


Abstract
Selective inactivation of α1B-adrenoceptor (AR) by the site-directed alkylating agent chlorethylclonidine (CEC) has been used as one of major pharmacological criteria to subclassify α1-AR; however, the mechanism for the differential CEC sensitivity of the two subtypes is uncertain, and the extent of CEC inactivation varies depending on the treatment employed. In this study, we examined the correlation between the subcellular localization of α1-AR subtypes (α1A and α1B) and CEC sensitivity. Constructing α1-AR tagged with the FLAG epitope at the amino terminus and/or green fluorescent protein (GFP) at the carboxyl terminus, we examined the subcellular distribution of α1-ARs expressed in COS-7 cells. Flow cytometry analysis showed that most populations of GFP-expressing α1B-AR cells, but very few GFP-expressing α1A-AR cells, were detected by the anti-amino terminus antibodies. The immunocytochemical and GFP-fluorescence confocal micrographs showed that α1A-ARs predominantly localize intracellularly, whereas α1B-ARs localize on the cell surface. Furthermore, CEC (10 μm) treatment of intact cells resulted in an inactivation of approximately 42% of α1A-ARs and 93% of α1B-ARs, whereas treatment of the membrane preparations resulted in an inactivation of approximately 83% of α1A-ARs and 88% of α1B-ARs, respectively. Together, the results showed that a hydrophilic alkylating agent CEC preferentially inactivates α1-AR on the cell surface irrespective of its subtype, and that the subtype-specific subcellular localization rather than the receptor structure is a major determinant for CEC inactivation of α1-AR. Subtype-specific subcellular localization suggests an additional class of functional properties that provide new insight into drug action.
α1-ARs play critical roles in the regulation of a variety of physiological processes, such as smooth muscle contraction, myocardial inotropy and chronotropy, and hepatic glucose metabolism (1, 2). Increasing evidence suggests that these physiological responses are regulated by multiple receptor subtypes that are structurally homologous (3, 4). Heterogeneity of α1-ARs (α1A and α1B) was first suggested by pharmacological studies based on differential affinity of a variety of agonist and antagonists, and differential sensitivity to the alkylating agent CEC (5-11). Molecular cloning identified three distinct cDNA encoding α1-AR subtypes (α1a, α1b, and α1d) (12-17). More recent studies provide evidence supporting the idea that the α1a-AR (formerly α1c-AR) cDNA encodes the pharmacological α1A-AR subtype, whereas the α1b-AR cDNA clone appears to encode the natively expressed, pharmacologically defined α1B-AR subtype (18-20).
A selective inactivation of α1B-AR by CEC has been used as one of major criteria to subclassify the α1-AR binding sites and functional responses; however, the mechanism for the preferential inactivation of α1B-AR by CEC is uncertain (4). Also, the extent of CEC inactivation has been shown to be varied depending on the CEC treatment employed (intact cell or membrane preparation), and the relative proportion of each subtype in native tissues determined by CEC inactivation does not correlate well with that delineated by subtype-selective competitive antagonists (21, 22). With these problems, it is difficult at present to elucidate the functional role of each receptor subtype. Possible mechanisms that could explain these CEC-related problems include the subtype-specific difference in cellular localization, because CEC is known to bind equally well to both α1A- and α1B-ARs and is highly hydrophilic (23), thereby alkylating only the α1-AR that can be accessed. In support of the hypothesis, our recent flow cytometry analysis of α1-AR subtypes with fluorescent ligand BODIPY-FL prazosin indicated that α1A- and α1B-ARs may distribute differently at steady state in stably expressing Chinese hamster ovary cells (24). In the present study, therefore, we examined the subcellular localization of α1A- and α1B-ARs, and compared it with their CEC sensitivity of each subtype. Flow cytometry analysis and immunocytochemical and GFP-fluorescent localization analysis by confocal microscopy show that α1A-ARs predominantly localize intracellularly, whereas most of α1B-ARs localize on the cell surface, and that CEC preferentially inactivates the cell surface α1-AR irrespective of the subtype. This study indicates that subtype-specific subcellular localization constitutes a new class of properties, in addition to ligand binding and G protein-coupling selectivity, which may pharmacologically distinguish structurally homologous subtypes of α1-AR (as well as other G protein-coupled receptors).
