


Abstract
β1- and β3-adrenergic receptors (AR) are the predominant β-AR subtypes in adipocytes, and analysis of native and recombinant β-AR has revealed several pharmacological and biochemical differences between these subtypes. This study used chimeric and mutated rat β-AR expressed in Chinese hamster ovary cells to examine the basis of certain characteristic differences in the agonist properties of catecholamines and prototypic β3-AR agonists. The exchange of sequence beyond transmembrane (TM) region 6 between the β-AR subtypes had dramatic and reciprocal effects on the affinity and efficacy of the prototypic β3-AR agonists BRL 37,344 and CL 316,243, without affecting the interactions with catecholamines. Mutation of Phe350 and Phe351 in TM7 of the β1-AR to Ala and Leu found in the β3-AR was sufficient to allow activation by prototypic β3-AR agonists. Interestingly, this mutation did not affect catecholamine action and it did not impair the ability of propranolol to block the actions of isoproterenol or the selective β3-AR agonists. β1-AR containing β3-AR sequence from predicted TM5 through TM6 exhibited reduced affinity for catecholamines without altering agonist potency, suggesting enhanced coupling efficiency. Inclusion of the homologous β1-AR sequence in the β3-AR, however, did not produce reciprocal effects. These results are the first to define a major determinant of β3-AR subtype-selective agonism in TM7 and demonstrate that the determinants of selective phenethanolamines, catecholamines, and propranolol action are distinct.
β1- and β3-AR are the predominant β-AR subtypes in adipocytes, and analysis of these subtypes has revealed several important pharmacological and biochemical differences [for review, see Granneman (1995)]. For example, catecholamines, the natural ligands of these receptors, exhibit a much higher binding affinity for β1-AR. β3-AR have a relatively low affinity for catecholamines, yet that low affinity is largely compensated by a much higher degree of coupling efficiency. Thus, β3-AR fully activate adenylyl cyclase when relatively few receptors are occupied, as indicated by the large difference between binding Kd and EC50 values for cAMP accumulation.
In addition, β3-AR, which are highly expressed in adipose tissues, have received considerable attention as a target for antiobesity and antidiabetes therapeutics (Granneman, 1995). In this regard, several substituted phenethanolamines have been described that potently activate the β3-AR, yet have little or no activity at β1-AR. These agents have proven to be very effective in animal models of obesity and diabetes, owing to their action on adipocyte β3-AR (Bloom et al., 1992; Susulicet al., 1995; Grujic et al., 1997).
The structural bases for these characteristic pharmacological differences are not completely known. Chimeric receptors have been used successfully to explore the structure/function relationships among adrenergic receptor subtypes. Previous analyses of the β3-AR subtype have used chimeras with β2-AR and have focused largely upon analysis of receptor desensitization and sequestration (Liggett et al., 1993; Nantel et al., 1993; Jockers et al., 1996). Although certain differences between the “atypical” β3-AR and the “typical” β1- and β2-AR have been noted (Granneman, 1992; Liggett et al., 1993; Nantelet al., 1993; Chaudhry and Granneman, 1994; Jockers et al., 1996), certain functional differences among these subtypes are unique or more robust between β1- and β3-AR subtypes. Therefore, we have performed direct comparisons between rat β1- and β3-AR and among chimeric and mutated receptors composed of these subtypes. These experiments have focused upon characteristic pharmacological properties of rat β1- and β3-AR subtypes, including catecholamine binding affinity, coupling to cAMP generation, and interaction with β3-AR-selective phenethanolamines.
Materials and Methods
Construction of chimeric and mutated receptors.
