


Abstract
Chronic nicotine up-regulates the number of high affinity nicotinic acetylcholine receptors (nAChRs) in mammalian brain. Here, we studied up-regulation of the nAChR composed of α4 and β2 subunits in the M10 cell line by using [3H]epibatidine to measure nAChR in cells in situ and in membrane preparations. Cultures were exposed to drugs for 2 days before assay. All agonists up-regulated [3H]epibatidine binding sites with EC50 values typically 10–100-fold higher than their respective Ki values from competition binding assays. Maximum up-regulation ranged from 40% to 250% above control values. Maximally effective concentrations of the less efficacious agonists methylcarbamylcholine or (±)-epibatidine together with nicotine resulted in less up-regulation than that produced by nicotine alone, showing that they are partial up-regulatory agonists. The antagonists dihydro-β-erythroidine, methyllycaconitine, d-tubocurarine, hexamethonium, decamethonium, and mecamylamine either failed to up-regulate [3H]epibatidine binding sites or up-regulated mildly at high concentrations. When tested at non-up-regulating concentrations, only d-tubocurarine significantly inhibited agonist-induced up-regulation; this inhibition seemed to be noncompetitive. Comparison of [3H]epibatidine displacement in intact M10 cells and membrane preparations by membrane-impermeant ligands indicated that 85% of [3H]epibatidine binding sites are intracellular. On chronic treatment with agonist, the proportion of surface receptors did not change significantly, indicating that most up-regulated [3H]epibatidine binding sites are internal. However, up-regulation is mediated at the cell surface because the impermeant ligand tetramethylammonium was as efficacious as nicotine in eliciting up-regulation, and methylcarbamylcholine (i.e., impermeant but with low efficacy) blocked nicotine induced up-regulation. Thus, agonists elicit up-regulation (mainly of intracellular receptors) by interacting with cell surface nAChRs that are not compatible with either an active or high affinity desensitized conformation.
Molecular cloning has disclosed a diversity of nAChR subunits that are expressed in the vertebrate central nervous system: to date, 11 subunits (α2–9 and β2–4) have been found (Sargent, 1993; Elgoyhen et al., 1994). Immunoprecipitation studies indicate that >90% of high affinity nicotinic agonist binding in the rat brain corresponds to a receptor composed of α4 and β2 subunits (Flores et al., 1992). Chronic nicotine treatment in vivoup-regulates the numbers of brain nAChRs identified by high affinity tritiated agonist binding in mouse (Marks et al., 1985) and rat (Flores et al., 1992; Schwartz and Kellar, 1985). Chronic nicotine treatment over 5–21 days produced concentration-dependent increases of 50–100% above control. The extent of the response varied in a brain region specific manner (Markset al., 1985, 1992; Sanderson et al., 1993). After cessation of nicotine treatment for 4–8 days, nAChR levels returned to control values (Marks et al., 1985;Schwartz and Kellar, 1985), demonstrating the reversibility of this phenomenon. Up-regulation of [3H]nicotine binding sites also has been observed in human brain tissue from tobacco smokers compared with controls from nonsmokers (Breese et al., 1997). This phenomenon may be relevant to tolerance, sensitization, and withdrawal, features considered to contribute to the development and/or maintenance of nicotine dependence. The up-regulation of nicotinic binding sites by nicotine has been termed paradoxical (Wonnacott, 1990) because chronic agonist exposure has traditionally been predicted to down-regulate receptor numbers (Creese and Sibley, 1981). This model was based on the down-regulation of G protein-coupled receptors but has been found to apply to certain ligand-gated ion channels, notably γ-aminobutyric acidA receptors: for example, in cortical neurons, both subunit polypeptide and mRNA are down-regulated by chronic treatment with γ-aminobutyric acid (Mhatre and Ticku, 1994).
Up-regulation of neuronal nAChR has also been demonstrated in in vitro systems, including cells transfected with α4 and β2 subunits (Peng et al., 1994; Zhang et al., 1994;Bencherif et al., 1995; Gopalakrishnan et al., 1997). The M10 cell line consists of mouse fibroblasts stably transfected with chick α4 and β2 subunits under the control of a dexamethasone-sensitive promoter (Whiting et al., 1991): treatment of M10 cells with nicotine for 2–3 days increases α4β2 nAChR by 2–3-fold, as judged by [3H]nicotine binding to immunoisolated material (Peng et al., 1994) or membrane preparations (Zhang et al., 1994; Bencherifet al., 1995). Up-regulation of human α4β2 nAChR expressed in human embryonic kidney 293 cells (Gopalakrishnan et al., 1997) (measured by [3H]cytisine binding to membrane preparations) is similar to that seen in the M10 cell line. This phenomenon is not unique to the α4β2 subtype of nAChR: α-bungarotoxin binding sites, considered to correspond to α7-type nAChRs, are up-regulated after chronic nicotine treatmentin vivo (Pauly et al., 1991) and in vitro (Barrantes et al., 1995), and α3-type nAChRs in SHSY-5Y human neuroblastoma cells have recently been shown to exhibit the same response to chronic agonist (Peng et al., 1997). Up-regulation of α7- and α3-type nAChRs requires higher nicotine concentrations and therefore may be less relevant to changes produced by smoking doses of nicotine.
Studies in vivo (Marks et al., 1985; Schwartz and Kellar, 1985; Sanderson et al., 1993) and in vitro (Peng et al., 1994) have established that up-regulation of [3H]nicotine binding sites reflects an increase in receptor numbers rather than a change in receptor affinity for the ligand. This increase is probably not the result of increased transcription because mRNA levels of the constituent subunits are unchanged (Marks et al., 1992; Penget al., 1994; Zhang et al., 1994; Bencherifet al., 1995). This result implies a post-transcriptional mechanism: altered translation rates (Gopalakrishnan et al., 1997), a decrease in receptor turnover (Peng et al., 1994), recruitment from a finite pool of preexisting receptors (Bencherifet al., 1995), and improved efficiency of receptor assembly from constituent subunits (Rothhut et al., 1996) have been proposed. As well as these conflicting views of the mechanism underlying up-regulation, how chronic nicotine initiates the response remains unclear. Originally it was proposed that up-regulation might be linked to desensitization of the nAChR (Marks et al., 1985;Schwartz and Kellar, 1985). Under the functional model of Katz and Thesleff (1957), chronic nicotine treatment would convert the receptor from an active to a desensitized state. Up-regulation would then be an adaptive mechanism to compensate for this loss of function, but the fact that up-regulation can be provoked in cell lines with no opportunity for natural cholinergic stimulation, and hence no function to “lose,” tends to refute this argument. Up-regulation must be an intrinsic molecular response, as opposed to a specifically neuronal cellular response. Indeed, Peng et al. (1994) showed that nicotine concentrations that promote up-regulation are considerably greater than those needed for desensitization of the α4β2 nAChR.
Here, we conducted a detailed pharmacological evaluation of the up-regulation of α4β2 nAChR in M10 cells, with respect to a wide spectrum of agonists and antagonists, which were analyzed over a broad concentration range. This homogeneous in vitro system facilitates a quantitative dose-response analysis that is untenable in vivo. Agonists showed a diversity of up-regulatory effects that were mediated at the cell surface. The conditions producing up-regulation are consistent with a state of the receptor that is different from either the high affinity desensitized or activated states.
Experimental Procedures
Materials
Tissue culture.
