32), in which the authors made a similar conclusion, came to our attention. Based on functional studies in X. laevis oocytes and on binding studies of the serine mutant of the tyrosine α209, it was concluded, similar to the current report, that this amino acid residue may be involved in binding of benzodiazepines.
Structure of the binding site.
The following amino acid residues on the α1 subunit, or homologous residues on other α isoforms, and on the γ2 subunit have been shown to affect functional alterations to the response of ligands of the benzodiazepine binding site: αY159, αY161, αT206, αY209, γF77, γM130, and γT142 (14–17, 32, current report). The binding of these ligands is affected by αH101, αY159, αG200, αT206, αY209, γF77, and γM130 (16, 17, 32–35; current report). These seven residues have been shown to strongly affect the binding properties. Unless these amino acid residues exert distal effects, they might take part in the formation of the binding pocket for ligands of the benzodiazepine binding site. These amino acid residues are located in five different areas of the protein complex, which may be located next to each other. This means that both the α subunit and the γ subunit may contribute to the formation of the binding site, each folding back after > 50 residues. The proper three-dimensional arrangement of these residues remains to be established.
Acknowledgments
We are grateful to Prof. H. Reuter, in whose institute this work was carried out, for continuous encouragement; to Prof. W. Sieghart (University of Vienna, Austria) for providing the α1 subunit-specific antibody in the context of the collaboration funded by the European Union; to Dr. K. Kannenberg for expert help with microscopic techniques; and to Dr. V. Niggli for careful reading of the manuscript.
Footnotes
- Received April 21, 1997.
- Accepted June 20, 1997.
-
Send reprint requests to: Dr. Erwin Sigel, Department of Pharmacology, University of Bern, Friedbühlstr. 49, CH-3010 Bern, Switzerland. E-mail:sigel{at}pki.unibe.ch
-
This work was supported by Grant 31–37192.93 from the Swiss National Science Foundation, EU Grant BIO4-CT96–0585 (BWW 96.0010), and the Foundation for the Promotion of Scientific Research at the University of Bern.
Abbreviations
- GABA
- γ-aminobutyric acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- DMCM
- methyl-6,7-dimethoxy-4-ethyl-β-carboline-3-carboxylate
- HEK
- human embryonic kidney
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}