The cloning of the rat β1- and β3-AR cDNAs has been described (Grannemanet al., 1991, Chaudhry and Granneman, 1992). The first chimera was constructed using the technique of Moore and Blakely (1994). This construct (β1/β3-O3) contained amino acids encoded by the β1 receptor to Val334, at the end of TM region 6, followed by β3-AR sequence beginning at Leu311. The complementary chimera (β3/β1-O3) was made by polymerase chain reaction mutagenesis and encoded the β3-AR to Pro317, followed by β1-AR sequence beginning at Arg339. Chimeras that substituted a region from TM5 through TM6 (including the third intracellular loop, I3) were constructed from the above chimeras using the common BsmBI site to replace codons before β1-AR Ser232 in TM5 with β3-AR sequence to form β3(β1TM5–6), and β1 sequence to β3-AR Ser209 to form β1(β3TM5–6). The TM5–6 chimeras also contained a binding site for the M2 monoclonal antibody (IBI) on the amino terminus of the receptors. Epitope tagging had no effect on the pharmacological properties of native rat β1- and β3-AR. For the final construct, Phe350 and Phe351 of the β1-AR were changed to alanine and leucine found in the β3-AR by replacing the HindIII toBglI fragment with a synthetic double-stranded oligonucleotide encoding the desired mutation to form β1-F350A,F351L. All mutations and amplified sequences were verified by dideoxynucleotide sequencing.
Mammalian cell expression.
Constructs were cloned into the mammalian expression vector pRc/CMV or the closely related derivative pcDNA3 (Invitrogen). These vectors contain the cytomegalovirus promoter to drive expression and the neomycin resistance gene for selection of transformed cells. CHO-k1 cells were transfected by CaPO4 precipitation (Maniatis et al., 1982) or with the LipofectAMINE liposome reagent (Gibco/BRL). Clonal cells were obtained by dilution and screened by ligand binding and adenylyl cyclase activation (Granneman et al., 1993). Cell lines used in the study had similar levels of receptor expression (1–2 pmol/mg of protein) and adenylyl cyclase activation.
Radioligand binding.
Ligand binding was performed with125I-CYP (DuPont NEN, Boston, MA) as described previously (Emorine et al., 1989). Briefly, culture medium was removed and cells were washed in phosphate-buffered saline, then harvested in 25 mm HEPES, pH 8.0, buffer containing 2 mm MgCl2 and 1 mm EDTA. Cells were lysed and centrifuged at 48,000 × g for 15 min to obtain crude membranes. Membrane pellets were resuspended by homogenization and used directly, or frozen at −80° until used. Freezing did not affect binding. Membranes were resuspended in 75 mm Tris, pH 7.4, 12.5 mmMgCl2, 2 mm EDTA, and 1 mm ascorbic acid. Saturation analysis was performed with concentrations of 125I-CYP ranging from 65 pm to 4 nm, with 1 mm ISO used to define nonspecific binding. For competition studies, 100 μm desmethylimipramine was included in the incubation to reduce nonspecific binding (Emorine et al., 1989). Incubations were carried out in volume of 150 μl for 1 hr at 30°, and were terminated by vacuum filtration over glass fiber filters.Ki values were calculated from IC50 values that were determined by nonlinear regression analysis of three to six experiments, each performed in triplicate.
cAMP accumulation assay.
cAMP accumulation was performed in duplicate as previously described (Chaudhry et al., 1994). Briefly, cells grown in 24-well plates were washed two times in Ham’s F-12 medium containing 1 mm IBMX and 0.1 mmascorbic acid. After a 15-min preincubation in the above medium, cells were challenged with various agonists. Reactions were terminated after 30 min by the addition of perchloric or trichloroacetic acid. After neutralization, accumulated cAMP was determined by radioimmunoassay (Fransen and Krishna, 1976) or protein binding assay (Brown et al., 1971).
Adenylyl cyclase assay.
Membrane adenylyl cyclase assays were performed by a modification (Granneman et al., 1991) of the method of Salomon et al. (1974). Briefly, membranes (5–15 μg of protein) were preincubated at 4° in a volume of 40 μl with the specified drugs for 15 min. Adenylyl cyclase reactions were initiated by addition of 10 μl of substrate mix and terminated after 30 min at 30°.
Data analysis.
Ligand binding and adenylyl cyclase data were analyzed with Enzfitter software (Enzfitter; Elsevier Biosoft, Cambridge, UK). EC50 values in cAMP accumulation assays were determined graphically. Antagonist binding affinity (KB ) was determined according to the equation KB = [B]/[DR −1] where [B] is the antagonist concentration and the dose-ratio (DR) is the EC50 value of the agonist in the presence of antagonist divided by the control EC50 value. Values reported are mean ± standard error. Planned comparisons between means were evaluated with Bonferroni t test, with critical values of p< 0.05 (two-tailed) judged significant.
