


Abstract
Activation of ionotropic glutamate receptors causes increases in intracellular Ca2+ concentration ([Ca2+]i) and intracellular Na+concentration in neurons. It has been suggested that reversal of the plasma membrane Na+/Ca2+ exchanger (NCE) may account in part for the rise in [Ca2+]i. Recently, KB-R7943 (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulfonate) was reported to selectively inhibit the reverse mode of the NCE in non-neuronal cells. We investigated the effects of KB-R7943 on glutamate-stimulated increases in [Ca2+]i. In cultured rat forebrain neurons loaded with indo-1 acetoxymethyl ester, KB-R7943 inhibited the reverse mode of NCE (IC50 = 0.7 μm). When tested against kainate- (100 μm),N-methyl-d-aspartate- (30 μm), glutamate- (3 μm), or KCl- (50 mm) induced [Ca2+]i transients (15 sec, in the presence of Na+ and Ca2+), KB-R7943 inhibited these transients with IC50 values of 6.6, 8.2, 5.2, and 2.9 μm, respectively. [Ca2+]iincreases caused by a higher concentration of glutamate (100 μm) also were inhibited by KB-R7943 (10 μm). However, KB-R7943 had no effect on peak [Ca2+]i changes caused by prolonged application of glutamate and did not inhibit glutamate-induced neuronal injury. KB-R7943 did not inhibitN-methyl-d-aspartate- or kainate-induced whole-cell currents, nor did it substantially inhibit voltage-sensitive Ca2+ currents, excluding a direct inhibition of these ion channels. These results suggest that reverse NCE contributes to the immediate rise in [Ca2+]i resulting from glutamate receptor activation. However, reverse NCE becomes less important as the stimulus time is increased, and Ca2+ entry by this route is not critical for the expression of excitotoxic injury.
Neuronal Ca2+ entry can be mediated directly by NMDA- and Ca2+-permeable (±)-α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid/KA receptors and activated indirectly by depolarization-induced activation of voltage-gated Ca2+ channels (Mayer and Miller, 1990). Because excessive Ca2+ loading induced by glutamate receptor activation is acutely toxic to neurons (Michaels and Rothman, 1990; Hartley et al., 1993), significant effort has been directed toward understanding the mechanism underlying [Ca2+]i increases and subsequent Ca2+ buffering. In addition to Ca2+ influx through the glutamate receptors and/or activation of Ca2+ channels, it has been proposed that the plasma membrane NCE may contribute to the glutamate-induced [Ca2+]iincrease (Kiedrowski et al., 1994; Courtney et al., 1995; Kirischuk et al., 1997). The NCE electrogenically exchanges 3 Na+ for 1 Ca2+ and can function to cause Ca2+ accumulation (reverse mode) or Ca2+ extrusion (forward mode) depending on the concentrations of each ion on either side of the membrane and on the membrane potential (Blaustein, 1988; DiPolo and Beauge, 1988; Yu and Choi, 1997). Because glutamate receptor activation causes a large increase in [Na+]i, it is possible that this process, along with membrane depolarization, favors the influx of Ca2+ via the NCE during the stimulus. The studies that have investigated the putative contribution of reversed NCE have been limited by the lack of selective inhibitors of NCE (Kiedrowski et al., 1994; Courtney et al., 1995; Storozhevykh et al., 1996; Kirischuk et al., 1997). As a consequence of this, the role of reverse NCE during glutamate stimulation with physiological Na+ concentrations is poorly understood. However, it is clear that forward NCE, along with mitochondrial Ca2+ uptake, contributes to the buffering of cytosolic [Ca2+]i after a glutamate stimulus because recovery is significantly slowed in the absence of extracellular Na+ (Wang et al., 1994; Kiedrowski and Costa, 1995; White and Reynolds, 1995). It also has been suggested that extrusion of Ca2+via the NCE serves an important neuroprotective function because inhibition of this process increases neuronal death after a toxic glutamate stimulus (Hartley and Choi, 1989; Andreeva et al., 1991).
Recently, KB-R7943 (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulfonate) was shown to selectively inhibit the reverse mode of the NCE. (Iwamoto et al., 1996; Watano et al., 1996). KB-R7943 inhibits reverse NCE (IC50 = 1.2–2.4 μm) in a number of cell types, including cardiomyocytes, smooth muscle cells, and NCX1-expressing fibroblasts (Iwamoto et al., 1996). The affinity for inhibition of the reverse mode of NCE is ≥10-fold higher than that required to inhibit the forward mode (Iwamoto et al., 1996). KB-R7943 does inhibit dihydropyridine-sensitive Ca2+channels but at concentrations ≥10-fold higher than those required to inhibit reverse NCE (Iwamoto et al., 1996).
In this study, we used KB-R7943 to elucidate the contribution of reverse NCE to the increase in [Ca2+]i after glutamate stimulation of cultured forebrain neurons. We developed a stimulation paradigm to assay reverse NCE in intact neurons and confirmed the activity of KB-R7943 on this process and report that reverse NCE makes a substantial contribution to the initial increase in [Ca2+]i caused by glutamate receptor activation. However, this contribution is diminished progressively during longer glutamate exposures, with the result that inhibition of reverse NCE does not seem to be neuroprotective in vitro.
Experimental Procedures
Primary neuronal culture.
For [Ca2+]i measurements, forebrains from e17 Sprague-Dawley rats were removed, dissociated, and plated at a density of 3 × 105 cells/ml onto 31-mm glass coverslips in 35-mm six-well culture plates, as described in detail previously (White and Reynolds, 1995). All procedures using animals were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Use Committee of the University of Pittsburgh. Dissociated neurons initially were plated in DMEM with 10% fetal bovine serum, 24 units/ml penicillin, and 24 μg/ml streptomycin. Twenty-four hours after plating, the media were removed and replaced with DMEM with horse serum instead of fetal bovine serum, and the coverslips were inverted. Coverslip inversion suppresses proliferation of non-neuronal cells. Neuronal cultures were kept at 37° in a 5% CO2 incubator until use (12–18 days).