M10 cells were provided by Dr. Paul Whiting (MSD Research Center, Harlow, Essex, UK). Tissue culture reagents were obtained from GIBCO BRL (Paisley, Renfrewshire, Scotland). Tissue cultureware was purchased from Becton Dickinson UK (Oxford, UK) or Sterilin (Stone, Staffs, UK). Maintenance medium consisted of DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine, and 0.5 mg/ml geneticin as a selection agent. Induction medium was composed of DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mm glutamine, and 1 μm dexamethasone to induce receptor expression.
Drugs and reagents.
(±)-[3H]Epibatidine (57 Ci/mmol in ethanol) was obtained from DuPont NEN (Stevenage, Herts, UK) and stored at −20°. (±)-Anatoxin-a was provided by Prof. T. Gallagher (School of Chemistry, University of Bristol, UK). ABT-418 [(S)-3-methyl-5-(1-methyl-2-pyrrolidinyl)isoaxole] was provided by Dr. S. Arneric (Abbott Laboratories, Chicago, IL). All other drugs were purchased from Sigma Chemical (Poole, Dorset, UK) or RBI (Natick, MA). Drugs were dissolved in distilled water or ethanol as appropriate and stored at −20°. All chemical reagents used were supplied by BDH/Merck (Poole, Dorset, UK), Sigma Chemical, or FSA Supplies (Loughborough, UK).
Methods
Cell culture.
Routine culture of M10 cells was carried out as described by Whiting et al. (1991). Briefly, cells were grown at 37° in a humidified incubator (95% O2/5% CO2 atmosphere). M10 cell stocks were maintained in 75-cm2 flasks containing 25 ml of maintenance medium. Cells were seeded onto 75-cm2 flasks or 24 × 16-mm well plates at ∼20% confluency (15,000 cells/cm2). Receptor production was induced when cells had reached ∼70% confluency (typically 2–3 days) by replacement of maintenance medium with induction medium (in which geneticin was replaced with 1 μm dexamethasone).
Quantification of [3H]epibatidine binding sites in M10 cells in situ.
[3H]Epibatidine binding was performed on cells in 24-well plates. All steps were carried out at 37° unless otherwise stated. Medium was removed by aspiration, and each well was washed by the addition and aspiration of 1 ml of sterile PBS (150 mmNaCl, 8 mmK2HPO4, 2 mmKH2PO4, pH 7.4). [3H]Epibatidine (500 pm in maintenance medium; 1 ml/well) was added. Nonspecific binding was assayed in the presence of 100 μm (−)-nicotine. Samples were incubated for 2 hr with gentle agitation before aspiration of the assay solution and washing with four changes of PBS to remove unbound ligand. To measure bound ligand, the cells were dissolved overnight in 1 ml/well of Markwell reagent A [2% (w/v) Na2CO3, 1% (w/v) sodium dodecyl sulfate, 0.1 m NaOH, 0.16% (w/v) Na/K tartrate]. Samples (700 μl) were mixed with 5 ml of Optiphase Safe liquid scintillant and counted for tritium (Tricarb 1600 liquid scintillation counter; Packard, Meriden, CT; counting efficiency, ≈45%). Protein was determined by the method of Markwell et al. (1978) using 200 μl of the remaining cell solution. Specific cpm [3H]epibatidine bound/well were converted to fmol bound/mg protein. [3H]Epibatidine binding to M10 cells induced for 48 hr gave 326 ± 46 fmol/mg (protein) (mean ± standard error, 10 independent assays).
Kinetic analysis of [3H]epibatidine binding to M10 cells in situ.
On and off rates were determined at 4°, 20°, and 37°. At equilibrium, bound [3H]epibatidine (200 pm) accounted for <5% of total ligand added, so ligand depletion effects were not significant.
For off-rates, cells induced for 2 days were preequilibrated with 200 pm [3H]epibatidine (total binding) or 200 pm [3H]epibatidine plus 100 μm (−)-nicotine (nonspecific binding) for 2 hr at 37°. After cooling to the chosen temperature for 30 min where necessary, the [3H]epibatidine solution was aspirated, and 2 ml of 100 μm (−)-nicotine in maintenance buffer was added. At defined intervals, the (−)-nicotine solution was removed from the wells by rapid aspiration and washing (achieved in <4 sec). Bound ligand was measured as described above. At the zero time point, the wells were washed as described above, with no addition of (−)-nicotine.
Dissociation of ligand from the α4β2 nAChR may be described by a single exponential decrease model, corresponding to dissociation from the desensitized (high affinity) isomer of the receptor (Lippielloet al., 1987). The averaged data obtained at each temperature were fit to eq. 1, using the nonlinear least-squares curve fitting facility of SigmaPlot for Windows V2.0.

Time dependence of [3H]epibatidine association to and dissociation from M10 cells in situ. Experiments were performed at 4°(○), 20° (•) and 37° (□). a, Association data were derived from cultures incubated with 200 pm [3H]epibatidine in the presence and absence of 100 μm (−)-nicotine for the times indicated. Specific binding was calculated as a percentage of equilibrium (maximum) binding determined by incubation at 37o for 2 hr. Values were fit to a double exponential increase model (see Experimental Procedures). b, Dissociation data were derived from cultures pre-equilibrated with 200pm[3H]epibatidine in the presence and absence of 100 μm (−)-nicotine, followed by incubation with excess unlabelled (−)-nicotine for the times indicated. Specific binding was calculated as a percentage of the initial (equilibrium) binding and values were fit to a single exponential decrease model (see Experimental Procedures). Logarithmic transformation of the data (insets) shows good agreement between the data and the model, confirming the validity of this approach. Points, mean ± standard error of at least three independent determinations.
For on-rates, cells induced for 2 days were equilibrated at the chosen temperature for 30 min. The cells were washed with one change of PBS; then, total and nonspecific binding of [3H]epibatidine [200 pm, in the absence or presence of 100 μm (−)-nicotine, respectively] were determined at intervals over a period of 90 min (or 3 hr at 4°). Equilibrium binding was determined in parallel after incubation for 2 hr at 37°. Binding was stopped by aspiration and rapid washing.
Association of ligand with the α4β2 nAChR may be modeled as a double exponential increase (eq. 2). The faster process describes binding to the high affinity, desensitized form of the receptor (which dominates under equilibrium binding conditions), whereas the slower rate represents isomerization of the receptor to the high affinity state from the resting state (Lippiello et al., 1987). This model was fit to the averaged association data at each temperature using the nonlinear least-squares curve fitting facility of SigmaPlot for Windows V2.0.
The equilibrium dissociation constant (Kd
) at each temperature was calculated by substitution of the derived values into eq. 3 (Lippielloet al., 1987).
Chronic treatment of M10 cells with nicotinic drugs.
To examine the effects of chronic exposure to nicotinic drugs, cells were incubated for 48 hr in induction medium supplemented with the desired concentrations of nicotinic agonist or antagonist. To assess the effects of antagonists or partial agonists on agonist-induced up-regulation, both compounds were added simultaneously. A control, consisting of cells treated with induction medium alone, was included in each experiment.
[3H]Epibatidine binding was determined by thein situ binding assay described above, except for the inclusion of a more rigorous washing procedure, performed at 37°. The supplemented medium was removed by aspiration, the cells were washed with 1 ml of sterile PBS followed by the addition of 1 ml of maintenance medium. This washing process was repeated twice at hourly intervals, with the cells incubated between washes. This extended washing process is necessary to completely remove drugs from cultured cells (Barrantes et al., 1995). Individual agonist-induced up-regulation profiles were analyzed according to the logistic equation, describing both up-regulatory and inhibitory phases of the up-regulation response, using the nonlinear least-squares curve fitting facility of SigmaPlot for Windows, ver. 2.0.