Results
The characteristic pharmacological properties of rat β1- and β3-AR that were evaluated are illustrated in Fig. 1. ISO, used as the reference catecholamine agonist, had a 100-fold higher affinity for β1- versus β3-AR in binding assays. Despite differences in binding afinity, ISO exhibited nearly similar potency in the stimulation of cAMP accumulation. Comparison of the binding and cAMP accumulation curves indicates that ISO fully activated adenylyl cyclase at concentrations that did not fully saturate the receptors. Although a discrepancy between Ki and EC50 values would be expected in cells expressing high levels of receptors (Wilson et al., 1996; Leby et al., 1993), the difference was at least 50 times greater for the β3-AR versus the β1-AR in cells expressing similar levels of receptors (3.4 log units versus 1.7, respectively). The high efficiency of β3-AR coupling to cAMP generation as indicated by large discrepancy between Ki and EC50 occurs at various levels of receptor expression and is present in cells that natively express the receptor (Granneman, 1995; Wilson et al., 1996). Thus, like the β2-AR (Leby et al., 1993), the β3-AR has a high degree of coupling efficiency that is not matched by the β1-AR (see alsoGreen and Liggett, 1994).
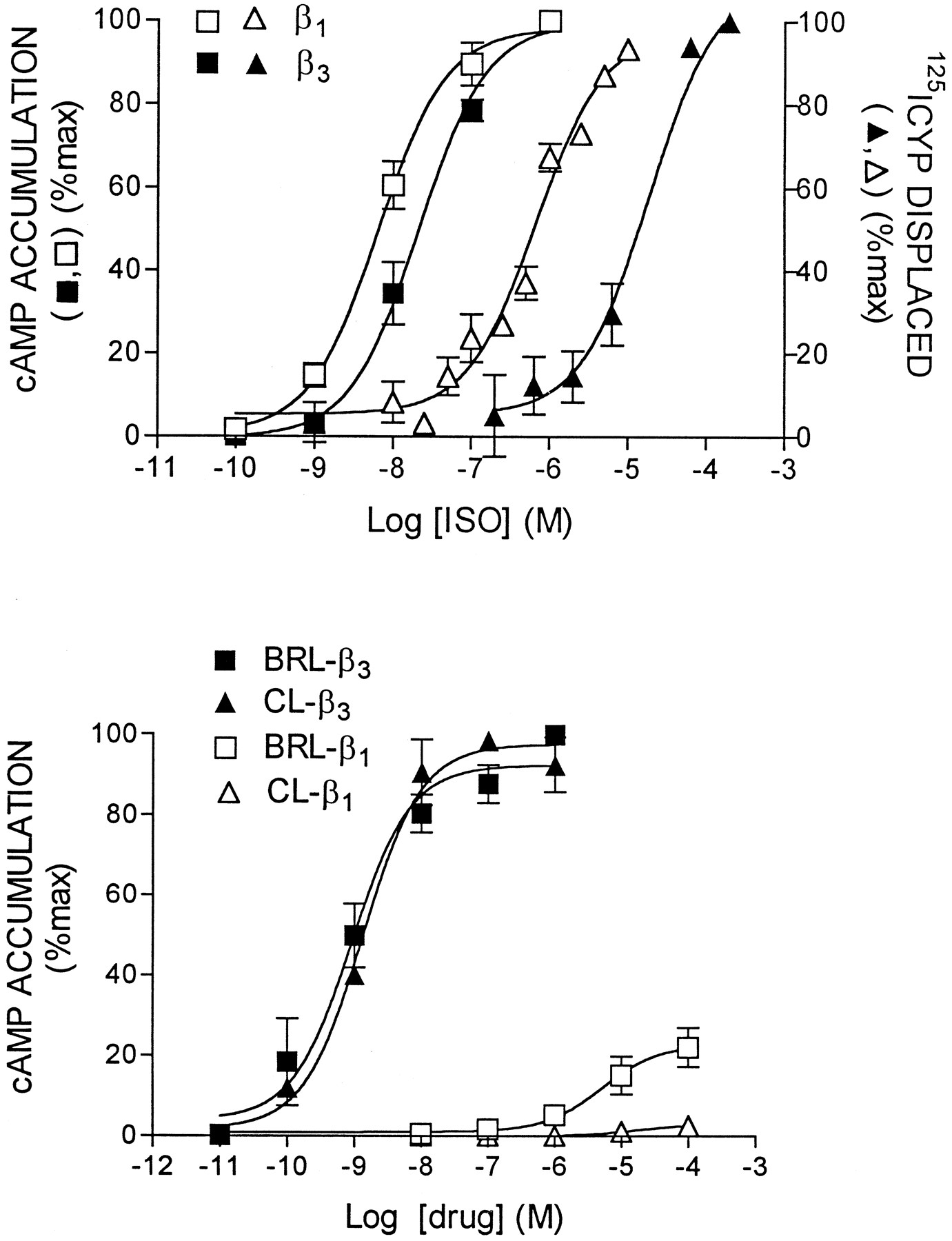
Pharmacological properties of rat β1- and β3-AR expressed in CHO cells. Top, Interaction of ISO with β1- and β3-AR in binding and cAMP accumulation assays. Competition assays were performed in the presence of 100 pm (β1-AR) or 500 pm (β3-AR) 125I-CYP.Bottom, Activation of cAMP accumulation by CL and BRL. Values are means ± standard error (3 experiments) and are given as a percentage of the maximal response elicited by ISO.
β3-AR have received attention as a therapeutic target for antiobesity agents and certain phenethanolamines have been synthesized that selectively bind and activate this receptor. CL and BRL are prototypic phenethanolamine agonists that bound the rat β3-AR with moderate affinity and activated the receptor with very high potency (Fig. 1, bottom). In contrast, BRL had moderate affinity for the β1-AR but exhibited little agonist activity. CL did not bind or activate the β1-AR at the concentrations employed.