For whole-cell recordings, cerebral cortices were obtained from e-16 Sprague Dawley C-D rats and dissociated according to methods described previously (Hartnett et al., 1997). Briefly, cortices were incubated in MEM solution containing 0.03% trypsin for 2 hr at 37°. Dissociated cells were plated at a density of 3–5 × 105 cells/ml in growth medium (v/v 80% MEM, 10% Ham’s F-12 nutrient mixture, 10% heat-inactivated iron-supplemented bovine calf serum, 25 mm HEPES, 24 units/ml penicillin, 24 μg/ml streptomycin, and 2 mm l-glutamine) into 35-mm tissue culture dishes containing five 12-mm poly-l-lysine-coated glass coverslips each. Cells were maintained at 37° in 5% CO2. Growth medium was changed three times per week. After 15 days in culture, non-neuronal cell proliferation was inhibited with 2 μm cytosine arabinoside, after which the growth medium contained 2% serum and no Ham’s F-12. Cells were used between 21 and 28 days in vitro.
[Ca2+]i measurements.
Recordings of [Ca2+]i in individual neurons were made in indo-1-loaded neurons as described previously (White and Reynolds, 1995). Cells were rinsed with HBSS that contained 137 mm NaCl, 5 mm KCl, 0.9 mmMgSO4, 1.4 mmCaCl2, 3 mmNaHCO3, 0.6 mmNa2HPO4, 0.4 mmKH3PO4, 5.6 mmglucose, and 20 mm HEPES, pH adjusted to 7.4 with NaOH, and then incubated in HBSS containing the Ca2+-sensitive fluorescent dye indo-1-AM (5 μm) and 5 mg/ml bovine serum albumin at 37° for 50 min. The cells were washed with dye-free HBSS and returned to the 37° incubator for an additional 20 min. The coverslip containing dye-loaded neurons was mounted in a recording chamber and placed on the stage of a Nikon Diaphot fluorescence microscope. Neurons were perfused with HBSS or drug solutions at a rate of 20 ml/min. Indo-1 fluorescence was measured by isolating a single neuron with a rectangular diaphragm in the emission pathway. Ratios of indo-1 fluorescence obtained at 405 and 490 nm (excitation, 340 nm) were used to calculate [Ca2+]i using in situ calibration parameters as described previously (White and Reynolds, 1997). For Na+-free HBSS experiments, the Na+ was replaced withN-methyl-d-glucamine, the NaHCO3 was replaced with KHCO3, and the KCl was omitted. For IC50 determinations of averaged data, data were fit with a four-parameter logistic equation using Prism Version 2.01 (GraphPAD Software, San Diego, CA).
Magfura-2 fluorescence measurements were made as described previously (Brocard et al., 1993). Neurons were loaded with 5 μm magfura-2-AM in HBSS with 5 mg/ml bovine serum albumin for 15 min at 37°. Ratios of fluorescence values (510-nm emission) from a single neuron were collected after sequential excitation at 340 and 380 nm. Data are presented as the ratio of magfura-2 fluorescence values obtained at 340 and 380 nm. The magfura-2 signal probably reflects a combination of both relatively high [Ca2+]i and physiological intracellular [Mg2+] and is an important tool in light of the suggestion that high affinity dyes underestimate [Ca2+]i during excitotoxic glutamate receptor activation (Stout and Reynolds, 1996;Hyrc et al., 1997).
Whole-cell recording.
Electrophysiological measurements were obtained using the whole-cell patch-clamp configuration. Results are expressed as the mean ± standard error. Whole-cell recording reagents were purchased from Sigma Chemical (St. Louis, MO) except as noted.
For ligand-gated ion channels (NMDA and KA), the methods of data acquisition and analysis have been described previously (Aizenmanet al., 1992; Tang and Aizenman, 1993). The external recording solution contained 150 mm NaCl, 2.8 mm KCl, 1.0 mm CaCl2, 10 mm HEPES, pH 7.2, 10 μm glycine, 0.03–0.1% dimethylsulfoxide, and 0.25 μm tetrodotoxin (Calbiochem, La Jolla CA). Patch electrodes (2–4 MΩ) were filled with 140 mm CsF, 10 mm EGTA, 1 mmCaCl2, and 10 mm HEPES, pH 7.2. NMDA and KA were dissolved in extracellular solution and applied onto cells by a multibarrel fast-perfusion system (Warner Instrument, Hamden, CT).
For voltage-gated Ca2+ channels, currents were evoked by a depolarizing step from −80 to 0 mV. Series resistance was compensated and leak currents were subtracted with the use of a prepulse protocol. Depolarizing steps were made once every 5 or 10 sec to avoid inactivation of Ca2+ currents. The external recording solution contained 140 mm NaCl, 5 mm CaCl2, 2 mmMgCl2, 10 mm HEPES, pH 7.2, 5 mm glucose, 10 mm tetraethylammonium·HCl, 1 μm tetrodotoxin, and 5 mm KCl. Patch electrodes (2–3 MΩ) were filled with 108 mm CsCl, 9 mm EGTA, 4.5 mm MgCl2, 9 mm HEPES, pH 7.2, 4 mm Mg·ATP, 0.3 mm Na3·GTP, and 14 mmNa2·phosphocreatine. Electrodes were “tip dipped” for ≈20 sec in intracellular solution that did not contain ATP, GTP, or phosphocreatine.
Materials.
Indo-1-AM and magfura-2-AM were purchased from Molecular Probes (Eugene, OR). DMEM, MEM, penicillin, and streptomycin were purchased from GIBCO BRL (Grand Island, NY). Glutamate, glycine, NMDA, and KA were purchased from Sigma Chemical (St. Louis, MO). KB-R7943 was a generous gift of Dr. Tomokazu Watano (Kanebo, Osaka, Japan).