Experimental Procedures
cDNA Construction
Expression constructs used in the present study are shown in Fig. 1. For tagging FLAG and HisX6 epitope to the amino terminus of α1A-AR, primer 1 (gtactatggactacaaggacgacgatgacaagcatcaccatcaccatcacgc) and 2 (acgtcatgatacctgatgttcctgctgctactgttcgtagtggtagtggtagtgcggtac) were annealed and ligated into the NcoI- andPstI-digested pME-α1A expression vector [previously pME-α1c (16)], which designated pME-α1A-FH. The amino acid sequence coded by the primers are also shown in Fig. 1.

Constructs for epitope-tagged and GFP-fused α1-ARs. As described in Experimental Procedures, we constructed epitope-tagged and GFP-fused α1A- and α1B-ARs. ▨, putative membrane spanning domains; ▪, epitope regions for anti-peptide antibodies or FLAG-tagged region. Epitope sequences are also shown.
For tagging GFP to carboxyl terminus of α1-ARs, the coding region of GFP mutant S65T (kind gift from Dr. H. Takahashi, Mitsubishi Kasei Inst. Life Sciences, Tokyo, Japan) was amplified by primer 3 (aaagggcccatgagtaaaggagaagaacttttc) and primer 4 (aaaactagttttgtatagttcatccatggc), which produce the 5′-ApaI site for ligation. All α1A- and α1B-AR expression vectors [pME-α1A, pME-α1B(25), and pME-α1A-FH] were digested byApaI and XbaI, to which the enzyme-digested GFP-polymerase chain reaction products ligated, and resulting constructs were designated pME-α1A-GFP, pME-α1B-GFP, and pME-α1A-FH-GFP, respectively (Fig. 1). Modified regions of these constructs were confirmed by sequencing with an ABI 373A DNA sequencer (Applied Biosystems, Foster City, CA).
We chose the ApaI site in the carboxyl terminus for the GFP to be integrated because the region distal to the ApaI site varies in α1A-AR splice variants with similar pharmacological properties (17), and deletion of the region in α1B-AR was shown not to affect binding and signal transduction properties (26). All experiments were performed with wild-type receptors in parallel whenever possible to confirm that the observation of subcellular localization is not an artifact of the epitope tagging and/or GFP fusion technique (i.e., that the expressed receptors fold and sort properly).
Cell Culture and Transfection
COS-7 cells, obtained from the American Type Culture Collection (Rockville, MD), were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum. The constructs were transfected into COS-7 cells by the electroporation method using Cell-Porator (BRL Life Technologies, Bethesda, MD) according to the manufacturer’s instructions, and cells were assayed 48–72 hr after transfection.
Antibody Preparation
Generation of an anti-peptide antibody (designated as 1B-N1-C) was described previously (23). Briefly, peptide was synthesized corresponding to amino acids 12–27 [peptide: 1B-N1; (C)SAPAQWGELKDANFTG] of the published hamster α1B-AR sequence (12), conjugated to the carrier protein keyhole limpet hemocyanin and injected to rabbits. Antisera were screened against the peptides by using cross-dot systems (Sebia, Moulineaux, France) and visualized by ABC system (Vector Laboratories, Burlingame, CA). By immunoblotting and immunoprecipitation studies, we confirmed that the antibody detect the α1B-AR (24).
Antiserum was purified on 1 ml of a protein A-Sepharose CL-4B column (Pharmacia Biotech, Tokyo, Japan) equilibrated with 20 mmphosphate buffer, pH 7.5, and eluted with glycine-HCl buffer (100 mm, pH 2.2), into 1-ml fractions, which were immediately neutralized with 1 m Tris·HCl buffer, pH 8.5. The resulting antibody fractions were concentrated by Centricon 30 microconcentrator (Amicon, Danvers, MA) and stored at −20°.
Flow Cytometry Analysis
For the flow cytometry analysis with specific antibodies, attached cells were trypsinized and washed twice with PBS. Cells were then incubated for 30 min at 4° with the primary antiserum (1B-N1-C, diluted 1/100) or anti-FLAG monoclonal antibody (10 μg/ml; Eastman Kodak, New Haven, CT), washed three times with PBS, and incubated with Cy3-conjugated goat anti-rabbit or goat anti-mouse IgG (Chemicon International, Temecura, CA) for 60 min at 4°.