The β-AR subtypes and chimeric receptors were characterized with various adrenergic agonists in ligand binding and cAMP accumulation assays, and the results are summarized below and in Table1.
Pharmacological properties of native and chimeric rat β1- and β3-AR expressed in CHO cells
Agonist binding affinity.
Exchange of the amino acids beyond TM6 had no effect on the binding of catecholamines. Thus, the β1/β3 O3 chimera retained high affinity for NE, ISO, and DO characteristic of β1-AR, whereas the β3/β1-O3 chimera retained low affinity characteristic of β3-AR. In sharp contrast, substitution of sequence beyond TM6 (importantly TM7) had dramatic and reciprocal effects on the affinity of β1- and β3-AR for the prototypic β3-AR agonists BRL and CL. The β1/β3-O3 chimera exhibited a much higher affinity for BRL (20-fold) and CL (>80-fold) compared with native β1-AR. The complementary chimera that substituted β1-AR sequence in the β3-AR had a 20-fold reduced affinity for BRL and a 125-fold lower affinity for CL.
Substitution of the region from TM5 through TM6 affected catecholamine binding in a receptor-specific fashion. Thus, the affinity of catecholamines for the β1(β3TM5–6) chimera was 5-fold lower than the affinity of catecholamines for the β1-AR (p < 0.05), whereas the affinity for prototypic β3-AR agonists was not altered. The difference in catecholamine binding affinity between β1-AR and β1(β3TM5–6) chimeras was observed in side-by-side assays and was not influenced by the presence of GTP (data not shown). In contrast, the β3(β1TM5–6) receptor was indistinguishable from the native β3-AR.
cAMP accumulation.
Catecholamines were equally potent in activating native β1 AR and the β1/β3-O3 chimera. In sharp contrast, this substitution profoundly affected the potency of β3-AR-selective ligands. Compared with β1-AR, the β1/β3-O3 chimera was 100 times more sensitive to the agonist actions of BRL, whereas the instrinsic activity relative to ISO was increased from 0.2 to 0.64 ± 0.11. The effects of the β3-O3 substitution on CL action were more dramatic, with agonist potency being increased by more than 2500 times. Indeed, CL was essentially inactive at the β1-AR, but was nearly as potent and efficacious (intrinsic activity = 0.96 ± 0.28) as norepinephrine in activating the β1/β3-O3 chimera.