Competition assays of [3H]epibatidine binding to M10 cells in situ.
[3H]Epibatidine displacement binding assays were performed using a modified version of the standard binding protocol. [3H]Epibatidine (200 pm) was prepared in DMEM, and this was used to make serial dilutions of nicotinic drugs. All steps were carried out at room temperature to reduce metabolically dependent transport of ligands, and incubations were extended to 3 hr to ensure that equilibrium binding of [3H]epibatidine was attained. The IC50 value for each drug was calculated by fitting to Hill equation, using the nonlinear least-squares curve fitting facility of SigmaPlot for Windows
Preparation of M10 cell membranes.
M10 cell membranes were prepared by a modification of the procedure described by Whitinget al. (1991). After induction of M10 cells in 75-cm2 flasks (48–72 hr), medium was removed, followed by washing with two changes (5 ml, 37°) of PBS. The cells were harvested in 5 ml of homogenization buffer (ice-cold PBS containing 10 mm EDTA, 10 mm EGTA, 1 mm phenylmethylsulfonyl fluoride) and collected by gentle centrifugation (5 min, 500 × g). After resuspension into 5 ml of homogenization buffer, the cells were disrupted by sonication (three times for 10 sec). The resulting membrane preparation was collected by centrifugation (100,000 × g, 15 min), and the supernatant fraction was discarded. The membrane pellet was resuspended in an additional 5 ml of homogenization buffer (hand-held glass homogenizer, six strokes) before recentrifugation (100,000 × g, 15 min), resuspension in homogenization buffer containing glycerol (1:1 v/v; 1 ml/75-cm2 flask of cells), and storage at −20° in 1-ml aliquots.
[3H]Epibatidine binding to M10 membranes.
Competition assays of [3H]epibatidine binding to membrane preparations were terminated by filtration. Membranes (duplicate 25-μl samples) were incubated with [3H]epibatidine (200 pm in DMEM, 2 ml) for 2 hr at 37° in the presence and absence of nicotinic ligands. Nonspecific binding was assayed in the presence of 100 μm(−)-nicotine. Samples were filtered and washed on Whatman GFA/E filters soaked in polyethyleneimine (0.3% w/v, 24 hr), using a Brandel (Semat, St. Albans, Herts, UK) cell harvester. Protein was determined according to the method of Markwell et al. (1978). The mean density of [3H]epibatidine binding sites in M10 membranes was 712 ± 49 fmol/mg of protein (mean ± standard error, three independent assays). IC50 andKi values were determined as for M10 cells in situ.
Estimation of surface [3H]epibatidine binding sites.
The contribution of surface nAChRs to the total population of [3H]epibatidine binding sites was determined in M10 cells in situ using a modified version of the protocol for cells chronically treated with nicotinic ligands. Cells in 24-well plates were induced in the presence or absence of up-regulating concentrations of agonists and then extensively washed as described above. [3H]Epibatidine binding (200 pm) was performed either in plain medium (to measure the total population) in the presence of ACh (3 μm, a sufficient concentration to completely block [3H]epibatidine binding to M10 membrane preparations and thus cell surface sites) or in the presence of (−)-nicotine (3 μm, to measure nonspecific binding).
Time course of nicotinic ligand entry by M10 cells in situ.
To determine the extent to which ligands that were impermeant in the acute binding protocols might permeate M10 cells during the standard up-regulation protocol (48 hr at 37°), competition binding experiments were performed at time points during the up-regulation process on M10 cells in situ. Cells were induced in 24-well plates for 72 hr to ensure equilibrium levels of nAChR expression. Spent induction medium was aspirated from the cells and replaced with fresh induction medium (0.5 ml). At intervals during the next 48 hr, the induction medium was replaced by drug solutions prepared by dilution in induction medium (0.5 ml). A control of cells grown in induction medium alone for an additional 48 hr was included in each experiment.
At the 46-hr time point, the induction medium was supplemented with [3H]epibatidine (final concentration, 200 pm). Nonspecific binding was determined by addition of 100 μm (−)-nicotine. For the 1-hr uptake time point, the [3H]epibatidine was added at 46 hr [with or without (−)-nicotine], and this solution was in turn replaced with induction medium supplemented with both the drug of interest and 200 pm [3H]epibatidine [with or without (−)-nicotine] at 47 hr. At 48 hr, unbound [3H]epibatidine was removed by rapid washing and aspiration before quantification of specific binding as described above for M10 cells in situ. By discounting the contribution of the relatively small cell-surface receptor population, approximate values of intracellular drug concentrations could be calculated by substitution into eq. 7, a rearrangement of eq. 5:
Statistical analysis.
Up-regulation and surface population data were examined by one- and two-factor analyses of variance, using SigmaStat for Windows V2.0. The Tukey-B test was used for multiple pairwise comparisons, whereas comparisons versus control were performed using Dunnett’s test.
Results
Kinetic studies.
α4β2 nAChRs in membrane preparations and M10 cells in situ were quantified by binding of [3H]epibatidine. Its binding to M10 cells gave an excellent signal-to-noise ratio and reproducible data (Whiteakeret al., 1996), but its low picomolar affinity for α4β2 nAChR (Houghtling et al., 1995) poses a problem of ligand depletion at low concentrations, which can distortKd determinations from saturation binding experiments. This prompted derivation of theKd value for [3H]epibatidine binding to M10 cells in situ by an independent method, based on binding rate constants. Rates of association and dissociation were measured at 4°, 20°, and 37° (Fig. 1, Tables 1 and2).
Association kinetics for [3H]epibatidine binding to M10 cellsin situ
Dissociation kinetics for [3H]epibatidine binding to M10 cells in situ
Association of [3H]epibatidine to the α4β2 nAChR conformed to a double exponential process (see Methods) as described by Lippiello et al. (1987) for (−)-[3H]nicotine binding to rat brain membranes; the kinetics of [3H]epibatidine binding to M10 cell membrane preparations were reported to be similar (Bencherif et al., 1995). The averaged values ofkfast and kslow(describing association with the high affinity desensitized state of the receptor and isomerization of resting receptors to this state, respectively) are given in Table 1, together with the corresponding half-times (t1/2, equal toln2/kfast andkslow) of each process.
Dissociation of [3H]epibatidine could be described as a single exponential decrease (Fig. 1b), again in agreement with Lippiello et al. (1987). The rate of [3H]epibatidine dissociation from the M10 cell surface nAChR was rather slow, with a half-time of 19 min even at 37° (Table 2). This has practical implications: first, removal of unbound [3H]epibatidine from M10 cells in situ by rapid washing (∼4 sec) will lead to negligible loss of specific binding. Second, it underlines the importance of allowing sufficient time for equilibration at the receptor. Third, it demonstrates the requirement for a prolonged washing procedure to remove epibatidine from chronically treated cells.
