


Abstract
We stably transfected human kidney embryonic 293 cells with the rat neuronal nicotinic acetylcholine receptor (nAChR) α3 and β4 subunit genes. This new cell line, KXα3β4R2, expresses a high level of the α3/β4 receptor subtype, which binds (±)- [3H]epibatidine with aK d value of 304±16 pm and a B max value of 8942 ± 115 fmol/mg protein. Comparison of nicotinic drugs in competing for α3/β4 receptor binding sites in this cell line and the binding sites in rat forebrain (predominantly α4/β2 receptors) revealed marked differences in theirK i values, but similar rank orders of potency for agonists were observed, with the exception of anatoxin-A. The affinity of the competitive antagonist dihydro-β-erythroidine is >7000 times higher at α4/β2 receptors in rat forebrain than at the α3/β4 receptors in these cells. The α3/β4 nAChRs expressed in this cell line are functional, and in response to nicotinic agonists, 86Rb+ efflux was increased to levels 8–10 times the basal levels. Acetylcholine, (−)-nicotine, cytisine, carbachol, and (±)-epibatidine all stimulated86Rb+ efflux, which was blocked by mecamylamine. The EC50 values for acetylcholine and (−)-nicotine to stimulate 86Rb+ effluxes were 114 ± 24 and 28 ± 4 μm, respectively. The rank order of potency of nicotinic antagonists in blocking the function of this α3/β4 receptor was mecamylamine >d-tubocurarine > dihydro-β-erythroidine > hexamethonium. Mecamylamine, d-tubocurarine, and hexamethonium blocked the function by a noncompetitive mechanism, whereas dihydro-β-erythroidine blocked the function competitively. The KXα3β4R2 cell line should prove to be a very useful model for studying this subtype of nAChRs.
Neuronal nAChRs are ligand-gated cation channels composed of α and β subunits. Eight different α subunits (α2–α9) and three different β subunits (β2–β4) have been identified in vertebrate neuronal tissue, which allows for the possibility of multiple nAChR subtypes that display a range of functional and pharmacological characteristics. In rat brain, the two most abundant nAChR subtypes (as measured by the density of binding sites) are one composed of α4 and β2 subunits (Whiting et al., 1991; Flores et al., 1992) and another that may be composed of α7 subunits only (Schoepfer et al., 1990; Seguela et al., 1993). The relative abundance of the mRNA encoding nAChR subunits in brain is consistent with the prevalence of these two receptor subtypes (Wada et al., 1989; Seguela, et al., 1993). However, the CNS expresses mRNA for all of the nAChR subunits, and despite their lower abundance in brain overall, the receptors that contain α3 subunits may mediate important effects of acetylcholine and nicotine in specific regions of the brain (Mulle et al., 1991; Connolly et al., 1995; Clarke and Reuben, 1996; Albuquerque et al., 1997; Kulak et al., 1997), in the retina (McKay et al., 1994), and in the spinal cord (Flores et al., 1997).
In addition to their roles in the CNS, nAChRs comprised of α3 subunits in combination with either β2 or β4 subunits may play fundamental roles in peripheral neuronal tissues, including neurons of mammalian sympathetic ganglia (Covernton et al., 1994;Mandelzys et al., 1994), parasympathetic ganglia (Pothet al., 1997), sensory neurons such as the trigeminal ganglia (Flores et al., 1996), and adrenal chromaffin cells (Rogers et al., 1992; Campos-Caro, 1997). Nicotinic receptors containing α3 subunits also may predominate in chick ciliary ganglia (Boyd et al., 1988; Couturier et al., 1990), superior cervical ganglia (Couturier et al., 1990; Vernallis et al., 1993; Conroy and Berg, 1995), and dorsal root ganglion cells (Boyd et al., 1991).
Compared with the α4/β2 nAChR in the CNS, receptors that contain α3 subunits seem to have much lower affinity for most nicotinic agonists; therefore, until recently, they could not be reliably measured or studied with the available radiolabeled ligands. This fundamental obstacle has hindered studies of the pharmacology and regulation of the binding sites of these receptor subtypes. EB, in contrast to most other nicotinic agonists, has high affinity for ganglionic-type receptors (Badio and Daly, 1994); in fact, [3H]EB binds with high affinity to several different defined nAChR subtypes in stably transfected cell lines (Xiaoet al., 1996), as well as to the receptor or receptors in rat adrenal gland (Houghtling et al., 1995) and trigeminal ganglia (Flores et al., 1996). Furthermore, the iodinated analog of EB, [125I]EB (formerly [125I]IPH), labels receptors in the superior cervical ganglia (Dávila-Garcı́a et al., 1997). These ganglionic tissues seem to contain nicotinic receptors composed predominantly of α3 subunits in combination with either β2 or β4 subunits, and in some cases, there may be an additional α5 subunit (Conroy et al., 1992; Conroy and Berg, 1995).
Another critical obstacle to the characterization of the pharmacology and regulation of nAChR subtypes containing α3 subunits is that the tissues in which they are found in high concentration, such as autonomic ganglia, adrenal gland, and specific nuclei within the brain, may contain more than one subtype of nicotinic receptor, making the assignment of characteristics to any one subtype difficult. Furthermore, these tissues provide only a very limited amount of tissue for study because of their relatively small size.