Like the β1/β3-O3 chimera, β1-O3 replacement in the β3 receptor did not alter the potency of catecholamines. The magnitude of the effects of β1-O3 replacement on the action of the β3-AR-selective ligands, however, was greater than that seen with the β3-O3 substitution: the potency of BRL was reduced by 600 times and that of CL by more than 10,000 times (pEC50 5.7 versus 9.8 in β3-AR). The intrinsic activity of BRL was reduced to 0.69 ± 0.13, whereas CL remained a full agonist (intrinsic activity, 1.12 ± 0.18).
Substitution of the sequence from TM5 through TM6 had no significant impact on overall agonist potency. The potency of full agonists (ISO and norepinephrine) tended to be increased in β1(β3TM5–6) and, taking into account the reduced binding affinity, it seems that coupling efficiency (i.e.,Ki -EC50) was improved by 10–20-fold in these cells. With the exception of BRL, replacement of β1-AR TM5–6 in the β3-AR did not significantly affect agonist potency. Thus, the effects of TM5–6 substitutions on coupling efficiency did not seem to be reciprocal.
The data above strongly indicate that sequence beyond TM6 has dramatic and reciprocal effects on the affinity and efficacy of prototypic β3-AR agonists. Ligand binding is thought to involve interactions with TM regions (Strader et al., 1987,1988; Dixon et al., 1988; Strosberg et al., 1993). Alignment of the predicted TM7 (Fig.2) of the β1- and β3-AR indicates that 16/20 residues are identical, with the differences clustered near the beginning of TM7. Of these, two adjacent phenylalanines (Phe350 and Phe351) in the β1-AR have bulky aromatic side chains absent in alanine and leucine found in the homologous position of the β3-AR and thus could influence accessibility of ligands to the hydrophobic binding pocket. Therefore, the effects of changing Phe350 and Phe351 of the β1-AR to alanine and leucine were examined in ligand binding and cAMP accumulation assays.