Results
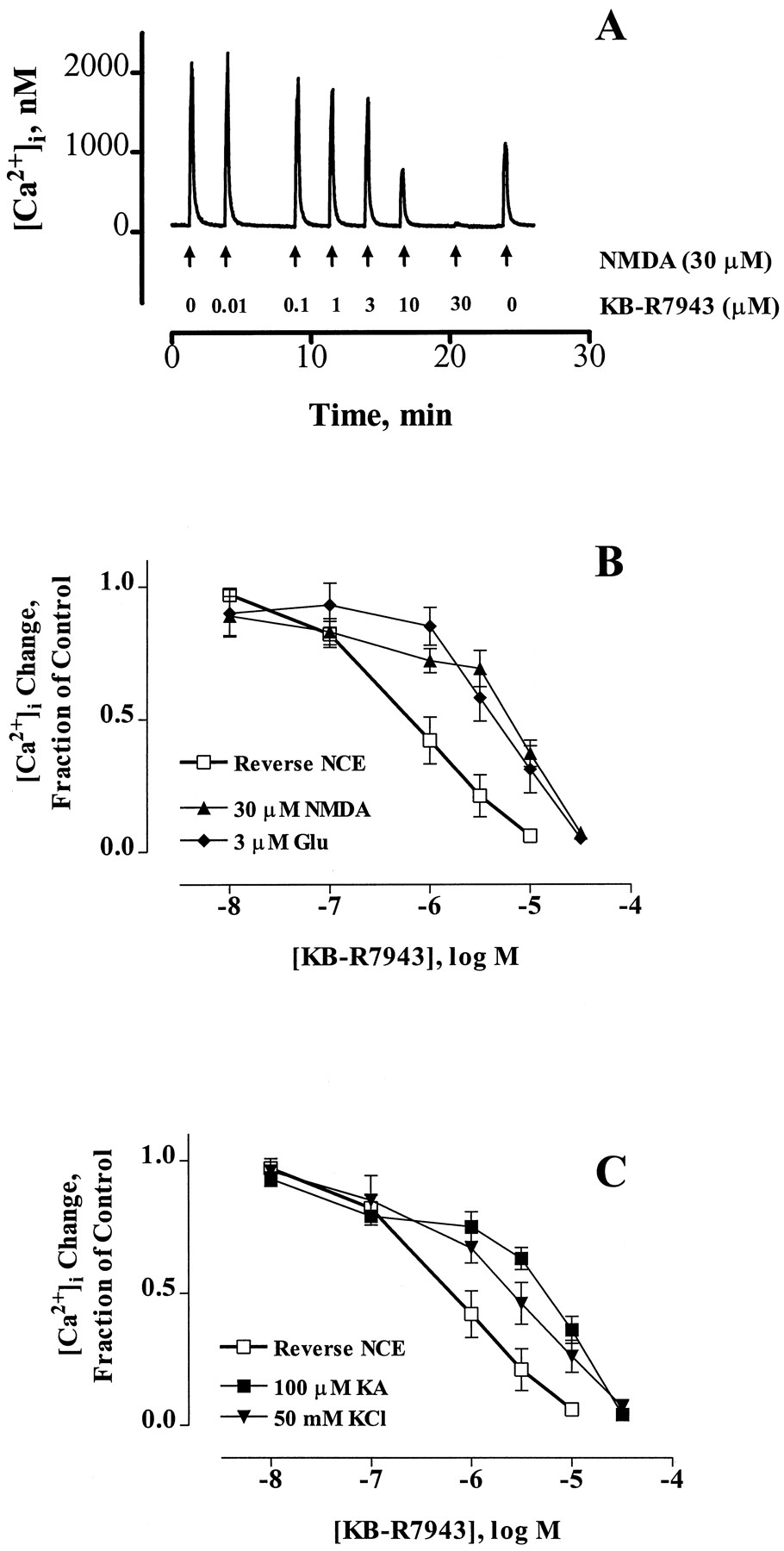
To assay the putative activity of KB-R7943, we first established a paradigm to selectively activate Ca2+ influx into indo-1-loaded neurons via reverse NCE. This paradigm is illustrated in Fig. 1A. Neurons were loaded with intracellular Na+ in the absence of extracellular Ca2+ using NMDA receptor activation (30 μm NMDA/1 μm glycine for 1 min) (Stoutet al., 1996). NMDA was removed, and the cells were washed with Ca2+-free HBSS for 2 min. The subsequent reintroduction of extracellular Ca2+ and the removal of extracellular Na+ (replaced withN-methyl-d-glucamine) resulted in a large increase in [Ca2+]i that presumably was the result of reverse NCE. This influx was inhibited effectively by treatment with 1 μm KB-R7943 (Fig. 1A). We tested the effects of increasing concentrations of KB-R7943 (0.01–10 μm) on this reverse NCE paradigm (Fig. 1B), and these data are summarized in Fig. 1C. The effect of KB-R7943 was partially reversible after washout of the drug at the end of the experiment (Fig.1B). The calculated IC50 value for inhibition of reverse NCE by KB-R7943 was 0.70 ± 0.08 μm, which is comparable to values obtained in cardiomyocytes (Iwamoto et al., 1996). We did not compensate for the partial reversibility of KB-R7943 in calculation of the IC50 because it is a relatively small effect. In addition, a similar degree of inhibition and reversibility was seen in cells that were treated with only 10 μm KB-R7943, which indicates that the inhibition is unlikely to result from cumulative incomplete reversal.

KB-R7943 blocks the reverse mode of the NCE in intact cultured forebrain neurons. A, In this [Ca2+]i trace from an indo-1-loaded cultured neuron, reverse NCE was activated as follows. Neurons were exposed to Ca2+-free HBSS (+20 μm EGTA) for 30 sec and then 30 μm NMDA/1 μm glycine in Ca2+-free HBSS for 1 min to load the neurons with Na+, followed by a 2-min wash with Ca2+-free HBSS to remove agonist. Then, Na+-free HBSS (with 1.4 mm Ca2+) was added for 1.5 min. The removal of extracellular Na+ and reintroduction of Ca2+ led to a large Ca2+influx via reverse-mode NCE. The addition of KB-R7943 (1 μm) for 30 sec before and during the Na+removal caused inhibition of the Ca2+ influx caused by activation of reverse NCE. B, Effect of increasing concentrations of KB-R7943 on reverse NCE. Reverse NCE was activated exactly as shown in A, and the effect of 0.01–30 μm KB-R7943 was tested on this Ca2+ influx. The effect of KB-R7943 was not irreversible, as shown by the last untreated Ca2+ peak caused by reverse NCE. C, Summary of the effects of KB-R7943 on reverse NCE represented as the mean ± standard error of 8–10 neurons from at least three different culture dates. Data are expressed as the fraction of the untreated Ca2+ response.