To study the pharmacological and functional characteristics of an important nAChR subtype that contains α3 subunits, we transfected HEK 293 cells with the genes encoding rat α3 and β4 nicotinic receptor subunits. A stable clonal cell line has been established, and it expresses this nAChR subtype at a very high density. Here, we report the characteristics of the α3/β4 nAChR binding site labeled by [3H]EB and the activation of this receptor ligand-gated ion channel by nicotinic agonists.
Experimental Procedures
Materials and drugs.
Tissue culture medium, serum, antibiotics, restriction endonucleases, modifying enzymes, and molecular size standards were obtained from GIBCO BRL (Gaithersburg, MD). [3H]EB and86RbCl were supplied by New England Nuclear Research Products (Boston, MA). [α-35S]dATPs, [α-32P]CTP, and [γ-32P]ATP were obtained from Amersham (Arlington Heights, IL). DNA sequencing reagents were purchased from United States Biochemicals (Cleveland, OH). Electrophoresis reagents were purchased from BioRad Laboratories (Melville, NY). All other chemicals were purchased from Sigma Chemical (St. Louis, MO) unless otherwise stated. A-85380, (+)-anatoxin-A, DHβE, and EB were from Research Biochemicals International (Natick, MA). (−)-Nicotine, acetylcholine, carbachol, d-tubocurarine, and cytisine were from Sigma Chemical. Mecamylamine was from Merck Sharp & Dohme Research Lab (Rahway, NJ). Two plasmids, pPCA48E-3 and pZPC13, which carry the cDNA clones of rat neuronal nAChR subunit α3 and β4 genes, were described previously (Boulter et al., 1987; Duvoisin et al., 1989) and were generously provided by Dr. J. Boulter (Salk Institute, La Jolla, CA). HEK 293 cells (CRL 1573; American Type Culture Collection, Rockville, MD) were a gift from Y.-H. Wang and B.B. Wolfe (Georgetown University, Washington, D.C.).
Construction of pKXα3RC1 and pKXβ4RC1.
To constitutively express the rat neuronal nAChR α3 subunit in mammalian cells, a 1.7-kb HindIII/EcoRI fragment was isolated from the pPCA48E-3 and subcloned into the eukaryotic expression vector pcDNA3 (InVitrogen, San Diego, CA). The resulting plasmid with the α3 coding sequence in the sense orientation was designated pKXα3RC1. For generation of the eukaryotic expressable β4 gene, a 2-kbEcoRI/XbaI fragment was isolated from the pZPC13 and inserted into the pcDNA3 vector. The new construct with the β4 coding sequence in the sense orientation was denoted pKXβ4RC1. pKXα3RC1 and pKXβ4RC1 were restriction mapped and sequenced to confirm correct ligation and orientation.
Cell culture and stable transfection.
HEK 293 cells were maintained at 37° with 5% CO2 in a humidified incubator. Growth medium for the HEK 293 cells was minimum essential medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin G, and 100 μg/ml streptomycin. For transfection experiments, plasmids were linearized by restriction digestion within their prokaryotic elements. Transfection was conducted according to the calcium phosphate method (Chen and Okayama, 1987). Briefly, exponentially growing HEK 293 cells were plated onto 100-mm dishes containing 10 ml of the growth medium 24 hr before transfection. For a single transfection experiment, 1 ml of transfection mixture was made. The mixture was composed of 10 μg of linearized DNA, 125 mm CaCl2, 25 mm HEPES, 140 mm KCl, 6 mm glucose, and 0.75 mm Na2HPO4, pH 7.05. The mixture was added to the dish of cells in dropwise fashion. The cells were incubated with the transfection mixture for 16 hr in the incubator, after which the mixture was removed, and the cells were grown in fresh growth medium for 24 hr. The cells then were collected and plated at a range of densities onto six-well plates in the selection medium, which consisted of the growth medium containing 0.7 mg/ml geneticin (G418). The cells were grown in the selection medium for 3–4 weeks before G418-resistant clones were picked up by cloning cylinders. The stably transfected cells were maintained in the selection growth medium.
RNA isolation and RNase protection assay.
Total cellular RNA was isolated from cells using RNA-STAT-60, an RNA isolation reagent (Tel Test B, Friendswood, TX). DNA templates for antisense riboprobes were prepared as described previously (Xiao et al., 1995). The size of the full-length probes and the expected protected fragments of the probes are for rat α2, 421 and 332 bases; rat α3, 308 and 231 bases; rat α4, 497 and 408 bases; rat α5, 450 and 388 bases; rat β2, 328 and 266 bases; rat β4, 258 and 174 bases; and human GAPDH, 224 and 154 bases, respectively. The probes were synthesized using T7 RNA polymerase (Ambion, Austin, TX) and [α-32P]CTP. Specific activities of [α-32P]CTP used for synthesizing the probes of rat nAChR subunit genes and the probe of GAPDH were 800 and 32 Ci/mmol, respectively. Approximately 50 μg of total RNA was hybridized with probes overnight at 42°. Nonprotected probes were digested with a mixture of RNase A and RNase T1, and the samples were processed using the RPA II kit (Ambion). The sizes of protected fragments were determined by electrophoresis on a 6% denaturing polyacrylamide gel.