Alignment of the seventh TM segment of the rat β1- and β3-AR.
With respect to catecholamines, β1-F350A,F351L receptors exhibited hallmark features of β1-AR, including relatively high binding affinity and low coupling efficiency to cAMP generation (Table 2). However, unlike β1-AR, the mutant receptor bound selective β3-AR agonists and was potently activated by them. Indeed, the β1-F350A,F351L mutant exhibited all of the features found in the β1/β3-O3 chimera, which contains the β3-AR sequence from all of TM7, as well as the nonconserved intracellular tail.
Pharmacological properties of β1-F350A,F351L mutant receptors
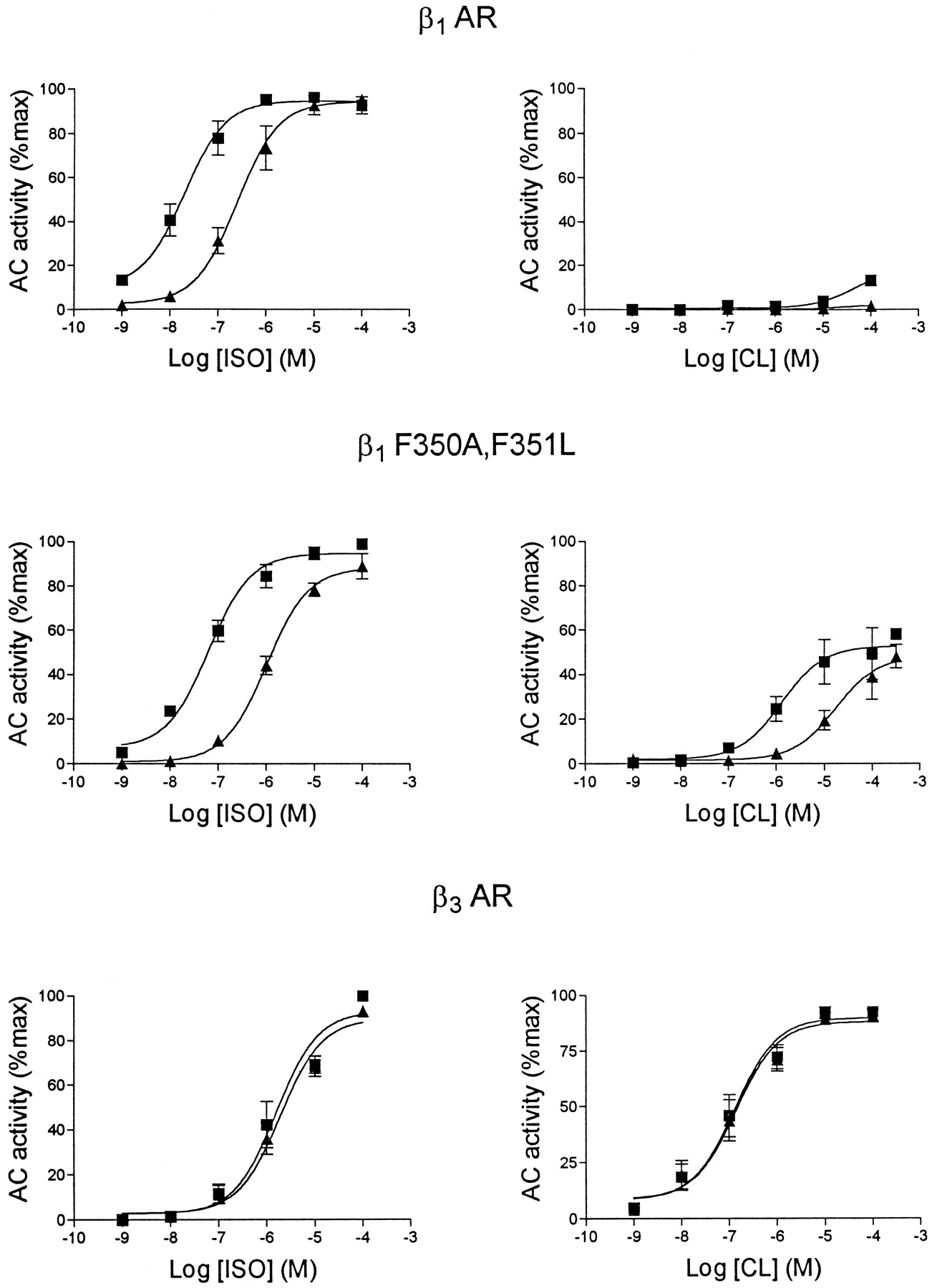
In addition to activation by CL, a hallmark feature of the β3-AR is its low affinity for typical β-AR antagonists such as propranolol. Therefore, the interaction of propranolol with β1-F350A,F351L receptors was examined in membrane adenylyl cyclase assays. For this purpose, ISO and CL concentration-response curves were generated in the absence and presence of a fixed concentration (100 nm) of (−)-propranolol (Fig. 3). As expected, ISO activated β1-AR with high potency and (−)-propranolol shifted the ISO concentration-response curve to the right. The affinity (KB ) of (−)-propranolol for β1-AR, calculated from the magnitude of the dextral shift (see Materials and Methods), was 7.0 ± 0.7 nm (three experiments). CL had virtually no effect on β1-AR in membrane adenylyl cyclase assays. In contrast, β3-AR were activated by CL, and by ISO with low potency. As expected, (−)-propranolol (100 nm) failed to significantly antagonize either agonist at the β3-AR. β1-F350A,F351L receptors were potently activated by ISO, and this activation was potently blocked by (−)-propranolol (KB = 6.1 ± 0.8 nm, three experiments). Unlike β1-AR, β1-F350A,F351L receptors were activated by CL. Nonetheless, this activation was potently blocked by (−)-propranolol with aKB of 10.6 ± 1.3 nm (three experiments).