Analysis of the cells was performed by using FACScan flow cytometer (Becton Dickinson, Mountain View, CA). To avoid artifacts arising from auto-fluorescence of dead cells and cellular debris, data acquisition was electrically gated for large particles presumably representing intact cells by forward and side scatter. We have used flow cytometry standard setting adjusted to routine double staining techniques employing GFP-associated fluorescence (FL-1) and Cy3 (FL-2) using operating program LYSIS-II. Routinely, data from green fluorescence of 104 cells were subjected to histogram and dot-plot analysis, and FL-2 fluorescence of GFP-positive cells were compared with the control.
Laser Scanning Microscope Analysis
Immunofluorescence detection.
Transfected and untransfected COS-7 cells were seeded at 1 × 105 per well of the 8-well Lab tek chamber slide (Nunc, Naperville, CT) in 0.5 ml of medium. Fixation was performed in 80% acetone for 5 min. Cells were then incubated with 0.05% Triton X-100 in PBS. The primary antibodies, 10 μg/ml of anti-FLAG monoclonal antibody (Eastman Kodak) and 5 μg/ml affinity-purified anti-α1B-AR antibody (1B-N1-C) (24), were brought in PBS containing 10% goat serum and 0.05% Triton X-100, and applied to cells, which were subsequently kept in a humidified chamber for 1 hr at room temperature. Fluorescein isothiocyanate-conjugated goat anti-mouse or anti-rabbit IgG (Chemicon International, Temecura, CA) was diluted 1/200 in PBS containing 2% bovine serum albumin and 0.05% Triton X-100, and applied to cells for 1 hr at room temperature. Cells were then washed twice with PBS, and coverslips were applied using Gel/Mount (Biomeda, Foster City, CA). After immunocytochemical staining, cells were examined by using LSM-GB200 laser scanning microscope (Olympus, Tokyo, Japan) with argon-ion laser set at 488 nm for excitation of fluorescein isothiocyanate.
GFP detection.
Transfected and untransfected COS-7 cells were seeded at 1 × 105 per well of the cover-glass bottom culture dish (MatTek, Ashland, MA) in 2.0 ml of medium. Two days after transfection, cells were washed with PBS and examined using LSM-GB200 within 30 min at room temperature.
125I-HEAT-Binding Assay
Membrane preparation.
Membrane preparation of the cells was performed as described previously (16, 17, 27). Briefly, the cells were collected and disrupted by the sonicator (model SONIFIER 250, setting 5 for 8 sec) in ice-cold buffer A (250 mm sucrose, 5 mm Tris·HCl, 1 mmMgCl2, pH 7.4) and centrifuged at 3,000 ×g at 4° for 10 min to remove nuclei. The supernatant fraction was centrifuged at 35,000 × g for 20 min at 4°. The resulting pellet was resuspended in binding buffer (50 mm Tris·HCl, 12.5 mmMgCl2, 10 mm EGTA, pH 7.4), and was frozen at −80° until assay. All buffers contain protein inhibitors of 1 mm phenylmethylsulfonyl fluoride, 100 μmbenzamidine, 1 μg/ml pepstatin A, and 1 μg/ml leupeptin.
125I-HEAT binding.
125I-HEAT-binding assay was performed as described previously (16, 17, 27). Briefly, membrane aliquots (∼10 μg of protein) were incubated with 125I-HEAT in a final volume of 250 μl of binding buffer for 60 min at 25°. The incubation was terminated by adding the ice-cold buffer B and immediately filtering through Whatmann GF/C glass-fiber filters with a Brandel cell harvester (model-30; Brandel, Gaithersburg, MD). Each filter was collected, and the radioactivity was measured. Binding assays were always performed in duplicate, and specific125I-HEAT binding was determined experimentally from the difference between counts in the absence and presence of 10 μm phentolamine. B max andKd values were obtained by fitting rectangular hyperbolic functions to the experimental data, using computer-assisted iterative nonlinear regression analysis. The protein concentration was measured using the BCA protein assay kit (Pierce, Rockford, IL). Values are expressed as the mean ± standard deviation.
CEC treatment.