Ligand binding studies.
Cultured cells at >70% confluence were harvested in 50 mm Tris·HCl, pH 7.4, and homogenized with a Polytron homogenizer (Brinkmann Instruments, Westbury, NY). Homogenates were centrifuged at 35,000 × g for 10 min, and pellets were washed twice with fresh buffer. Membrane pellets were resuspended in fresh buffer, and aliquots equivalent to 60–200 μg of protein were used for binding assays. Ligand binding was measured as described previously (Houghtling et al., 1995) with minor modifications. Briefly, membrane preparations were incubated with [3H]EB for 4 hr at 24° in a final volume of 2.5 ml. Bound and free ligands were separated by vacuum filtration through Whatman GF/C filters treated with 0.5% polyethylenimine. The filter-retained radioactivity was determined by liquid scintillation counting. Total binding and nonspecific binding was determined in the absence and presence of (−)-nicotine (300 μm), respectively. Specific binding was defined as the difference between total binding and nonspecific binding. Typically, total binding was measured in duplicate, and nonspecific binding was measured in singlet. In saturation binding experiments, receptor densities (B max) and dissociation constants (Kd ) were determined by nonlinear least-squares regression analyses (Accufit Saturation Two-Site Program; Beckman Instruments, Fullerton, CA). The one-site model was accepted unless the two-site model gave a statistically better fit of the data (p < 0.05 by F test). Hill coefficients (nH ) of specific binding curves were determined by linear regression analyses of those specific binding values that fell between 10% and 90% ofB max. The affinities of drugs at the receptors were determined from binding inhibition curves, in which a series of concentrations of each drug was incubated with a single concentration of [3H]EB. In some studies, the type of binding inhibition mechanism was investigated by including the drug in a [3H]EB binding saturation assay. Hill coefficients (nH ) and IC50 values of binding inhibition curves were determined by linear regression analyses. The inhibition constants (Ki ) were calculated according to the Cheng-Prusoff equation (Cheng and Prusoff, 1973).
To determine the fraction of nAChRs on the cell surface, [3H]EB binding was measured in intact cells according to a method that was adapted from one used for other receptors (Koenig and Edwardson, 1997) with minor modifications. Briefly, cells were plated onto 24-well plates coated with poly-d-lysine and grown in selection medium for 14–20 hr to reach −30% confluency. The medium was removed and replaced by 20 mm HEPES buffer, pH 7.4, containing minimum essential medium and 3 nm [3H]EB. The intact cells were incubated with [3H]EB for 2 hr, after which the buffer was removed by aspiration and the cells were washed rapidly three times with buffer. The cells then were lysed by the addition of 1 ml of 0.1 m NaOH, and the lysate in each well was counted in a liquid scintillation counter. Nonspecific binding was determined in parallel samples incubated with [3H]EB in the presence of either 300 μm nicotine, which crosses cell membranes and thus has access to both cell surface and intracellular receptors, or 30 mm carbachol, a quaternary ammonium nicotinic agonist that does not easily cross membranes and thus has access only to cell surface receptors. In this method, aggregate specific binding is defined as total binding minus nonspecific binding measured in the presence of nicotine, whereas cell surface specific binding is defined as total binding minus nonspecific binding measured in the presence of the nonpermeant carbachol.
86Rb+efflux assay.
The function of nAChRs expressed in the transfected cells was measured using a86Rb+ efflux assay as described by Lukas and Cullen (1988), with modifications. In brief, identical aliquots of cells in the selection growth medium were plated onto 24-well plates coated with poly-d-lysine. The plated cells were grown at 37° for 18–24 hr to reach 70–95% confluence. The cells then were incubated in growth medium (0.5–1 ml/well) containing 86RbCl (2 μCi/ml) for 4 hr at 37°. The loading mixture was aspirated, and the cells were washed three times with 1 ml aliquots of buffer (15 mm HEPES, 140 mm NaCl, 2 mm KCl, 1 mmMgSO4, 1.8 mmCaCl2, 11 mm glucose, pH 7.4) for 30 sec, 5 min, and 30 sec, respectively. One milliliter of buffer, with or without drugs, was added to each well. After incubation for 2 min, the assay buffer was collected, and the amount of86Rb+ in the buffer was determined. Cells were lysed by the addition of 1 ml of 0.1m NaOH to each well, and the lysate was collected for determination of the amount of86Rb+ that was in the cells at the end of the efflux assay. Radioactivity of the assay samples and lysates was measured by liquid scintillation counting. Total loading (cpm) was calculated as the sum of the assay sample and the lysate of each well. Values of total loading were 100,000–200,000 cpm/well. The amount of 86Rb+ efflux was expressed as a percentage of86Rb+ loaded. Stimulated86Rb+ efflux was defined as the difference between efflux in presence and absence of nicotinic agonists. The EC50, IC50, and nH values were estimated by linear regression analysis. Experiments with antagonists were done in two different ways. For obtaining an IC50 value, inhibition curves were constructed in which different concentrations of an antagonist were included in the assay to inhibit efflux stimulated by 100 μm nicotine. For determination of the mechanism of antagonist blockade, concentration-response curves for activation by nicotine were constructed in the absence or presence of one or more concentrations of an antagonist.