We then tested the effect of KB-R7943 on the forward mode of NCE. Because it has been demonstrated that forward NCE is responsible, in part, for the recovery of [Ca2+]i after a glutamate stimulus (White and Reynolds, 1995), we tested the effect of KB-R7943 on this recovery. In this series of experiments (Fig.2), we exposed neurons to two short (15-sec) pulses of 3 μm glutamate and manipulated the recovery during the 2 min immediately after the second glutamate stimulus. Fig. 2A is a control that shows that the responses are reproducible within a given cell. Fig. 2B shows that removal of extracellular Na+ (to inhibit forward NCE) significantly slows Ca2+ recovery, which is in agreement with our previous results (White and Reynolds, 1995). A drug that inhibits forward NCE would be expected to mimic the effects of Na+ removal in this paradigm. KB-R7943 (30 μm) did tend to slow Ca2+ recovery (Fig. 2C) but not to the same degree as Na+removal, suggesting that KB-R7943 is not as an effective inhibitor of forward NCE as Na+ removal at the concentration tested. These data are summarized in Fig. 2D, where the recovery time is expressed as time to twice basal [Ca2+]i levels. The limited inhibition of forward NCE observed with a relatively high concentration of KB-R7943 does not allow us to determine the precise ratio of the potency of KB-R7943 for the forward compared with the reverse mode because we cannot determine an IC50value for the forward mode.

Effect of KB-R7943 on recovery from a glutamate-induced [Ca2+]i increase as a measure of its effect on forward NCE. Indo-1-loaded neurons were exposed to two 15-sec stimulations with 3 μm glutamate/1 μm glycine performed 15 min apart. The first glutamate exposure serves as an untreated control for comparison purposes. All experimental manipulations were made during the 2 min immediately after glutamate removal. Neurons were treated with (A) HBSS control, (B) Na+-free HBSS, or (C) KB-R7943 (30 μm) after glutamate removal. D, Time to twice basal [Ca2+]i (♦) after glutamate removal. Although KB-R7943 (C) was able to delay the recovery from a glutamate-induced Ca2+ load, it is not nearly as effective an inhibitor of forward NCE as is Na+ removal. Values are from three or four neurons/condition from at least three different culture dates. ∗, Statistically different from untreated control (p < 0.05, analysis of variance followed by Dunnett’s multiple comparison test).
We next sought to determine whether stimuli in normal HBSS result in Ca2+ influx that is partially or wholly mediated through reverse NCE. We tested the effect of KB-R7943 on NMDA-, non-NMDA-, and depolarization-induced [Ca2+]i increases (Fig.3). Fig. 3A shows a representative [Ca2+]i trace from a neuron exposed repeatedly to 30 μm NMDA/1 μm glycine for 15 sec (in HBSS containing Na+ and Ca2+). Pretreatment with increasing concentrations of KB-R7943 (0.01–30 μm) results in inhibition of these NMDA-induced [Ca2+]i increases, and the inhibitory effect of KB-R7943 is partially reversible after washout of drug at the end of the experiment. These data are summarized in Fig.3B, which also shows that KB-R7943 has a similar inhibitory effect if 3 μm glutamate is used as the [Ca2+]i-increasing agonist. This concentration of glutamate causes a Ca2+ influx that is mostly mediated by NMDA receptor activation (Rajdev and Reynolds, 1994). Fig. 3C shows the effect of KB-R7943 on KA-induced (100 μm) and depolarization-induced (50 mm KCl) [Ca2+]i increase when tested in the same paradigm as Fig. 3A. Overall, KB-R7943 inhibited 30 μm NMDA-, 3 μm glutamate-, 100 μm KA-, and 50 mm KCl-induced [Ca2+]i increases with IC50 values of 8.2 ± 1.7, 5.2 ± 0.6, 6.6 ± 1.7, and 2.9 ± 0.5 μm, respectively, which suggests that a significant component of the acute [Ca2+]i change in response to each of these stimuli occurs via reverse NCE.

KB-R7943 inhibits acute [Ca2+]i increases caused by NMDA and non-NMDA receptor activation and by depolarization. A, Representative [Ca2+]i trace from a neuron treated with 30 μm NMDA/1 μm glycine (in Na+- and Ca2+-containing HBSS) for 15 sec (arrows). The neuron was pretreated with the indicated concentration of KB-R7943 (0.01–30 μm) for 30 sec before agonist exposure. B and C, Summary of data collected as in A using 15-sec exposure to the indicated agonist for 15 sec [i.e., NMDA, glutamate (Glu), KA, or KCl] at the indicated concentration. The effect of KB-R7943 on reverse NCE (from Fig. 1) is plotted here for comparison purposes. Data represent the mean ± standard error of 4–11 neurons from at least three different culture dates and represent [Ca2+]i changes normalized to the untreated control.
The effects of KB-R7943 on reverse NCE were established under conditions in which the agonist had been washed out, so direct receptor inhibition could not reasonably account for the effects seen. However, when the inhibitor was applied in normal buffer, direct inhibition of glutamate receptors could underlie the inhibition of Ca2+ transients. To address this possibility, we tested the effect of this drug on whole-cell currents activated by NMDA or KA (Fig. 4). KB-R7943 (30 μm), at a concentration that almost completely inhibited agonist-induced Ca2+ transients, was added to neurons in the continuous presence of either KA (30 μm; Fig. 4A) or NMDA (30 μm; Fig. 4B). It is apparent from these representative traces that KB-R7943 does not appreciably inhibit these currents. For KA-induced currents, 10 or 30 μmKB-R7943 potentiated the response by 7.6 ± 2.5% (p = 0.02, significantly different from untreated control, Student’s t test; seven neurons in total) or 1.8 ± 0.8% (p > 0.05, not different from untreated control, Student’s t test; five neurons/condition), respectively. For NMDA-induced currents, 10 or 30 μm KB-R7943 did not significantly alter responses [decreased by 1.9 ± 1.6% (seven neurons/condition) or 2.4 ± 0.9% (three neurons/condition) respectively, p > 0.05, not different from untreated control, Student’s ttest]. These results suggest that direct receptor inhibition is not responsible for the effects of KB-R7943 on [Ca2+]i increases caused by NMDA or KA receptor activation.