The equilibrium dissociation constants (Kd ) for [3H]epibatidine binding to M10 cells in situ, derived from the kinetic constants describing association to and dissociation from the high affinity (desensitized) form of the receptor (see Methods), were 7.7, 5.0, and 19.7 pm at 4°, 20°, and 37°, respectively. Kinetic analysis of [3H]nicotine binding to a putative α4β2 nAChR in rat brain membranes (Lippiello et al., 1987) demonstrated no temperature dependence for theKd . Therefore, the Kd values derived for [3H]epibatidine binding to M10 cells at each temperature are assumed to reflect experimental variability, giving a mean Kd value of 10.8 ± 3.7 pm. This is in good agreement with the reported value of 4.5 pm for [3H]epibatidine binding to M10 cell homogenates and with values of 15 and 1 pm for the predominant site labeled by [3H]epibatidine (and proposed to correspond to α4β2-type nAChR) in rat and human brain, respectively (Houghtling et al., 1995).
Agonist-induced up-regulation.
The abilities were compared of a wide spectrum of nicotinic agonists to up-regulate numbers of [3H]epibatidine binding sites in M10 cells. (−)-Nicotine, (±)-epibatidine, MCC, (±)-anatoxin-α, (−)-cytisine, ABT-418, and TMA were coadministered with 1 μmdexamethasone for 48 hr. All of the agonists provoked dose-dependent up-regulation (Fig. 2), which exhibited an inverted U-shaped profile (except in the case of ABT-418, which did not reach a distinct maximum up-regulation value within the concentration range tested). The dose-response profiles vary among compounds; differences in profile show no correlation with agonist potency. For instance, the slopes of the up and down phases of (±)-epibatidine-induced up-regulation are extremely shallow compared with those produced by (±)-anatoxin-α or TMA. Averaged up-regulation data were fit to a logistic model (Fig. 2): the calculated values of maximum up-regulation, EC50 (concentration giving half-maximum up-regulation) and IC50(concentration at which up-regulation declines to half the maximum) are recorded in Table 3.
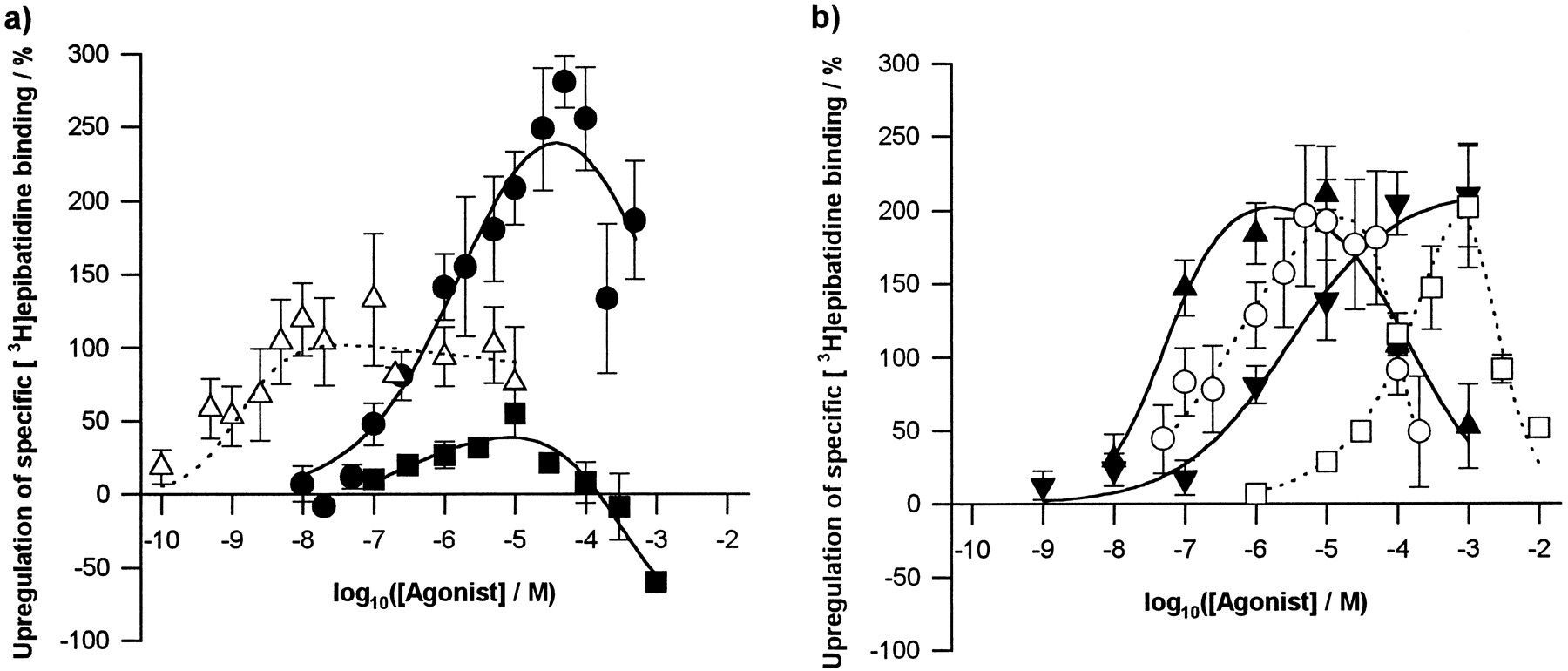
Agonist induced up-regulation of [3H]epibatidine binding sites in M10 cells. Nicotinic agonists were administered during nAChR induction (48 hr). After extensive washing to remove nicotinic ligands, numbers of [3H]epibatidine binding sites were determined by binding to cells in situ. a, Epibatidine (▵) and MCC (▪) are partial agonists for up-regulation compared with (−)-nicotine (•). b, All other compounds tested [(−)-cytisine (▴), ABT-418 (▾), (±)-anatoxin-a (○) and TMA (□)] up-regulate with full efficacy compared with nicotine. Points, mean ± standard error of at least three independent determinations, using separate cultures. For each culture, percentage up-regulation was determined relative to a control of cells induced for 2 days with no agonist present. Data were fitted to a logistic model (see Experimental Procedures).
Parameters for agonist-induced up-regulation
The maximum up-regulation provoked by each of the agonists tested was similar, with the exception of MCC and (±)-epibatidine (Fig. 2a, Table3): MCC produced only 16% of the maximum up-regulation observed for (−)-nicotine, whereas the corresponding figure for (±)-epibatidine is 38%. To confirm that these two compounds were acting as partial agonists for up-regulation, up-regulation produced by fixed concentrations of (−)-nicotine was determined in the presence of varying doses of MCC (Fig. 3a) and (±)-epibatidine (Fig. 3b). Two (−)-nicotine concentrations were studied: one approximated to its EC50 value for up-regulation (1 μm; Table 3) and the other corresponded to a concentration producing maximum up-regulation (100 μm; Fig. 2a). With 1 μm (−)-nicotine, peak concentrations of both MCC and (±)-epibatidine had little or no additional effect on up-regulation, whereas these concentrations depressed the up-regulation produced by 100 μm(−)-nicotine. Coadministration of higher concentrations of either drug with (−)-nicotine resulted in up-regulation indistinguishable from that produced by MCC or (±)-epibatidine alone, suggesting that the (−)-nicotine had been fully displaced from the up-regulatory site or sites.