Ca2+ and Na+ imaging assays.
Effects of nicotinic drugs on changes in [Ca2+]i or [Na+]i were evaluated by Ca2+ and Na+ imaging assays. These assays were performed on an Attofluor Fluorescence Imaging System (Attofluor, Rockville, MD) with an Axiovert 135 fluorescence microscope (Zeiss, Germany). Cells were plated onto 25-mm glass coverslips (Fisher, Pittsburgh, PA) or 35-mm plastic tissue culture dishes (Nunc, Roskilde, Denmark), both precoated with poly-d-lysine. The plated cells were grown for 36–72 hr at 37°. Culture medium was removed, and cells were rinsed three times with Locke’s buffer with the following composition: 140 mmNaCl, 5.6 mm KCl, 3.6 mmNaHCO3, 1.3 mmCaCl2, 1 mmMgCl2, 5.6 mm glucose, and 10 mm HEPES, pH 7.4.
For measuring [Ca2+]i, cells were loaded with 5–10 μm Fura-2/AM (Molecular Probes, Eugene, OR) in Locke’s buffer supplemented with 0.1% Pluronic F-127 (Molecular Probes) to facilitate dye loading. The cells were incubated with the loading solution for 30 min at room temperature and then washed three times with Locke’s buffer. Dishes and coverslips were mounted onto the fluorescence microscope stage and then incubated with the Locke’s buffer for 15–20 min to complete Fura-2/AM deesterification. Buffer with or without drugs was delivered at a flow rate of 1 ml/min. Fluorescence was measured at room temperature at excitation wavelengths of 334 and 380 nm using dry objective 40/0.6 LD Achroplan for cells plated onto plastic dishes or oil immersion objective 40/1.3 Fluar Ph3 for cells seeded onto glass coverslips. Emission wavelengths, monitored with intensified CCD cameras, were >420 nm for objectives 40/0.6 LD Achroplan and >510 nm for objectives 40/1.3 Fluar Ph3. The [Ca2+]i was calculated from the ratio (R) of fluorescence signals obtained at the two excitation wavelengths as described by Grynkiewicz et al. (1985). Rmin (ratio F334/F380) values were measured in each experiment by incubation of cells in Locke’s buffer supplemented with 10 mm EGTA and 10 μmionomycin (Calbiochem, San Diego, CA) for 5–10 min in the absence of Ca2+. Rmax values were obtained by subsequent incubation of the cells in Locke’s buffer supplemented with 10 mm CaCl2 and 10 μm ionomycin for 5–10 min. The background fluorescence then was measured in the presence of 10 mmMnCl2. The Kd value of the Fura-2/Ca2+ complex used for the calculation was 386 nm without correction for room temperature.
Imaging of Na+ was carried out as described above for Ca2+ imaging with the following differences. Cells were loaded with 10 μm SBFI/AM (Molecular Probes) in Locke’s buffer supplemented with 0.1% Pluronic F-127 for 45–60 min at room temperature. To estimate [Na+]i, cells were permeabilized with 5 μm Gramicidine A (Molecular Probes). Rmax values (F334/F380 ratio) were obtained when cells were permeabilized in regular Locke’s buffer (154 mmNa+), whereas Rmin values were measured when sodium ions were completely replaced by K+ ions.
Results
Stable transfections of HEK 293 cells.
Four sets of plasmid DNA were used for transfection experiments independently: (1) pcDNA3 (vector only), (2) pKXα3RC1 (α3 subunit gene), (3) pKXβ4RC1 (β4 subunit gene), and (4) the combination of pKXα3RC1 and pKXβ4RC1 (α3 and β4 subunit genes). From each of the transfections, 36–72 stable, G418-resistant cell clones were isolated after cultivation in selection medium for 3–4 weeks. These clonal cell lines were then grown in selection medium for an additional 4 weeks.
Initial screening of the G418-resistant clonal cells was carried out by using multiple-probe RNase protection assays to measure mRNA in the total RNA preparation isolated from each clone. Stably transfected cell lines representing each of the transfection experiments were selected and designated as (1) KXC1 (transfected with the vector pcDNA3 only), (2) KXα3R1 (expressing the α3 subunit gene only), (3) KXβ4R1 (expressing the β4 subunit gene only), and (4) KXα3β4R2 (expressing both the α3 and β4 subunit genes). An assessment of the mRNA for six different rat nAChR subunits (α2–α5, β2, and β4) in the parent HEK 293 cells and in each of the four clonal cell lines is shown in Fig. 1. As expected, there was no detectable expression of mRNA for any of the six nAChR subunit genes in either HEK 293 or KXC1 cells. However, the mRNA levels for the appropriate subunit gene in KXα3R1 and KXβ4R1 were quite high, as were the mRNA levels of both subunit genes in KXα3β4R2 (Fig. 1).