Effects of KB-R7943 on KA- or NMDA-induced whole-cell currents and voltage-sensitive Ca2+ channels. Neurons were voltage-clamped at −60 mV and continuously exposed to (A) 30 μm KA or (B) 30 μm NMDA in the absence or presence of 30 μm KB-R7943 during the indicated time. KB-R7943 did not substantially affect either current. Voltage-sensitive Ca2+ currents (C) were activated by step depolarization from −80 to 0 mV. Exposure of neurons to 10 μm KB-R7943 resulted in a small, but statistically significant, inhibition of this Ca2+ current. However, this inhibitory effect on voltage-sensitive Ca2+ currents is not sufficient in magnitude to account for the substantial inhibitory effects of KB-R7943 on agonist-induced increases in [Ca2+]i. Data are representative of results obtained from 3–12 cells for each condition.
Another important Ca2+ influx pathway is the activation of voltage-sensitive Ca2+ channels. KB-R7943, at relatively high concentrations, has been shown previously to inhibit these channels in heart cells (35% inhibition at 30 μm) (Iwamoto et al., 1996). Such an inhibitory effect, if present, potentially could account for the inhibitory effect of KB-R7943 on the glutamate- or depolarization-induced [Ca2+]i changes. We therefore tested the effect of KB-R7943 on neuronal voltage-sensitive Ca2+ currents (Fig. 4C). Whole-cell voltage-sensitive Ca2+ currents evoked by a depolarizing step from −80 to 0 mV were inhibited 7.7 ± 2.4% by 10 μm KB-R7943 (Fig. 4C; p < 0.05, significantly different from untreated control, Student’s ttest; six neurons/condition). A higher concentration of KB-R7943 (30 μm) inhibited currents by 24.3 ± 1.1% (p < 0.05, significantly different from untreated control, Student’s t test; 12 neurons/condition). Clearly, although KB-R7943 did inhibit voltage-sensitive Ca2+ channels, the magnitude of this inhibition is not sufficient to explain the much more substantial inhibitory effect of KB-R7943 on [Ca2+]i increases caused by glutamate receptor activation or depolarization (see Fig. 3).
Additional predictions can be made based on the proposed action of KB-R7943. Previous work has shown that exposure of neurons to Na+-free HBSS for 15 min results in the depletion of intracellular Na+ (Stout et al., 1996). Reverse NCE should be inactive under conditions of low [Na+]i. However, KA still can increase [Ca2+]i in the absence of Na+ because of the presence of Ca2+-permeable KA receptors in these cells (Hoytet al., 1995). This KA-induced [Ca2+]i increase in the absence of Na+, although substantial (2.1 ± 0. 8 μm, four neurons/condition), is significantly smaller than KA-induced [Ca2+]i increases in the presence of Na+ (6.6 ± 1.3 μm; p < 0.05, Student’s ttest; five neurons/condition). Fig. 5A shows that KB-R7943 (10 μm) does not inhibit KA-induced [Ca2+]i increases in neurons that have been depleted of intracellular Na+. In fact, KB-R7943 potentiated this response (148 ± 16% of untreated control; p = 0.03, significantly different from control, Student’s t test; six neurons/condition). These data further support that the action of KB-R7943 is on reverse NCE and not a direct inhibitory effect on receptor activation. Also, these results suggest that KB-R7943 itself does not directly interfere with the ability of indo-1 to record [Ca2+]i in our cells.
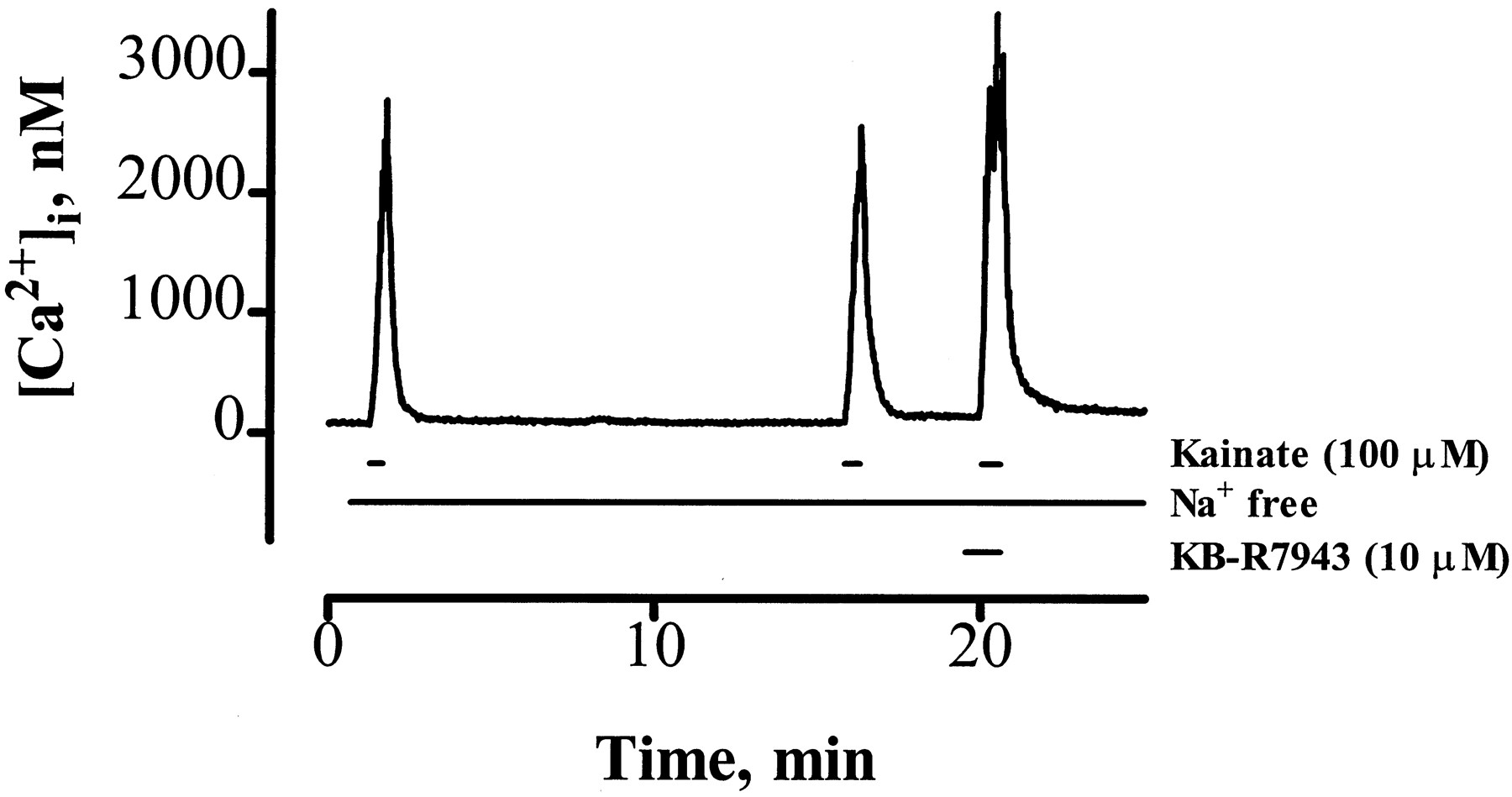