Adenylyl cyclase activation of CHO cell membranes containing rat β1-AR (top), β3-AR (bottom), and β1-F350A,F351L (middle). Concentration-response curves to ISO and CL were generated in the absence (▪) and presence (▴) of 100 nm(−)-propranolol. Values are mean ± standard error (3 experiments).
Discussion
β1- and β3-AR have distinct pharmacological properties, yet relatively little molecular genetic analysis has been performed that examines the binding specificity of β3-AR. Guan et al.(1995) investigated the binding affinity of BRL in a series of chimera composed of human β2- and β3-AR. However, selectivity of BRL for β3-AR was modest (about 10-fold), and effects on agonist activity were not addressed. In the present work, a series of chimeric and mutated β1- and β3-AR subtypes was constructed to identify regions of these molecules that confer subtype-specific signaling properties. This work took advantage of the greater differences between β1- and β3-AR in affinity for BRL, as well as the greater selectivity of CL, which has not been previously examined.
The major finding of this analysis is that the binding of and activation by prototypic phenethanolamine β3-AR ligands was dramatically affected by alterations in TM7. Specifically, replacing β1-AR TM7 with sequence derived from the β3-AR conferred the ability of β3-AR-selective ligands to bind and activate the receptor. Conversely, replacing β3-AR TM7 with sequence derived from the β1-AR dramatically reduced the affinity and potency of β3-AR-selective agonists. It is important to note that these substitutions did not alter binding or activity of catecholamine agonists.
The effects of TM7 substitutions on β3-selective agonist potency (EC50) were far greater than could be accounted for by changes in binding affinity, indicating that this region is also critical for receptor activation by these ligands. In general, the effects of TM7 substitutions were greatest for CL, which exhibits the highest degree of selectivity for the β3-AR. The improvement in CL action in the β1/β3-O3 chimera is difficult to quantify because the compound was essentially inactive at the β1-AR. Nevertheless, CL potency increased by more than 2500-fold in this chimera. The differential effects of TM7 substitution on affinity and potency are perhaps best illustrated in the β3/β1-O3 chimera, in which CL potency was reduced 100 times more than binding affinity. Thus, although CL and ISO bound this chimera with equal affinity, the ability of CL to activate the receptor was two orders of magnitude less than ISO.
Site-directed mutagenesis demonstrated that substitution of Phe350 and Phe351 in TM7 to alanine and leucine, respectively, was sufficient to produce the phenotype seen with complete TM7 and carboxyl tail substitution. Like the β1/β3-O3 chimera, the actions of catecholamines at the β1-F350A,F351L mutant were indistinguishable from the native β1-AR. Similarly, the mutation did not affect the ability of (−)-propranolol to antagonize catecholamine or β3-AR agonists. Nonetheless, mutation of Phe350 and Phe351 of the β1-AR to residues found in the β3-AR was sufficient to improve the affinity and potency of CL by more than 100- and 1000-fold, respectively.
How might mutation of Phe350 and Phe351 permit binding and activation by β3-AR ligands without affecting the actions of catecholamines or propranolol? Binding depends upon direct interaction of the ligand with specific amino acids, as well as interactions among amino acids that form and stabilize the ligand binding pocket, but do not directly contact the ligand. Catecholamines are relatively small molecules whose binding is thought to be stabilized by specific interactions with amino acid side chains in TM3, TM4, and TM5 that are conserved in each of the β-AR subtypes (Straderet al., 1987, 1988; Dixon et al., 1988; Strosberget al., 1993; Blin et al., 1993). In contrast, ligands that selectively activate β3-AR contain bulky alkylamine chains that presumably permit subtype-selective interactions. Strosberg et al. (1993) have used molecular modeling to suggest that extended conformations of selective β3-AR ligands mediate agonist properties by contacting residues within the TM regions, including potential interactions with TM7. Based upon models of related receptors, Phe350 and Phe351 are predicted to be present near the beginning of TM7 in the β1-AR and Phe351 is likely to face the ligand binding core of the receptor (Mizobe et al., 1996, Baldwinet al., 1997). It is therefore possible that Phe350 and Phe351 prevent activation of the β1-AR by denying extended conformations of β3-AR-selective ligands access to the ligand binding groove. Alternatively, the interaction of Phe350 and Phe351 with residues in other TM regions might alter the binding pocket available to the phenethanolamine agonists and thereby restrict activation indirectly (Mizobe et al., 1996). Such an affect on the binding pocket, however, would seem to be subtle, because the actions of catecholamines and propranolol were not affected in the F350A,F351L mutant. It is also conceivable that β3-AR agonists interact directly with alanine and/or leucine in TM7, although additional interactions must be required because the β2-AR has a similar sequence to the β3-AR in this region (leucine in both positions), yet is not activated by CL (Bloom et al., 1992). In any event, the present results provide experimental support for a model in which the selectivity of prototypic β3-AR ligands is conferred by the ability of these compounds to access a binding pocket formed with TM7.