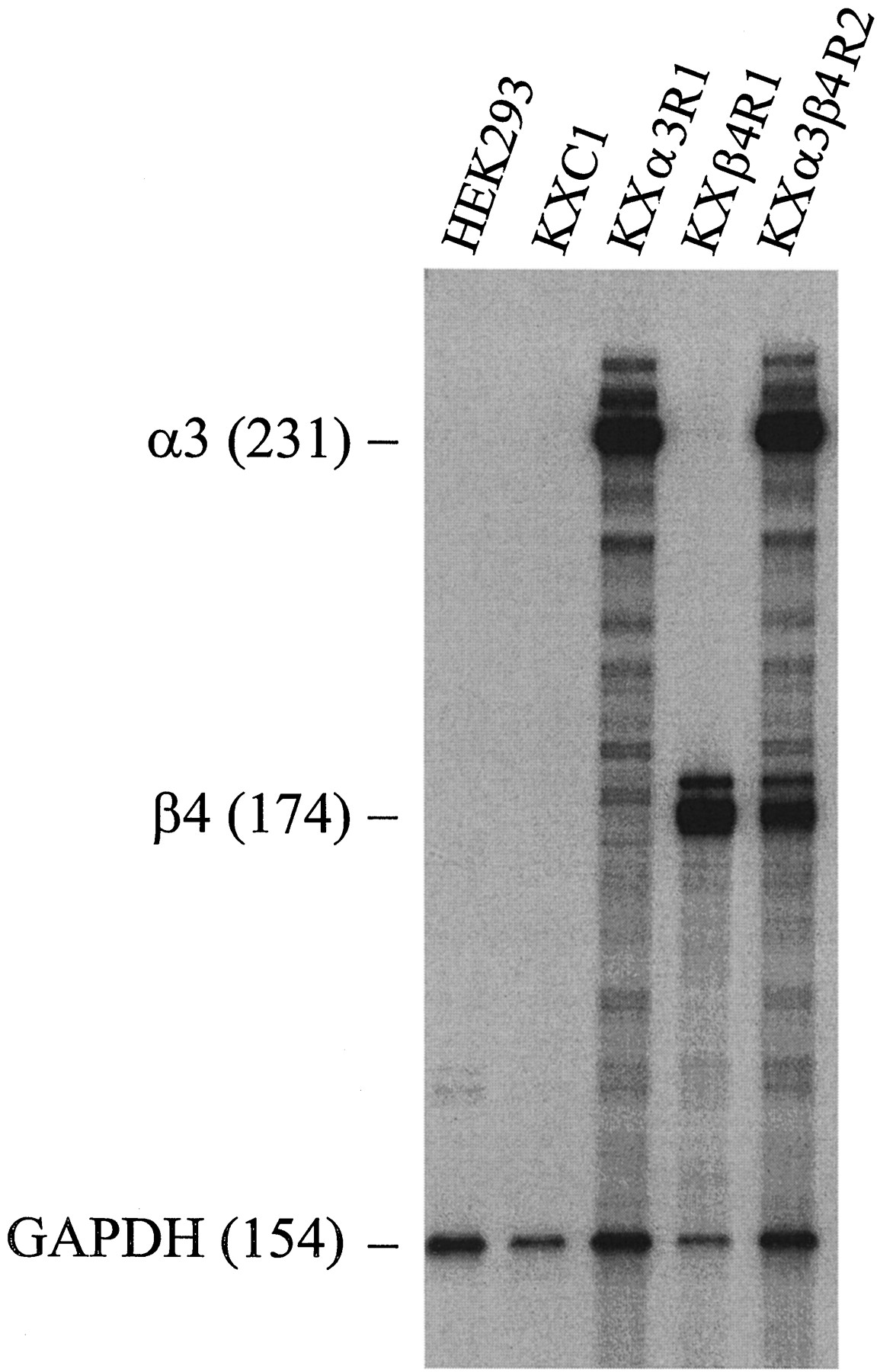
Multiple-probe RNase protection assay of expression of nAChR subunit genes in HEK 293 cells and transfected cell lines. RNase protection assay was carried out as described in Experimental Procedures. Total RNA from each cell line was hybridized with a mixture of antisense probes labeled with [32P] for six rat nAChR subunit genes (α2, α3, α4, α5, β2, β4) and the human GAPDH gene. Nonprotected probes were digested with a mixture of RNase A and RNase T1. The sizes of protected RNA fragments were determined by electrophoresis on a 6% denaturing polyacrylamide gel. HEK 293, the parental cell line for transfection experiments; KXC1, the cell line that was transfected with the vector, pcDNA3; KXα3R1, the cell line that was transfected with rat α3 subunit gene; KXβ4R1, the cell line that was transfected with rat β4 subunit gene; KXα3β4R2, the cell line that was transfected with both rat α3 and β4 subunit genes. Labels, specific protected fragments and their sizes (bases). The GAPDH was used as an internal and loading control in the experiments.