Up-regulation by fixed (−)-nicotine concentrations in the presence of (±)-epibatidine, MCC and dTC. Drugs were co-administered with dexamethasone during the nAChR induction period (48 hr). Specific [3H]epibatidine binding to M10 cellsin situ is expressed as a percentage above control values determined in parallel for cells induced in the absence of nicotinic ligands. a, Up-regulation caused MCC alone (○) and co-administered with (−)-nicotine (1 μm [(•) or 100 μm (▪)]. (−)-Nicotine alone increased [3H]epibatidine binding to induced M10 cells by 83.5 ± 14.2% (mean ± standard error, 1 μm) and 193 ± 29.3% (100 μm) above control. b, Up-regulation caused by (±)-epibatidine alone (○) and co-administered with (−)-nicotine [1 μm (•) or 100 μm (▪)]. Up-regulation by 1 μm(−)-nicotine alone was 85.0 ± 26.1%, by 100 μm(−)-nicotine alone was 171.7 ± 21.0% above control. c, Up-regulation produced by (−)-nicotine [1 μm (•) or 100μm (▪)] in the presence of dTC. To allow direct comparison of the effects of dTC on up-regulation by different concentrations of (−)-nicotine, up-regulation has been converted to a percentage of that produced by the appropriate concentration of (−)-nicotine in the absence of dTC. Points, mean ± standard error of at least three separate experiments, using separate cultures.
Antagonist-induced up-regulation.
The ability of the antagonists DHβE, MLA, dTC, HEX, DEC, and mecamylamine to induce up-regulation was also examined by coadministration with 1 μm dexamethasone for 48 hr. Generally, these compounds had very little effect on the density of [3H]epibatidine binding sites except at high concentrations (Fig. 4, a and b). In contrast to the up-regulation produced by agonists, there was no common pattern of dose response, so no attempt was made to fit these data to a model.
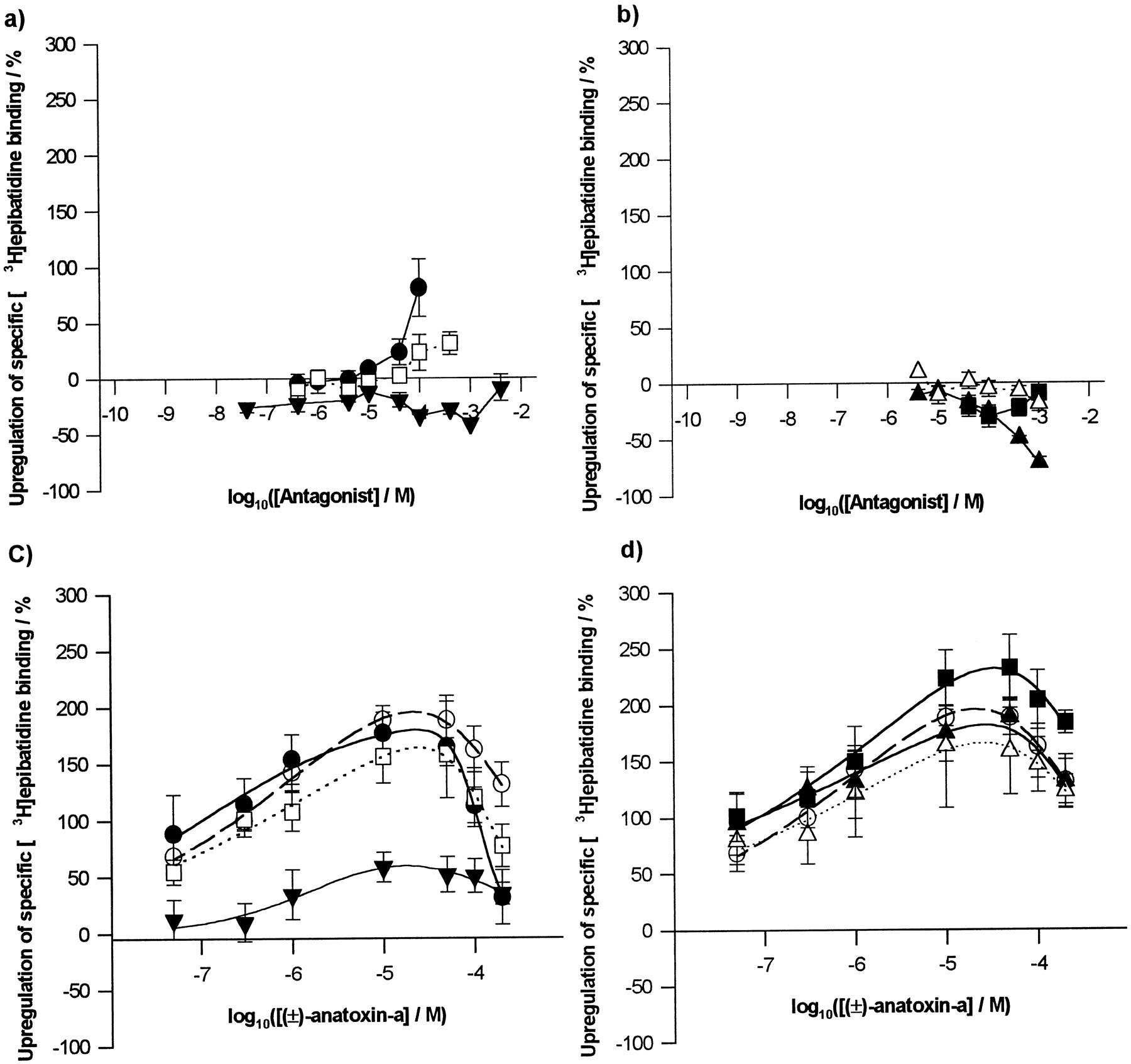
Effects of nicotinic antagonists on the up-regulation of [3H]epibatidine binding sites in M10 cells. Drugs were administered with dexamethasone during the 48-hr nAChR induction period. Specific [3H]epibatidine binding to M10 cells in situ is expressed as a percentage above control values determined in parallel in cells not exposed to any nicotinic ligand. a and b, Effects of antagonists alone. a, ‘Competitive’ antagonists: MLA (•) and DHβE (□) produced mild up-regulation at high concentrations, whereas dTC (▾) was ineffective. b, Noncompetitive antagonists: HEX (▪) and DEC (▵) have no effect below 1 mm, but mecamylamine (▴) is down-regulatory at high concentrations. c and d, Effects of antagonists on up-regulation produced by increasing concentrations of (±)-anatoxin-a (control, ○). c, MLA (30 μm; •), DHβE (100 μm; □) and dTC (100 μm; ▾). Only dTC blocked anatoxin-a induced up-regulation (p < 0.001, two-way analysis of variance). d, HEX (1 mm; ▪), DEC (1 mm; ▵) and mecamylamine (100 μm; ▴). Points, mean ± standard error of at least three independent determinations, using separate cultures. Data in c and d were fitted to a logistic model (see Experimental Procedures).
To determine whether any of the antagonists were able to block nicotinic agonist-induced up-regulation, each was coadministered at a fixed concentration with a range of (±)-anatoxin-α concentrations (Fig. 4, c and d). (±)-Anatoxin-α was chosen because its sharp up-regulation dose-response profile (Fig. 2) would facilitate identification of shifts in potency. The antagonist concentrations selected were the highest that did not themselves produce changes in [3H]epibatidine binding site density after chronic exposure. Two-way analysis of variance (the factors being antagonist treatment and anatoxin-α concentration) showed no significant effect by any of the antagonists on (±)-anatoxin-a induced up-regulation, with the exception of dTC, which significantly blocked up-regulation (p < 0.001, Tukey test). The lack of a sideways shift in the dose-response profile for (±)-anatoxin-α-induced up-regulation in the presence of dTC (Fig.4c) suggests a noncompetitive mode of antagonism of up-regulation. This was further examined by assessing the effects of increasing doses of dTC on up-regulation produced by fixed (−)-nicotine concentrations (1 and 100 μm; Fig. 3c). The ability of dTC to block (−)-nicotine-induced up-regulation was independent of the nicotine concentration used, supporting a noncompetitive mode of action for dTC.