β1-and β3-AR can be distinguished by differences in coupling efficiency to cAMP generation (Leby et al., 1993; Green and Liggett, 1994; Granneman, 1995; Wilson et al., 1996). The high degree of coupling efficiency of β3-AR requires intact cells, occurs over a large range of receptor expression levels and is independent of cell background (Granneman, 1995; Wilson et al., 1996). As generation of cAMP requires the interaction of multiple proteins in the context of an intact cell, modest changes in agonist potency can be difficult to interpret. Previous work by Green and Liggett (1994) attributed the low degree of coupling efficiency of β1-AR to a proline-rich sequence in the third intracellular (I3) loop. Consistent with these results, replacement of TM5 through TM6 sequence with β3-AR sequence, which contains I3, seemed to improve coupling efficiency of the β1-AR. Indeed, the impact of TM5–6 replacement on coupling observed the present experiment (10–20-fold) was somewhat greater than that observed by Green and Liggett for the replacement of the proline-rich sequence alone, suggesting that differences in coupling efficiency involve the proline-rich region as well as additional sequences in I3 and perhaps TM5 and 6. Nevertheless, β1-AR TM5–6 replacement failed to consistently suppress the coupling efficiency of the β3-AR, indicating that this region alone is insufficient to account for subtype differences in coupling efficiency.
Several amino acids in the β2-AR have been identified that are crucial for high affinity binding of and activation by catecholamines, including Asp113, Ser204, Ser207, Phe290, and Tyr326 (Strader et al., 1987, 1988; Dixon et al., 1988). These amino acids are conserved in the β1- and β3-AR and thus other determinants must be important in explaining the 30–50-fold difference in catecholamine binding affinity between these subtypes. However, the present study did not clearly identify these regions. Although replacement in β1-AR of the sequence from TM5–6 with the β3-AR sequence reduced catecholamine binding affinity by about 5-fold, including the homologous region of the β1-AR did not increase affinity in the β3-AR. Given that the catecholamine binding pocket is thought to be formed by the juxtaposing of TM regions, it is likely that several amino acids in the TM regions indirectly influence affinity by affecting the alignment or stabilization of the residues that interact directly with catecholamines. Thus, although β3-AR substitutions disturb catecholamine binding in the context of the β1-AR, the converse does not seem to be true.
In summary, the present work demonstrates that residues in TM7 are critical in conferring subtype-specific activation by β3-AR-selective phenethanolamines. The ability of these residues to dramatically influence activation by prototypic β3-AR-selective agonists without affecting catecholamine or propranolol action demonstrates that the sites critical for these interactions are distinct.
Footnotes
-
Send reprint requests to: Dr. James Granneman, 2309 Scott Hall of Basic Medical Sciences, Wayne State University School of Medicine, Detroit, MI 48201. E-mail:jgranne{at}med.wayne.edu
-
This work was supported by United States Public Health Service Grant DK46339.
- Abbreviations:
- AR
- adrenergic receptors
- ISO
- (−)-isoproterenol
- CL
- CL 316,243
- BRL
- BRL 37,344
- CYP
- cyanopindolol
- TM
- transmembrane
- CHO
- Chinese hamster ovary
- NE
- (−)-norepinephrine
- DOB
- (±)-dobutamine
- Received November 11, 1997.
- Accepted January 27, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}