The clones that expressed the highest levels of the expected nAChR subunit mRNA or mRNAs were tested for their ability to bind [3H]EB. At a [3H]EB concentration of ∼3 nm, essentially no specific binding (<3 fmol/mg of protein) was detected in membrane homogenates from HEK 293 cells, from cells transfected with pcDNA3 only, or from cells transfected singly with either the α3 or β4 subunit gene (Fig.2). In contrast, in membrane homogenates from KXα3β4R2 cells, which had been transfected with both α3 and β4 genes, specific binding of [3H]EB was quite high, displaying a density of binding sites >8000 fmol/mg of protein (Fig. 2). As a point of comparison, the density of nicotinic receptors labeled by [3H]EB in rat brain membrane homogenates (primarily α4/β2 receptors) in parallel assays was ∼88 fmol/mg protein, or ∼1% of the density of α3/β4 receptors in this cell line (Fig. 2, inset). The KXα3β4R2 cell line was selected for further study and has been continually cultured in our laboratory for >12 months with no significant change in the levels of expression of the mRNA encoding α3 or β4 subunits or in the binding to or function of the receptor.
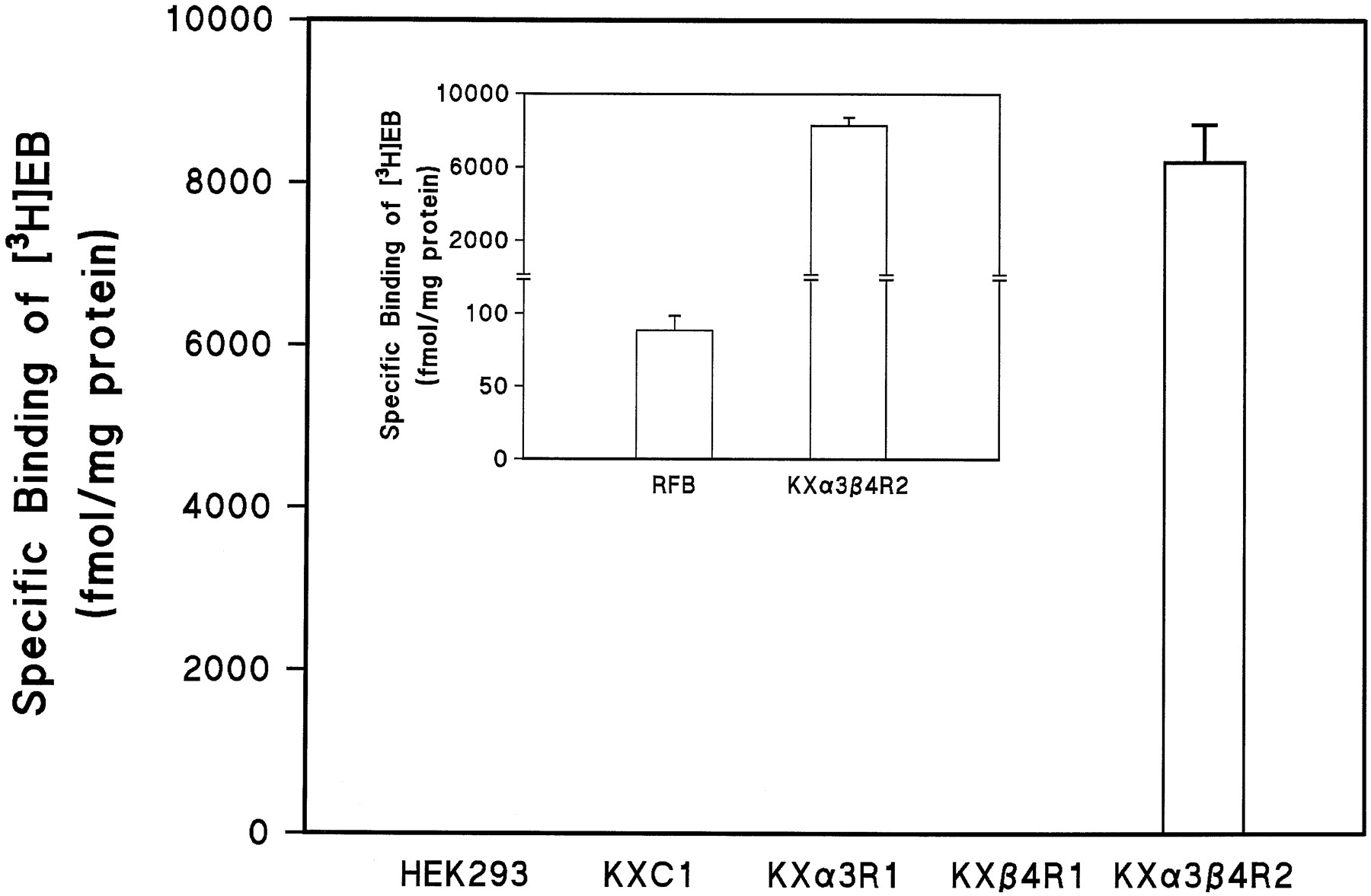
Specific binding of [3H]EB to membrane homogenates from HEK 293 cells, transfected cells, and rat forebrain (RFB). Binding assays were carried out as described in Experimental Procedures. The concentration of [3H]EB used in these experiments was ∼3 nm. Total binding and nonspecific binding was determined in the absence and presence of 300 μm (−)-nicotine, respectively. Specific binding was defined as the difference between total binding and nonspecific binding. See the legend for Fig. 1 for designations of cell lines. Values are the mean ± standard error from three to six independent experiments.
Analysis of [3H]EB binding to α3/β4 receptors in KXα3β4R2 cell membrane homogenates.