Intact cell treatment: 5–10 × 106 cells suspended in 1 ml of the buffered salt solution (140 mm NaCl, 4 mm KCl, 1 mm MgCl2, 1.25 mmCaCl2, 1 mmNaHPO4, 5 mm HEPES, 11 mmglucose and 0.1% bovine serum albumin, pH 7.4) was incubated with or without CEC (10 μm) at 37° for 30 min. After incubation, cells were washed three times with the buffered salt solution and used for the binding assay.
Hypoosmotic membrane treatment: Membranes prepared from cells were incubated in 1 ml volume of hypotonic buffer (5 mmTris·HCl, 5 mm EDTA, pH 7.6) with or without CEC (10 μm) at 37° for 30 min (8, 9). The reactions were stopped by adding 16 ml of ice-cold buffer, and the solution was centrifuged at 35,000 × g for 20 min at 4°. The membranes were washed and resuspended in buffer B, and residual125I-HEAT binding was assessed.
Materials
Materials were obtained from the following sources:125I-HEAT (specific activity, 2200 Ci/mmol; New England Nuclear, Boston, MA); prazosin HCl (Pfizer, Brooklyn, NY); phentolamine mesylate (Ciba-Geigy, Summit, NJ); fetal bovine serum and goat serum (Gibco, Gaithersburg, MD); and Dulbecco’s modified Eagle’s medium (Nissui, Tokyo, Japan). All other reagents were of the highest analytical grade.
Results and Discussion
We first compared ligand binding of the α1-AR conjugates we constructed with their wild-type α1-AR subtype. All constructs were expressed transiently in COS-7 cells to characterize their binding properties to the antagonist 125I-HEAT. The antagonist-binding isotherms for α1A-FH, α1A-GFP, α1A-FH-GFP and α1B-GFP conjugates were nearly identical to the wild-type α1A-AR or α1B-AR, respectively (Table1), confirming that epitope tagging and/or GFP fusion in this manner does not perturb normal ligand binding. Receptor subtypes displayed similar transfection efficiencies (assessed by the percentage of transfected cells expressing GFP-associated fluorescence; Fig. 2). Both wild-type and conjugated receptors expressed in COS-7 cells were found to be functional, as a norepinephrine (10−7 m)-induced intracellular free Ca2+ concentration response was observed by using Indo-1 (28) (data not shown). Although all the receptor conjugates showed similar ligand binding to 125I-HEAT, the tagging technique may have an artifactual effect on receptor folding and sorting. Hence, all experiments below were performed with wild-type receptors in parallel whenever possible.