Surface versus intracellular [3H]epibatidine binding sites.
As a prerequisite to determining whether up-regulation is initiated through cell surface or intracellular nAChR, we addressed the distribution of receptors in M10 cells. Because [3H]epibatidine is a readily cell-permeant radioligand that will access both populations, a quantitative evaluation of nAChR distribution was sought by comparing the abilities of permeant and impermeant ligands to displace [3H]epibatidine binding in intact cells and membrane preparations. Competition binding assays were carried out with all of the compounds examined in the up-regulation studies, plus ACh (Fig. 5, Table4). In the case of M10 membrane preparations, each of the compounds (with the sole exception of mecamylamine) was capable of fully displacing [3H]epibatidine binding, with Hill slopes of 1.0–1.5. Ki values are typical of these compounds at α4β2-type nAChR (Houghtling et al., 1995; Gopalakrishnan et al., 1996), with agonists generally giving Ki values in the nanomolar range, whereas antagonists yielded Ki values in the micromolar range. (±)-Epibatidine gave aKi value of 11 pm, in excellent agreement with theKd value of 10.8 pm derived from the kinetic analysis of binding to intact cells.

[3H]Epibatidine displacement binding. The ability of compounds to displace [3H]epibatidine (200 pm) binding from either intact M10 cells in situ (filled symbols) or M10 membrane preparations (open symbols) was assessed in equilibrium binding assays. Where displacement was observed, data were fitted to the Hill equation (see Experimental Procedures). a, Cell permeable ligands: (±)-epibatidine (▪, □), (−)-nicotine (•, ○) and dTC (▴, ▵). b, Cell impermeable ligands, MCC (▾, ▿), ACh (♦, ⋄) and tetramethylammonium (⬡, ⬡). Points, mean ± standard error of at least three independent determinations.
Binding affinities of nicotinic ligands at the α4β2 nAChR expressed in M10 cells
Fewer ligands were able to displace [3H]epibatidine binding to intact M10 cellsin situ. Of the agonists, (−)-nicotine, (±)-epibatidine, (−)-cytisine, (±)-anatoxin-a, and ABT-418 resulted inKi values very close to those derived from membrane assays (Table 4, Fig. 5a). However, agonists with quaternary ammonium groups (ACh, MCC, TMA) failed to displace [3H]epibatidine binding significantly (Fig.5b), consistent with most of the binding sites being inaccessible to these impermeant ligands. Of the antagonists examined, DHβE and MLA were effective competitors in both preparations (Table 4), whereas DEC and HEX seemed to be impermeant. The dTC competition curve was displaced to the right in the intact cells compared with M10 membranes (Fig. 5a), perhaps reflecting rather slow or incomplete permeation of the cells by dTC.
Having demonstrated the cell impermeance of the permanently charged compounds, ACh was used to quantify the surface population of [3H]epibatidine binding sites more precisely. Assays were carried out on intact M10 cells in situ, using a saturating concentration of [3H]epibatidine in the presence and absence of 3 μm ACh, a concentration that is capable of fully displacing [3H]epibatidine binding to membrane preparations (Fig. 5b) and thus can be assumed to fully displace [3H]epibatidine from surface nAChRs. ACh displaced 15 ± 3% of [3H]epibatidine binding (p < 0.001; Table5), suggesting that 85% of binding sites are intracellular. This is consistent with the presence of the comparatively small cell surface population of nAChRs indicated by confocal microscopy of intact versus permeabilized M10 cells, using mAbs directed against the β2 subunit (mAb270, mAb290, gifts of Dr. Jon Lindstrom; data not shown). For cells that had been exposed to a maximally up-regulating concentration of various agonists during the 48-hr induction period, the proportion of binding sites susceptible to inhibition by ACh ranged from 6.3 to 13% (Table 5). Statistical analysis (one-way analysis of variance) showed no significant difference between the proportions of receptors found on the surface of cells induced in the presence of dexamethasone alone and cells up-regulated by chronic agonist treatment (p = 0.31).
Surface and intracellular populations of [3H]epibatidine binding sites in basal and upregulated M10 cells defined by ACh
Is up-regulation mediated by cell surface or intracellular nAChRs?
Given the large proportion of intracellular binding sites and the freely permeant nature of drugs such as nicotine that have commonly been used to promote up-regulation, it is pertinent to inquire whether interaction with surface nAChRs provides the trigger for up-regulation or whether binding to intracellular sites can also effect this response. From the ability of ligands to access the intracellular pool (Fig. 5, Table 4), MCC and TMA can be defined as impermeant, yet both provoke up-regulation (Fig. 2, Table 3). Although MCC was found to be a partial up-regulator compared with (−)nicotine, TMA was as effective as the permeant agonists. However, it remained possible that over the extended period of chronic drug exposure in the up-regulation protocol, these “impermeant” ligands might gain access to the interior of the cell. Therefore, the rate of entry of nicotinic ligands into M10 cells was assessed by determining their ability to compete for [3H]epibatidine binding sites in M10 cellsin situ at intervals throughout the 48-hr exposure to the compounds: displacement of a large proportion of [3H]epibatidine binding would indicate entry of the ligand into the cell interior. Equilibrium [3H]epibatidine binding was determined in the presence of 10 μm TMA and 100 μm MCC, concentrations sufficient to displace [3H]epibatidine binding to M10 cell membranes (Fig. 5) while evoking minimal up-regulation themselves (Fig. 2). The antagonist dTC also was examined, at a concentration (1 mm) sufficient to displace all [3H]epibatidine binding to M10 cell membranes (Fig. 5).
After 48-hr exposure to TMA, [3H]epibatidine binding (in the continued presence of TMA) was 139 ± 21% of the untreated control (Fig. 6a): rather than displacement, there is a slight increase in binding attributable to up-regulation by 10 μm TMA (Fig. 2b). At all time points, [3H]epibatidine binding was the same as or greater than that measured in cells not exposed to TMA, and it can be concluded that no TMA entered the cell during the course of the experiment. Some displacement of [3H]epibatidine binding was observed during the time course of exposure to MCC and dTC (Fig. 6a). The extent of [3H]epibatidine binding displaced at each time point was used to estimate the intracellular concentration of the drug (Fig. 6b) by reference to its affinity for [3H]epibatidine binding sites in M10 membranes (Table 4; see Methods). This exercise indicates that intracellular MCC was sustained at or below 0.1 μm throughout the experiment, ∼0.1% of the external drug concentration. This concentration is subactivating, unlikely to promote up-regulation (see EC50 for up-regulation of 0.46 μm, Table 3) and too low to compete effectively with the intracellular concentrations of nicotine, which would be in the micromolar range. On the other hand, dTC seemed to be much more cell permeant, attaining up to 10% of its external concentrations. To determine whether any of these drugs were degraded during the 48-hr incubation with M10 cells, aliquots of medium-containing drug were removed at the end of this period and compared with freshly made solutions for their abilities to displace [3H]epibatidine binding to M10 cell membranes. No differences were seen (data not shown), indicating that degradation does not contribute to the apparently low permeation by these ligands. Thus, it can be concluded that the ability of MCC and TMA to induce up-regulation reflects their interaction with cell surface nAChR.