KB-R7943 potentiates KA-induced [Ca2+]i increases in neurons depleted of intracellular Na+. In this representative [Ca2+]i trace, neurons were pretreated with Na+-free HBSS for 30 sec before exposure to KA (100 μm) in Na+-free HBSS for 30 sec and then washed with Na+-free HBSS for 15 min to deplete intracellular Na+. After Na+ depletion, neurons again were exposed to KA in Na+-free HBSS. Finally, the neuron was exposed to 10 μm KB-R7943 in Na+-free HBSS for 30 sec, followed by 100 μmKA plus 10 μm KB-R7943 in the continued absence of Na+. KB-R7943 did not inhibit, and in fact potentiated, this KA-induced [Ca2+]i response, suggesting that KB-R7943 is indeed inhibiting reverse NCE and not simply inhibiting receptor activation. Data are representative of six neurons from three different culture dates.
We next looked at the effect of KB-R7943 on a higher concentration of glutamate to evaluate the contribution of reverse NCE to excitotoxic Ca2+ loads. KB-R7943 (10 μm, 30-sec pretreatment and included with agonist) inhibited the peak [Ca2+]i increase caused by 100 μm glutamate/1 μm glycine for 15 sec (41.0 ± 6.6% of untreated control; Fig.6A; five neurons/condition), and this effect was partially reversible. The time required for [Ca2+]i to return to twice basal levels after 100 μm glutamate (for 15 sec) removal was significantly decreased by KB-R7943 treatment (recovery time, 23.0 ± 5.6% compared with untreated control recovery time; five neurons/condition), which is consistent with a decreased [Ca2+]i load. We also investigated the effect of KB-R7943 on the [Ca2+]i changes caused by longer glutamate exposures. For these experiments, we used mag-Fura-2 (a low Ca2+ affinity [Ca2+]i indicator) as an indication of [Ca2+]iinstead of indo-1 because these prolonged glutamate exposures caused [Ca2+]i increases that frequently saturated the indo-1 signal. Fig. 6B demonstrates the effect of a low concentration of glutamate (3 μm for 15 sec, for comparison purposes) followed 15 min later by a 5-min exposure to 100 μm glutamate. The effect of KB-R7943 (10 μm) on this prolonged glutamate response is shown in Fig.6C. It is evident that although there is an initial attenuation of the [Ca2+]i response immediately after the addition of 100 μm glutamate in the presence of KB-R7943 (the ratio change at 15 sec is 69.2 ± 7.9% of the corresponding 3 μm glutamate response in the same cell), the peak ratio change during the 5-min exposure is not different from untreated control (0.24 ± 0.08 ratio units for untreated control versus 0.38 ± 0.11 ratio units for KB-R7943-treated neurons, eight neurons/condition). Also, the recovery time (to basal ratio) after removal of 100 μm glutamate was not significantly different in KB-R7943-treated neurons compared with untreated controls (KB-R7943-treated, 674.8 ± 43.1 sec; untreated controls, 787.7 ± 99.3 sec; eight neurons/condition). Finally, we considered whether KB-R7943 could protect neurons from glutamate excitotoxicity. We found that 10 μm KB-R7943 was not neurotoxic alone (88.2 ± 10.4% viability, four neurons/condition) and did not protect neurons from the cell death caused by 100 μm glutamate for 10 min [resulting in 53.7 ± 5.2% viability caused by glutamate alone compared with 46.0 ± 5.3% viability in the presence of KB-R7943, 20 hr after exposure (four neurons/condition) measured by retention of the vital dye calcein (Bozyczko-Coyne et al., 1993)]. Also, activation of reverse NCE (identical to the paradigm described in Fig.1) did not induce neuronal death (103 ± 5.8% viability, three neurons/condition) when measured 20 hr after exposure. These data suggest that although KB-R7943 is an effective inhibitor of short glutamate exposures, its inhibitory effect eventually is overcome by longer agonist exposure because the contribution made by direct Ca2+ influx through NMDA receptors [the receptor subtype predominately activated by glutamate (Rajdev and Reynolds, 1994) and responsible for glutamate neurotoxicity (Choi et al., 1987)] ultimately is more substantial than that meditated by reverse NCE.