Specific binding of [3H]EB in cell membrane homogenates was saturable and represented >98% of the total binding throughout most of the concentration range used (Fig.3A). The high density of nicotinic receptor binding sites in KXα3β4R2 cells and the very low nonspecific binding of [3H]EB allowed binding to be analyzed over a wide range of concentrations. Over this range, [3H]EB binding fit a model for a single site with a Hill coefficient (nH ) close to 1, a Kd value of ∼300 pm, and a site density of ∼8900 fmol/mg of protein (Fig. 3B, see figure legend for calculated values).

Saturation binding of [3H]EB to KXα3β4R2 membrane homogenates. Saturation binding assays were carried out as described in Experimental Procedures. Plots shown are a representative of four saturation binding experiments. A, Binding curves showing specific binding (•) and nonspecific binding (○).Inset, semilogarithmic plot of the specific binding. B, Scatchard plot of the specific binding. Inset, Hill plot of the specific binding. Data from saturation binding experiments were analyzed by nonlinear least-squares regression (Accufit Saturation Two-Site Program) for curve-fitting andK d andB max estimation. TheK d andB max values were 304 ±16 pm and 8942 ± 115 fmol/mg protein (mean ± standard error, four experiments), respectively. The Hill coefficient, estimated from Hill plots, was 1.06 ± 0.06 (mean ± standard error, four experiments).
Pharmacological characteristics of α3/β4 receptor binding sites in KXα3β4R2 cells.
The pharmacological characteristics of the α3/β4 nicotinic receptor binding sites in these cells were examined in binding competition assays in which drugs competed against ≈500 pm [3H]EB for binding sites in cell membranes (Fig. 4A, Table1). EB was by far the most potent drug in competing for α3/β4 receptor binding sites, with an affinity >2 orders of magnitude higher than that of any other drug tested. For example, among nicotinic agonist drugs, EB was >100 times more potent than anatoxin-A and A85380, >500 times more potent than cytisine, and >1000 times more potent than nicotine, acetylcholine, or carbachol (Fig. 4, Table 1). Among nicotinic antagonists,d-tubocurarine was nearly 10 times more potent than DHβE in competing for α3/β4 receptor binding sites (Fig. 4, Table 1). Neither mecamylamine nor hexamethonium at concentrations up to 1 mm competed effectively for [3H]EB binding sites.

Comparison of drug competition profiles of nAChRs in membrane homogenates from KXα3β4R2 cells and rat forebrain. Binding assays were carried out as described in Experimental Procedures. Competition for [3H]EB binding sites in KXα3β4R2 cell homogenates (A) and in rat forebrain membrane homogenates (B). Data shown are representative of five competition binding experiments. The concentration of [3H]EB used in competition experiment 5 was ∼500 pm. See Table 1 for a summary and analyses of data from all experiments.
Comparison of pharmacological properties of nAChR ligand binding in membrane homogenates from KXα3β4R2 cell and rat forebrain
For comparison, the affinities of these same drugs were examined in parallel at nicotinic receptor binding sites labeled by [3H]EB in rat forebrain membranes, which are predominantly the α4/β2 subtype of nicotinic receptor (Whitinget al., 1991; Flores et al., 1992). All of the drugs examined were more potent in competing for the α4/β2 receptor binding sites in rat forebrain than the α3/β4 receptors in KXα3β4R2 cells. This can be seen in the leftward shift of the curves in Fig. 4B compared with those in Fig. 4A. Furthermore, it should be noted that the affinity of [3H]EB is ∼7 times higher at the α4/β2 receptors in rat forebrain than at the α3/β4 receptors in KXα3β4R2 cells; therefore, the fraction of receptors occupied by [3H]EB in rat forebrain was greater than that in the cells (90% versus 57%). Thus, as shown in Table 1, after accounting for this difference in receptor occupancy (Cheng and Prusoff, 1973), the differences in affinity of these drugs at the two receptor subtypes is actually greater than they appear from the curves in Fig. 4. The drugs that show the greatest differences in affinity at the two receptor subtypes are DHβE and A85380, which show, respectively, 7565 and 759 times higher affinity for α4/β2 receptors than for α3/β4 receptors. Cytisine, nicotine, and acetylcholine, all of which have been used as radioligands to label nicotinic receptors in brain, also have much higher affinity for α4/β2 receptors than for α3/β4 receptors (Table 1).
Among agonists, the rank order of binding affinities at the two receptor subtypes is similar, with only the relative position of anatoxin-A changing. In contrast, the order of affinities of the two antagonists, d-tubocurarine and DHβE, is very different at the two receptor subtypes. Thus, at α3/β4 receptorsd-tubocurarine binds with almost 10 times higher affinity than DHβE, whereas at α4/β2 receptors, DHβE has 77 times higher affinity than d-tubocurarine (Table 1). Interestingly, the Hill coefficients for d-tubocurarine competing at the α3/β4 receptor binding sites and for both antagonists competing at the binding sites in brain are significantly <1 (Table 1). Hexamethonium competed weakly for [3H]EB binding sites in brain, whereas mecamylamine displayed virtually no affinity.
Assessment of α3/β4 receptor function in KXα3β4R2 cells.