Time course of nicotinic ligand uptake by M10 cells. a, The rate of entry of nicotinic ligands into M10 cells was assessed by determining their ability to compete for [3H]epibatidine binding in induced (1 μmdexamethasone, 72 hr) M10 cells in situ after increasing periods of time. Equilibrium binding in the presence of TMA (10 μm, ▪), MCC (100 μm, ▵) and dTC (1 mm, •) was measured over a period of 48 hr. Specific [3H]epibatidine binding was assessed as percentage of control (cells untreated with drugs). b, From the degree of inhibition observed, the intracellular concentration of drug could be estimated by reference to the affinity of the ligand for [3H]epibatidine binding sites in M10 membranes (see Experimental Procedures). Intracellular levels of TMA were undetectable. Points, mean ± standard error of at least three independent determinations, using separate cultures.
Discussion
In this study, we investigated whether up-regulation of α4β2 nAChRs is triggered at the cell surface and examined the state of the receptor coupled to up-regulation. To this end, we used M10 cells exposed to a variety of drug conditions and monitored receptor numbers, properties, and distribution using [3H]epibatidine binding to intact M10 cellsin situ and to M10 membrane preparations. Previous studies of α4β2 nAChR up-regulation in vitro have used [3H]agonist and [125I]mAb binding to immunoisolates (Penget al., 1994) or membrane preparations (Zhang et al., 1994; Bencherif et al., 1995) or subunit-specific [125I]mAb binding to intact cells (Rothhutet al., 1996) (or both) to quantify receptor expression. [3H]Agonist binding has the advantage of recognizing only fully assembled receptors, whereas study of cellsin situ allows cellular receptor populations to be quantified reliably because there is no possibility of changes in subpopulation proportions arising from processing of the sample. In this study, [3H]epibatidine binding to M10 cells in situ combines the advantages of both approaches; nAChRs expressed on the cell surface can be discriminated using an impermeant competing ligand.
Agonist induced up-regulation.
We examined up-regulation by a wide spectrum of drugs over a broader range of concentrations than has previously been documented; where there is overlap, the data presented here are in good agreement with those of previous reports (Penget al., 1994; Zhang et al., 1994). The bell-shaped dose-response curves for up-regulation are reminiscent of α4β2 nAChR activation profiles in M10 cells (Thomas et al., 1993) and Xenopus laevis oocytes (Vibat et al., 1995). Correlation plots (Fig.7) show that there is no correlation between the EC50 value for up-regulation and either maximum up-regulation observed (Fig. 7c) or the IC50 value for the downturn in the dose-response profile (Fig. 7d). There is, however, good correlation between the EC50 value for up-regulation and both the EC50 value for activation (Fig. 7a) and theKI value for binding (Fig. 7b). However, up-regulation of α4β2 nAChRs in M10 cells requires chronic exposure to agonist concentrations intermediate between those that produce receptor activation on acute application (typically in or near the micromolar range) and those producing desensitization without prior activation (typically nanomolar). Other considerations also make the hypothesis that activation may provoke up-regulation untenable. First, in contrast to their efficacy in activating nAChRs, (±)-epibatidine and MCC are partial agonists for up-regulation: the maximum up-regulation induced by (−)-nicotine is approximately double that elicited by (±)-epibatidine and over six times greater than that produced by MCC (Table 3). Both enantiomers of epibatidine behaved as potent full agonists at a number of nAChR subtypes composed of human or chicken nAChR subunits and expressed in X. laevis oocytes (Gerzanich et al., 1995), (±)-epibatidine was more efficacious than (−)-nicotine at chicken (Vibat et al., 1995) and human α4β2 nAChR (Gopalakrishnan et al., 1996) and at least as efficacious as (−)-nicotine in activating “type III” currents, attributed to α4β2-type nAChRs, in rat hippocampal cultures (Alkondon and Albuquerque, 1995). Marks et al. (1996) found both enantiomers of epibatidine to be approximately twice as effective as (−)-nicotine in stimulating86Rb+ efflux from mouse thalamic synaptosomes (a response considered to be mediated by α4β2-type nAChRs). These authors found MCC to be even more efficacious than epibatidine. Second, dose-response profiles for activation of nAChR in M10 cells do not exactly match the dose dependence of up-regulation. For instance,86Rb+ flux induced by (−)-nicotine is maximal at 3–5 μm (Thomaset al., 1993; Court et al., 1994), whereas maximum up-regulation is produced by 30–50 μm(−)-nicotine (Fig. 2).

Correlations between agonist potency for up-regulation and other parameters. EC50 values for up-regulation are plotted against (a) EC50 values for activation (Table 3); (b) Kivalues for binding to membranes (Table 4); (c) maximum up-regulation (Table 3) and (d) IC50 values for the downturn of the up-regulation dose-response profiles (Table 3). Linear regression gives correlation coefficients of 0.78 for EC50 (activation); 0.97 for Ki (binding); 0.20 (for maximum up-regulation); 0.52 for IC50 (up-regulation).
Antagonist effects on up-regulation.
If agonist activation of nAChR is the trigger for up-regulation, antagonists (particularly competitive antagonists) should block agonist-induced up-regulation while themselves having no effect. However, with the exception of dTC, none of the antagonists tested prevented agonist-induced up-regulation (Fig. 4). dTC was an effective antagonist of up-regulation, as previously reported (Peng et al., 1994; Zhang et al., 1994), but inhibition by dTC was independent of nicotine concentration (Fig. 3c) and the lack of a sideways shift in the (±)-anatoxin-α dose-response curve in the presence of dTC (Fig. 4c) suggests a noncompetitive mode of action. This is in contrast to the competitive antagonism by dTC of activation of chicken α4β2 nAChR in X. laevis oocytes (Bertrand et al., 1990). The lack of inhibition by DHβE is consistent with the findings of Zhanget al. (1994). Mecamylamine has been reported to up-regulate numbers of (−)-[3H]nicotine binding sites in M10 cells (Peng et al. 1994) and in vivo (Abdullaet al., 1996; Pauly et al., 1996). We found no effect of mecamylamine: similarly, chronic mecamylamine (alone or in combination with agonist) has no effect on the up-regulation of α3- and α7-type nAChRs expressed in the SHSY-5Y cell line (Peng et al., 1997) or α7-type nAChR in hippocampal neurons (Wonnacott and Rogers, 1996).
These observations indicate that the agonist-selective receptor conformation triggering up-regulation is different from that associated with activation and further supports the proposition that receptor activation is not required for up-regulation. This view is reinforced by the inability of chlorisondamine (which produces a long-lasting blockade of central nicotinic responses) to prevent agonist-induced up-regulation of [3H]nicotine binding sitesin vivo (El-Bizri and Clarke, 1994). Thus, it seems that up-regulation is triggered by an agonist-favoring conformation of the nAChR that is not the active state.
[3H]Epibatidine displacement binding studies.