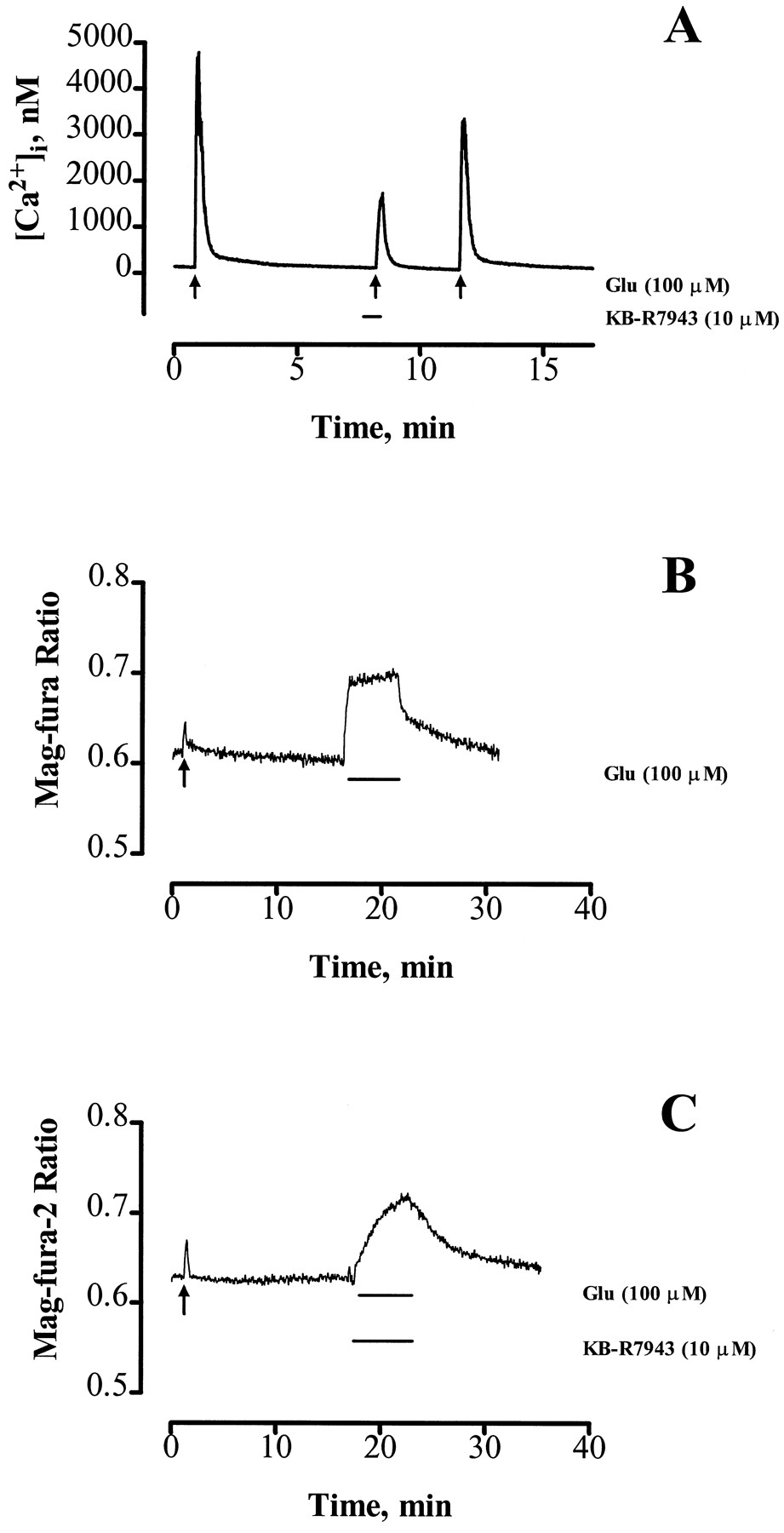
Effect of KB-R7943 on increases in [Ca2+]i caused by acute (15 sec) or prolonged (5 min) exposures to toxic concentrations of glutamate (100 μm). A, Indo-1-loaded neurons were exposed to 100 μm glutamate for 15 sec in the presence or absence of 10 μm KB-R7943 as indicated. KB-R7943 greatly inhibited this glutamate-induced [Ca2+]i response. B and C, In these representative traces from mag-Fura-2-loaded neurons, the cells initially were treated for 15 sec with a low concentration of glutamate (3 μm for 15 sec,arrow) and 15 min later were exposed to 100 μm glutamate for 5 min in the absence (B) or presence (C) of 10 μm KB-R7943. KB-R7943 (10 μm) attenuated initial increases in [Ca2+]icaused by 100 μm glutamate but did not affect the peak [Ca2+]i increase during the 5-min exposure. Data are representative of five to eight neurons from at least three different cultures.
Discussion
The findings described in this study show that reverse operation of the NCE contributes substantially to the increase in [Ca2+]i caused by glutamate receptor activation in central neurons in the presence of physiological extracellular [Na+] and extracellular [Ca2+]. It has been hypothesized previously that reverse NCE contributes to these changes in [Ca2+]i, but it has been difficult to test this hypothesis rigorously given the lack of specific inhibitors. The use of the selective inhibitor of reverse NCE KB-R7943 allowed us to evaluate the role of this exchanger in the [Ca2+]i changes caused by glutamate receptor activation. That KB-R7943 is an inhibitor of reverse NCE has been established in studies with cardiomyocytes (Iwamotoet al., 1996). We confirmed this action in cultured neurons (Fig. 1) and also demonstrated that it is not an effective inhibitor of forward NCE (Fig. 2) in these cells. Our observation that the initial [Ca2+]i change is completely blocked by KB-R7943 is surprising and argues that the [Ca2+]i change does not predominantly arise from direct Ca2+ entry. It is possible that there also is an effect related to local [Ca2+]i changes within cells, so the reverse NCE pathway may be more prominent in soma than dendrites. However, we cannot resolve this effect with the techniques used in this study.
Because KB-R7943 was such an effective inhibitor of NMDA-, non-NMDA-, and depolarization-induced increases in [Ca2+]i, we were concerned that KB-R7943 may have inhibitory effects on receptor function independent of effects on reverse NCE. However, this does not seem to be the case because KB-R7943 did not inhibit NMDA- or KA-induced whole-cell currents. The KA-induced whole-cell current was slightly, but significantly, increased by KB-R7943. In addition, KB-R7943 potentiated KA-induced [Ca2+]i increases (mediated by Ca2+-permeable (±)-α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid/KA receptors) when cells were depleted of intracellular Na+. It is unclear whether the small potentiation of KA-induced currents and the larger potentiation of KA-induced [Ca2+]i increases in the absence of [Na+]i are related because the mechanism of this potentiation is not understood at this point.