Comparison of Kd andB max of wild-type, epitope-tagged, and GFP-fused α1-ARs.

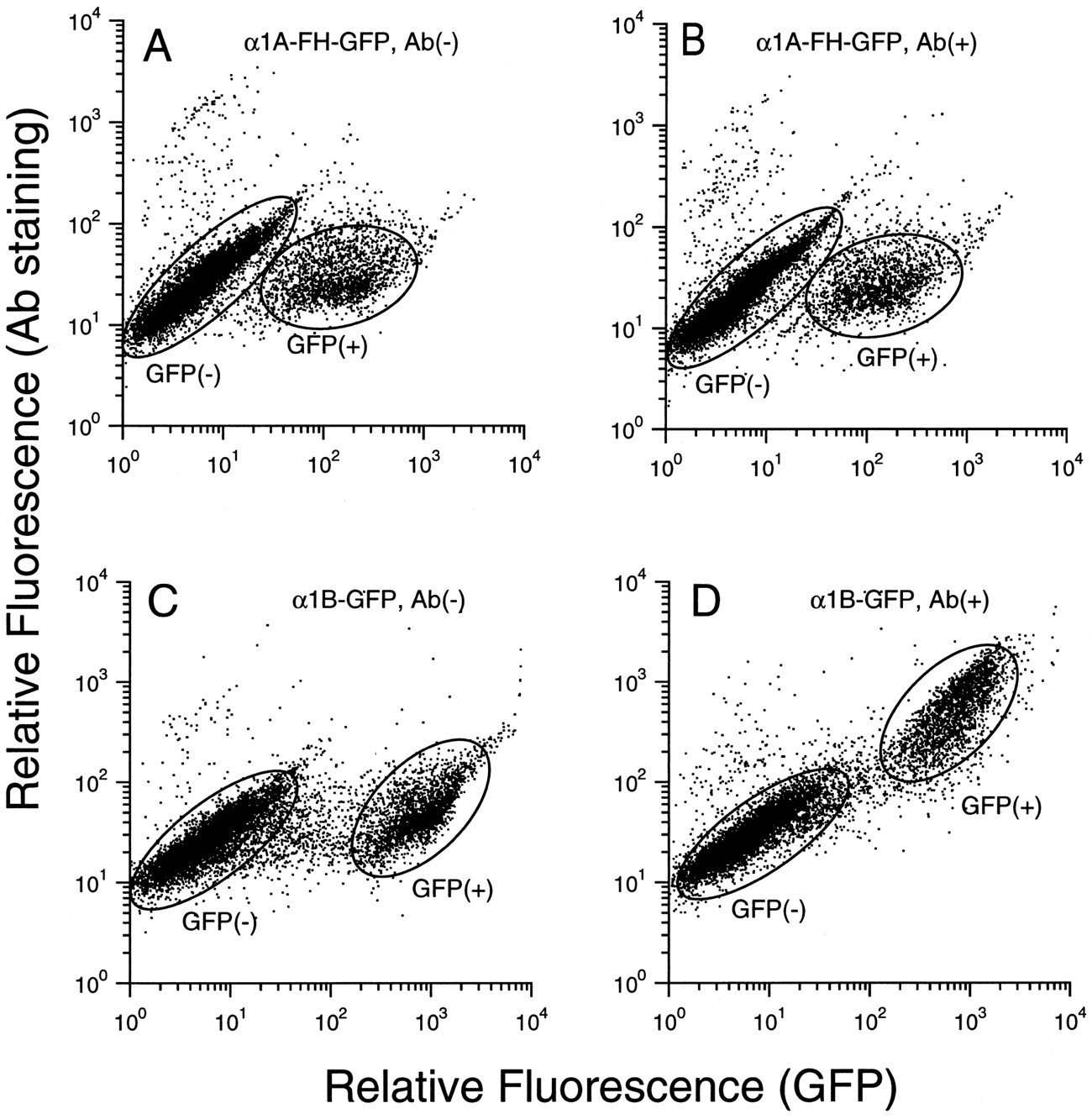
Flow cytometry analysis for α1-ARs. COS-7 cells transfected with pME-α1A-FH-GFP (A and B) and pME-α1B-GFP (C and D) were analyzed by fluorescence-activated cell sorter flow cytometer. The cells were stained with nonimmune serum (A and C), anti-FLAG antibody (B), or anti-peptide antiserum against the α1B-amino terminus (D). Results are showed by two-dimensional dot-plot using GFP fluorescence (FL-1: horizontal) and Cy3 labeled secondary antibody (FL-2: vertical).GFP+, cells expressing GFP-fused receptor;GFP−, cells that do not express GFP-fused receptor. Thex-axis is relative fluorescence of GFP;y-axis is relative fluorescence of Cy3.
Using the epitope- and/or GFP-tagged receptors, we characterized the subcellular localization of α1-AR subtypes. We used flow cytometry analysis and confocal microscopy analysis of immunolocalization and GFP fluorescence, which enable us to detect the cell surface receptor and also to visualize the subcellular distribution of α1-ARs, respectively. First, the cells transfected with α1A-FH-GFP and α1B-GFP conjugates were examined by flow cytometry. When transfected, approximately 10–20% of COS-7 cells were positively detected as GFP-associated fluorescence for α1A-FH-GFP and α1B-GFP receptors (Fig. 2, A and C). The mean value of fluorescence intensity of α1B-GFP receptor was noted to be stronger than that of α1A-FH-GFP or α1A-GFP (x-axis; Fig. 2, A versus C). Then, the cells transfected with α1A-FH-GFP and α1B-GFP conjugates were stained by the anti-FLAG monoclonal antibody and by the antibody 1B-N1-C, respectively. Interestingly, as shown in Fig. 2, B and D, most populations of the cells expressing GFP were further detected (an upward shift) by 1B-N1-C (Fig. 2D), whereas very few cells expressing GFP were detected by anti-FLAG antibody (no upward shift) (Fig. 2B). The failure for α1A-GFP-expressing cells to be detected by anti-FLAG monoclonal antibody is not due to the construct or the weak affinity of antibody, because α1A-FH and α1A-FH-GFP can be immunostained by the same antibody (as shown below). Additionally, when the empty vector pME18s was transfected, no cell was positively detected as GFP-associated fluorescence or stained by antibodies. The results show that amino terminus of the α1B-AR can be, but that of α1A-AR cannot be, detected by the antibody.