It is no longer tenable that up-regulation of nAChR is mediated by the high affinity desensitized state of the receptor as originally proposed (Marks et al., 1985; Schwartz and Kellar, 1985). Although there is a positive correlation between the rank order ofKI values from competition binding assays and EC50 values for up-regulation for nicotinic agonists (Fig. 7b), the concentrations required to promote up-regulation are 1 or 2 orders of magnitude greater than the binding affinities (which represent the desensitized state of the nAChR), in agreement with Peng et al. (1994) and Gopalakrishnanet al. (1997). At agonist concentrations at which the high affinity desensitized state predominates, very little or no up-regulation is observed. Moreover, this view is consistent within vivo evidence: Rowell and Li (1997) measured brain nicotine levels and numbers of [3H]nicotine binding sites in rats chronically treated with nicotine over 10 days. In treatment regimens that produced brain nicotine concentrations sufficient to achieve a complete desensitization of receptors in other studies (Marks et al., 1994), there was no statistically significant up-regulation of [3H]nicotine binding sites: up-regulation required higher concentrations of nicotine. This refutes the candidature of the classic desensitized state as an up-regulatory trigger.
To distinguish cell surface and intracellular populations of nAChR, affinity constants (Ki values) for each of the nicotinic drugs used in the up-regulation studies were determined in intact M10 cells and M10 membrane preparations. ACh was included as a potentially cell-impermeant ligand, although it was unsuitable for chronic treatment because of its vulnerability to the cholinesterases present in the serum needed to sustain the cells. The addition of anticholinesterases was discounted because they may up-regulate α4β2 nAChR themselves (Svensson and Nordberg, 1996). Of the compounds tested, only mecamylamine was incapable of fully displacing [3H]epibatidine binding to M10 cell membranes, which is consistent with its noncompetitive mode of action (Bertrand et al., 1990). TheKi values determined using M10 membranes are in excellent agreement with those previously obtained in transfected cells (Gopalakrishnan et al., 1996) or rat forebrain membranes (Houghtling et al., 1995).
The poor ability of the quaternary nitrogen containing compounds (MCC, ACh, TMA, DEC, and HEX) to displace [3H]epibatidine binding to intact M10 cells is consistent with a large proportion of the [3H]epibatidine binding sites being located internally, where they are not accessible to these ligands. Conversely, the other compounds studied seem able to access this intracellular pool of receptors freely, with no difference inKi values between binding to intact cells and membranes (Table 4). This suggests that the receptor populations on the cell surface and inside the cell have similar binding characteristics and that these compounds enjoy relatively unimpeded access to the intracellular milieu.
More detailed investigation showed the surface (ACh sensitive) pool of [3H]epibatidine binding sites in induced cells to be 15% of the total population (Fig.8). Rothhut et al. (1996) used [125I]mAb binding to measure the distribution of assembled α4β2 nAChRs between intracellular and cell surface locations in M10 cells, and arrived at a figure of “about 20%” for cell surface receptors, in good agreement with the current findings. Maximally effective concentrations of agonists (with respect to up-regulation) produced no significant difference in the ratio of [3H]epibatidine binding sites at the two locations (Table 5), despite large (up to 350%) increases in the total number of nAChR. If this increase was limited to cell surface receptors, one would have seen a massive change in the ACh-sensitive portion of [3H]epibatidine binding sites. Conversely, if the increase was entirely intracellular, this would have produced a ∼2-fold decrease in the proportion of binding sites found on the cell surface. Rothhut et al. (1996) demonstrated a modest increase (≈60%) in the cell surface nAChR population of M10 cells chronically treated with (−)-nicotine (100 μm): in the current experiments, given an overall increase of 350%, a 60% increase would have changed the extracellular population to ∼10% of the total (Fig. 8). Such an increase would be compatible with the data in Table 5. It is clear that the up-regulation of [3H]epibatidine binding sites does not occur exclusively or even predominantly in the cell surface population. This result is likely to be relevant in vivo because a large pool of intracellular nAChRs also occurs in neurons (Stolberg and Berg, 1987; Conroy and Berg, 1995; Nakayama et al., 1995).

Model of cellular up-regulation mediated by a maximally up-regulating concentration of (−)-nicotine. Left model, an M10 cell in the basal state with 15% of its total receptor population expressed on the cell surface. The permeable ligand, [3H]-epibatidine labels all the receptors, whereas cell impermeable ACh displaces [3H]-epibatidine from surface receptors only, thus enabling the two populations to be distinguished. Rightmodel, result of 48-hr exposure to 50 μm (−)-nicotine. The total population of receptors has increased by 250% over basal levels and surface and intracellular populations have increased by 60% and 280%, respectively.
Is up-regulation mediated at the cell surface?
Because it is predominantly the large intracellular population of nAChR that is up-regulated and permeant agonists like nicotine are typically used in chronic treatment regimens, in vivo and in vitro, it is important to establish the locus at which up-regulation is triggered. Peng et al. (1994) demonstrated up-regulation of immunoisolated [3H]nicotine binding sites in M10 cells by chronic exposure to the quaternary nitrogen-containing compounds carbamylcholine and 1,1-dimethyl-4-phenylpiperazinium, but the cell impermeance of these compounds during the prolonged exposure was not established. Similarly, although the effective up-regulators MCC and TMA were shown to be impermeant in the displacement binding assays (Fig. 5), the conditions (3-hr incubation) did not reflect the prolonged exposure to ligand (48 hr) in the up-regulation studies. Therefore, the rate of entry of MCC and TMA into M10 cells was assessed by measuring their ability to compete for [3H]epibatidine binding sites under conditions matching those used in up-regulation experiments. The failure of the agonists tested to attain high intracellular concentrations under these conditions confirmed the cell impermeance of both TMA and MCC, showing that both compounds must up-regulate [3H]epibatidine binding sites by acting on the cell surface population of nAChRs. Moreover, the ability of MCC to block the up-regulation by nicotine (Fig. 3a) suggests that (−)nicotine must also exert its effects at the surface. Thus, both cell-permeant and -impermeant agonists produce up-regulation by interacting with the surface population of nAChR in M10 cells. Furthermore, because TMA is a full agonist of up-regulation, with a sharp dose-response relationship, and MCC is a weak partial agonist of up-regulation displaying a much shallower dose-response relationship, the full gamut of up-regulatory effects may be attributed to interactions between ligands and surface receptors.
This study has demonstrated that up-regulation of α4β2 nAChRs in M10 cells, in response to chronic drug application, is mediated by a state of the receptor that does not conform to the classically defined active or desensitized states. Furthermore, up-regulation is produced by interaction with cell surface receptors, but the increase in nicotinic binding sites is largely confined to the intracellular nAChR population.
Footnotes
-
Send reprint requests to: Dr. S. Wonnacott, Dept. of Biology and Biochemistry, University of Bath, Bath, BA2 7AY, United Kingdom. E-mail: s.wonnacott{at}bath.ac.uk
-
↵1 Current affiliation: Institute for Behavioral Genetics, University of Colorado, Denver, CO 80309-0447.
-
This work was supported by Medical Research Council Grant 9619434 (S.W.). C.G.V.S. is the recipient of a postgraduate studentship from the Biotechnology and Biological Sciences Research Council.
- Abbreviations:
- nAChR
- nicotinic acetylcholine receptor
- DEC
- decamethonium
- DHβE
- dihydro-β-erythroidine
- dTC
- d-tubocurarine
- HEX
- hexamethonium
- MCC
- methylcarbamylcholine
- MLA
- methyllycaconitine
- TMA
- tetramethylammonium
- mAb
- monoclonal antibody
- DMEM
- Dulbecco’s modified Eagle’s medium
- PBS
- phosphate-buffered saline
- Received October 9, 1997.
- Accepted January 29, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}