We were concerned in the case of KA- and depolarization-induced Ca2+ increases that the inhibitory effect of KB-R7943 involved inhibition of voltage-sensitive Ca2+ channels because KB-R7943 can inhibit these channels in cardiomyocytes at relatively high concentrations (35% inhibition at 30 μm) (Iwamoto et al., 1996). When we tested the effect of KB-R7943 on neuronal voltage-sensitive Ca2+ currents, we found that it does have a relatively small inhibitory effect. The magnitude of this inhibition (e.g., 24% at 30 μm KB-R7943), however, cannot account for the nearly complete blockade of [Ca2+]i increases we demonstrated, further strengthening the conclusion that the action of KB-R7943 on glutamate receptor-mediated or depolarization-induced [Ca2+]i increases is inhibition of reverse NCE. Other potential [Ca2+]i regulatory mechanisms that could be affected by KB-R7943 include Ca2+-induced Ca2+ release and the mitochondrial NCE. We previously investigated the contribution of Ca2+-induced Ca2+release to glutamate-induced Ca2+ responses in our cell culture preparation (Reynolds, 1996). This process seems to make, at best, a small contribution to our responses and thus is unlikely to be the site of action of KB-R7943. We also do not believe that KB-R7943 inhibits mitochondrial NCE. We have shown previously that a drug (CGP-37157) that blocks mitochondrial NCE causes a rapid recovery from glutamate-induced Ca2+ increases due to blockade of mitochondrial Ca2+ efflux, which contributes to the Ca2+ measured during recovery (White and Reynolds, 1997). KB-R7943 had little effect on Ca2+ recovery (Fig. 2). Also, if KB-R7943 were blocking mitochondrial NCE, one would expect a rise in Ca2+ as the KB-R7943 is washed out, reflecting efflux of Ca2+ stored in the mitochondria. We never saw such an effect of KB-R7943.
When we extended the exposure time to glutamate, we found that although the initial [Ca2+]i increase was inhibited by KB-R7943, the ultimate levels to which [Ca2+]i rose during the longer glutamate exposure were not different from those of untreated controls. Also, the time for the cell to recover from the longer glutamate exposure was not shortened by including KB-R7943 with the glutamate, suggesting that the glutamate imposed a quantitatively similar [Ca2+]i load on the cell. For these prolonged glutamate exposures, we used magfura-2 instead of indo-1 because the indo-1 signal often became saturated during the stimulus. The use of magfura-2 allows us to more reliably estimate high [Ca2+]ibecause it has a lower affinity for Ca2+ than does indo-1 (Grynkiewicz et al., 1985; Raju et al., 1989; Hurley et al., 1992). Magfura-2 also detects [Mg2+]i, which has been reported to increase during glutamate exposure, but this Mg2+ increase mainly is dependent on Ca2+ influx (Rajdev and Reynolds, 1995) and therefore is an additional indication of the magnitude of Ca2+ increase in these cells. Overall, these data lead us to conclude that although reverse NCE is a prominent route of Ca2+ influx during short glutamate exposures, other routes of Ca2+ entry or Ca2+ buffering become more important as the stimulus duration increases, possibly because as Ca2+ accumulates in the cytoplasm, the gradient for Ca2+ influx through the NCE decreases or Ca2+ buffering simply becomes overwhelmed. This conclusion is supported by the recent report that exposure of neurons to glutamate for 3–5 min ultimately leads to an inhibition of reverse NCE when measured after glutamate removal (Yu and Choi, 1997).
Consistent with the finding that KB-R7943 does not block glutamate-induced [Ca2+]iincreases for the duration of a prolonged glutamate exposure, we found that KB-R7943 was not an effective inhibitor of glutamate-induced neuronal death. This suggests that the Ca2+influx through the NCE is not a substantial contributor to the Ca2+ load that ultimately leads to induction of cell death. Simple activation of reverse NCE also did not kill cells, supporting this conclusion. Therefore, the primary source of Ca2+ that causes neuronal death must be excessive influx through glutamate-activated channels.
KB-R7943 was somewhat more potent as an inhibitor of the reverse NCE paradigm (Fig. 1) than as a blocker of NMDA-, non-NMDA-, or depolarization-induced [Ca2+]i increases (Fig.3). The basis for the difference in potency between assay paradigms is not entirely clear. There clearly is a marked difference in the [Na+]i at the point of drug addition. There also may be a difference in the maximal [Na+]i reached during the different paradigms, as well as a differing degree of depolarization. Finally, in the reverse NCE paradigm, the [Ca2+]e is much lower when the inhibitor is first applied to the cells. If KB-R7943 inhibits reverse NCE by binding to the extracellular Ca2+recognition site, as proposed by Watano et al. (1996), the latter difference between the two assays could account for the difference in potency.
In summary, we conclude that reverse NCE is an important contributor to the early [Ca2+]iincrease caused by NMDA and non-NMDA receptor activation. KB-R7943 inhibited [Ca2+]iincreases caused by acute, but not prolonged, glutamate receptor activation and correspondingly did not inhibit glutamate-induced cell death. These findings suggest that although reverse NCE certainly is involved in some of the increase in [Ca2+]i caused by glutamate receptor activation, its role may be limited to short, more physiological stimuli and not prolonged pathophysiological ones.
Acknowledgments
We thank Kristi Rothermund and Chialin Cheng for preparing the neuronal cultures, Heather Raphael and Dr. Amy Stout for assistance with toxicity experiments, and Jim Dilmore for his generous assistance with recording voltage-sensitive Ca2+ currents. KB-R7943 was a generous gift of Dr. Tomokazu Watano (Kanebo, Osaka, Japan).
Footnotes
- Received September 11, 1997.
- Accepted December 12, 1997.
-
Send reprint requests to: Ian J. Reynolds, Ph.D., Department of Pharmacology, University of Pittsburgh School of Med., E1354 Biomedical Science Tower, Pittsburgh, PA 15261. E-mail:ijr{at}prophet.pharm.pitt.edu
-
This work was supported by National Institutes of Health Grants NS34138 and NS29365 and by the American Heart Association. S.R.A. is supported by National Institutes of Health Training Grant MH18273. I.J.R. is an Established Investigator of the American Heart Association.
Abbreviations
- NMDA
- N-methyl-d-aspartate
- NCE
- plasma membrane Na+/Ca2+ exchanger
- DMEM
- Dulbecco’s modified Eagle’s medium
- MEM
- minimal essential medium
- HBSS
- HEPES-buffered salt solution
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- DMEM
- Dulbecco’s modified Eagle’s medium
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- AM
- acetoxymethyl ester
- KA
- kainate
- [Na+]i
- intracellular Na+ concentration
- [Ca2+]i
- intracellular Ca2+ concentration
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}