We further visually examined the cellular distribution of receptors using fluorescent antibody (α1A-FH and α1B-AR) and also the endogenous receptor fluorescence (α1A-GFP and α1B-GFP) by fluorescent confocal microscopy. As seen in Fig. 3 (left), immunocytochemical analysis showed that the fluorescence distribution of α1A-AR is characteristic of the cytoplasmic distribution (enhanced perinuclear fluorescence), whereas that of α1B-AR is typical of a plasma membrane-labeling pattern. Furthermore, corresponding well with immunocytochemical localization, the fluorescence micrograph of GFP shows that a strong green fluorescence densely localizes in a perinuclear region for α1A-GFP, whereas fluorescence is diffuse in plasma membranes for α1B-GFP (Fig. 3,right). Additionally, a similar cytoplasmic distribution was observed for α1A-FH-GFP by using fluorescent antibody and also endogenous receptor fluorescence, and no fluorescence signal was detected in untransfected cells (data not shown). Together with the results of flow cytometry analysis, these confocal microscopy analyses show that α1A-AR predominantly localizes intracellularly, whereas most α1B-AR localizes on the cell surface.

The immunocytochemical and GFP fluorescence confocal micrographs of the α1A-AR and α1B-AR in COS-7 cells. Immunocytostaining with anti-peptide antiserum (1B-N1-C) and anti-FLAG antibody were performed in the pME-α1A-FH (left)- or pME-α1B (right)-transfected COS-7 cells. GFP fluorescence was also observed in the pME-α1A-GFP (left)- or pME-α1B-GFP (right)-transfected COS-7 cells in intact condition.Bottom, cells were also observed by electrical pseudo-differential interference contrast (DIC) image.Scale bar, 10 μm.
We showed previously that the transfected α1A-ARs could be inactivated easily by CEC when treated in membrane preparation, whereas they were relatively resistant to CEC inactivation compared with α1B-AR when treated in intact cells (17, 29). To examine the correlation between the subcellular localization and the extent of CEC inactivation, we further compared the extent of CEC inactivation when treated in intact cells with that when treated in membrane preparation. As summarized in Fig. 4, CEC (10 μm 30 min) treatment of intact cells reduced the number of125I-HEAT binding sites by 41.8 ± 12.3%, 27.1 ± 18.1%, 37.7 ± 17.7%, and 43.7 ± 17.1% (n = 3 each) in the wild-type α1A-AR, α1A-FH, α1A-GFP, and α1A-FH-GFP, respectively; on the other hand, by 86.0 ± 4.5% and by 92.6 ± 1.9% (n = 3 each) in the wild-type α1B-AR and α1B-GFP, respectively. In contrast to the intact cell treatment, membrane preparation treatment resulted in a marked reduction of 125I-HEAT binding sites in α1A-AR (by 83.3 ± 2.5%,n = 5); however, it did not cause any further decrease of 125I-HEAT binding sites in α1B-AR (by 88.2 ± 4.5%,n = 4). In all cells, theKd values to125I-HEAT obtained after CEC treatment were not much different from those obtained without CEC treatment (see Fig. 4legend). Together with the observation that the two α1-AR subtypes differentially localize, the results may suggest that a highly hydrophilic alkylating agent CEC inactivates only α1-AR on the cell surface irrespective of its subtype; thus, the subtype-specific cellular distribution rather than the receptor structure is a major determinant for CEC inactivation of α1-AR.
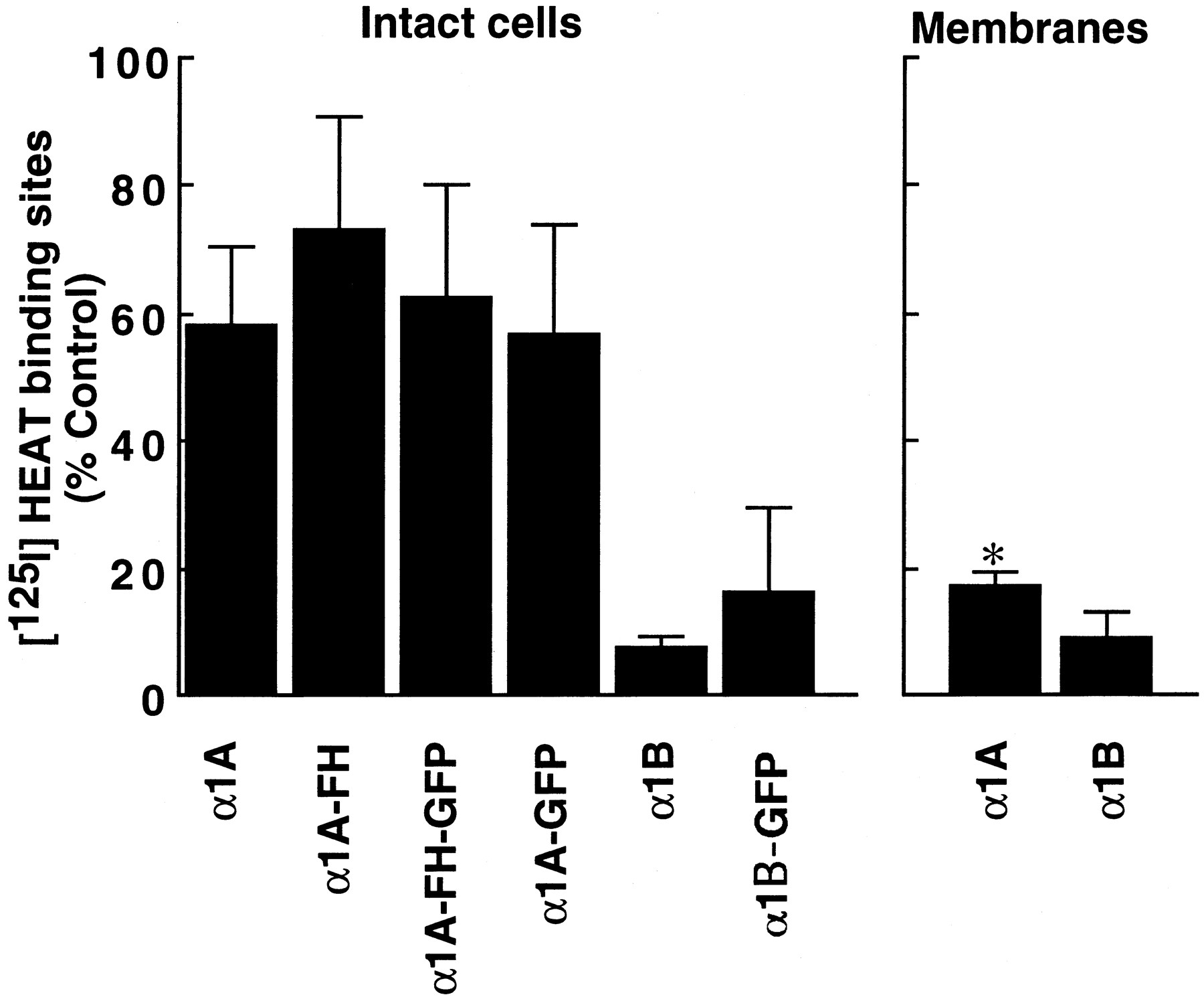
Comparison of CEC inactivation by intact cell treatment and by membrane preparation treatment. For the intact cell treatment, COS-7 cells incubated with or without CEC (10 μm) at 37° for 30 min. After incubation, cells were washed three times with the buffered salt solution and used for the binding assay. In the membrane preparation treatment, membranes prepared from cells were incubated in hypotonic buffer (5 mm Tris·HCl, 5 mm EDTA, pH 7.6) with CEC (10 μm) at 37° for 30 min and residual125I-HEAT binding was assessed. The meanK d values obtained in cells after CEC treatment were 48.5, 142, 33.0, 37.0, 28.8, and 67.0 nm, n = 2–3, in the wild-type α1A-AR, α1A-FH, α1A-GFP, α1A-FH-GFP, wild-type α1B-AR, and α1B-GFP, respectively. The results are expressed as the decrease in percentage of the control 125I-HEAT binding. The values correspond to the mean ± standard deviation of at least three independent experiments. ∗, CEC inactivation was significantly different from that obtained in the intact cell treatment (unpaired t test, p < 0.05).
As indicated in the introductory material, three α1-AR cDNA clones have been isolated, but their relationship to the pharmacologically defined α1A- and α1B-AR subtypes has been controversial. More recently, based on the comparison of the Ki values of native α1-ARs from tissues with those of cloned α1-ARs, and on the comparison of the tissue distribution of native α1-ARs assessed by using subtype-selective drugs with that of mRNA expression of cloned receptors (18, 19), the IUPHAR Committee on the Classification of Adrenoceptor recommended that the α1a-AR (formerly α1c-AR) cDNA encodes the pharmacological α1A-AR subtype, whereas the α1b-AR cDNA clone appears to encode the natively expressed, pharmacologically defined α1B-AR subtype (20). In this report, we demonstrated that α1A-AR predominantly localizes intracellularly, whereas most of α1B-ARs localize on the cell surface, and further indicate that the subtype-specific difference in receptor distribution appears to be reflected by inactivation of the highly hydrophilic alkylating agent CEC. The current results emphasize that one must be cautious in interpreting the data obtained by using CEC inactivation to study the functional role of each subtype.
Differential distribution of receptor subtypes appears to be generally observed for G protein-coupled receptors. von Zastrowet al. (30) and Saunders et al. (31) showed that subtype-specific different localization of α2-AR subtypes. Thus, subtype-specific sorting comprises another important parameter, in addition to ligand binding affinity and G protein-coupling specificity, which may functionally differentiate receptor subtypes, thereby increase signal diversity. It is postulated that subtype-specific sorting might segregate individual receptors, G proteins, and effectors into functionally specialized microdomains (compartmentalization) and specify the signal transduction (30). Subtype-specific sorting may play an important role not only in increasing the signal diversity but also in the regulatory properties of receptor. Subtype-specific differences in susceptibility to agonist-promoted desensitization (and receptor phosphorylation) appeared to be well correlated with the cellular localization of receptor subtype; thus, α2-C10-ARs and α1B-ARs, which localize in the plasma membrane, are more easily desensitized compared with α2-C4-ARs and α1A-ARs, which predominantly localize intracellularly (32, 33). It is also suggested that intracellular receptors could serve as a reserve pool for delivery to the plasma membrane (30). In support of this view, we had previously shown a sizable pool of spare receptor for α1A-AR and an absence of a receptor reserve for α1B-AR by using the phenoxybenzamine-inactivation method (9, 10). Such internal stores could contribute to the rapid up-regulation of G protein-coupled receptor observed in physiological and pathophysiological conditions (34, 35). Further studies are clearly required to clarify the functional role of the subtype-specific subcellular localization.
In conclusion, we demonstrate for the first time that α1-AR subtypes have differential subcellular localization, which appears to be a major determinant for CEC inactivation of α1-AR. Thus, receptor subtype-specific localization suggests an additional functional properties that may explain the signal and functional diversity, and moreover it is of importance in assessing drug action. With the availability of experimental system as exemplified in the present study, elucidation of the molecular mechanism and function of the subtype-specific subcellular distribution may provide new insight into signal transduction as well as drug action.
Acknowledgments
We thank Dr. M. Kataoka (Department of Molecular and Cell Pharmacology) and Dr. H. Takahashi for generously providing GFP cDNA and for helpful discussion.
Footnotes
- Received May 27, 1997.
- Accepted July 29, 1997.
-
Send reprint requests to: Gozoh Tsujimoto, M.D., Ph. D., Department of Molecular, Cell Pharmacology, National Children’s Medical Research Center, 3-35-31 Taishido, Setagaya-ku, Tokyo, 154 Japan. E-mail:gtsujimoto{at}nch.go.jp
-
This investigation was partially supported by research grants from the Scientific Fund of the Ministry of Education, Science, and Culture of Japan, the Japan Health Science Foundation and Ministry of Human Health and Welfare.
Abbreviations
- AR
- adrenoceptor
- CEC
- chlorethylclonidine
- HEAT
- (2-β-(4-hydroxyphenyl)-ethylaminomethyl)-tetralone
- PBS
- phosphate-buffered saline
- GFP
- green fluorescent protein
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}