To examine the function of the α3/β4 nicotinic receptor in these cells, we measured ion flux stimulated by nicotinic agonists. Three types of ion flux were measured: efflux of86Rb+ from preloaded cells, increases in intracellular Ca2+ as measured by Fura-2/Ca2+ imaging, and increases in intracellular Na+ as measured by SBFI/Na+ imaging.
Stimulation of 86Rb+ efflux.
As shown in Fig. 5A, nicotine stimulated86Rb+ efflux from KXα3β4R2 cells, which express both α3 and β4 subunits, but not from the parent HEK 293 cells or from HEK 293 cells transfected with either the vector only or the α3 or β4 subunit only. Both nicotine and acetylcholine stimulated86Rb+ efflux from KXα3β4R2 cells in a concentration-dependent manner (Fig. 5B), with EC50 values of 28 and 114 μm, respectively. Maximal 86Rb+efflux, 8–10 times greater than the basal efflux, occurred at a nicotine concentration of ∼300 μm and an acetylcholine concentration of 2 mm. Concentrations of >1 mmnicotine and 10 mm acetylcholine tended to decrease 86Rb+ efflux, possibly due to desensitization or agonist blockade of the channel (see Fig. 6B). The Hill coefficients for functional activation of the receptor by nicotine and acetylcholine were 1.6 and 1.5, respectively, and in both cases, these values were significantly different from 1 (p < 0.01). In addition to nicotine and acetylcholine, cytisine (30 μm), carbachol (300 μm), and EB (30 nm) all stimulated 86Rb+ efflux from KXα3β4R2 cells, and in each case, the efflux was nearly completely blocked by 10 μm mecamylamine (Fig. 5C).

Effects of nicotinic agonists and antagonists on86Rb+efflux from KXα3β4R2 cells.86Rb+ efflux was measured as described in Experimental Procedures. Cells were loaded with86Rb+ and then exposed to buffer alone or buffer containing nicotinic drugs for 2 min. A, Effects of nicotine on86Rb+ efflux from HEK 293 cells and transfected cells. Cells were exposed to buffer alone (basal efflux) or buffer containing 100 μm nicotine. The amount of86Rb+ efflux was expressed as a percentage of86Rb+ loaded. Data shown are the mean ± standard error from three separate measurements. B, Concentration-dependent activation of nAChR function in KXα3β4R2 cells by nicotine and acetylcholine. The amount of86Rb+ efflux was expressed as a percentage of86Rb+ loaded. The stimulated86Rb+ efflux was defined as the difference between efflux with and without nicotinic agonists (i.e., basal efflux, normally 4–8%, has been subtracted). Data shown are representative of five independent experiments. Parameters were estimated by Hill plot analyses. The EC50 values (mean ± standard error) for nicotine and acetylcholine were 28.2 ± 3.7 and 113.7 ± 24.2 μm, respectively. The Hill coefficients (mean ± standard error) for nicotine and acetylcholine were 1.59 ± 0.1 and 1.48 ± 0.04, respectively. These Hill coefficients are significantly different from 1 (p < 0.01). The efficacy of acetylcholine relative to nicotine was 98.8 ± 3% (mean ± standard error). C, Effects of nicotinic agonists on86Rb+ efflux from KXα3β4R2 cells. Cells were exposed to buffer alone (basal efflux) or buffer containing nicotinic agonists, with or without mecamylamine. The amount of86Rb+ efflux was expressed as a percentage of86Rb+ loaded. The concentrations of drugs used were 30 μm (−)-nicotine (Nic), 10 μm mecamylamine (Mec), 30 μmcytisine (Cyt), 300 μm carbachol (Carb), and 30 nm (±)-EB. Data shown are the mean ± standard error from at least two independent measurements, each carried out in quadruplicate. D, Concentration-dependent inhibition by nicotinic antagonists of nicotine-stimulated 86Rb+ efflux from KXα3β4R2 cells. Cells were exposed to buffer alone or buffer containing 100 μm nicotine with or without antagonists. Control equaled the amount of 86Rb+ efflux stimulated by 100 μm nicotine in the absence of antagonists. Parameters were estimated from Hill plots. The IC50 and Hill coefficients were, respectively, 1.2 ± 0.4 μm and 1.15 ± 0.1 (three experiments) for mecamylamine, 9.6 ± 1.4 μm and 0.78 ± 0.03 (four experiments) for d-tubocurarine, 100 ± 6 μm and 1.14 ± 0.16 (three experiments) for DHβE, and 208 ± 27 μm and 1.20 ± 0.14 (five experiments) for hexamethonium (values are mean ± standard error).

Effect of mecamylamine, hexamethonium, dihydro-β-erythroidine, and d-tubocurarine on the concentration-response relationship for nicotine-stimulated86Rb+efflux from KXα3β4R2 cells.86Rb+ efflux was measured as described in Experimental Procedures. Cells were loaded with86Rb+ and then exposed to buffer alone or buffer containing increasing concentrations of nicotine with or without antagonists for 2 min. The maximal stimulated86Rb+ efflux was defined as the difference between maximal efflux in the presence of nicotine and basal efflux measured in buffer alone. Data shown are representative of five independent experiments. A, Effects of mecamylamine and hexamethonium.Control, concentration-response curve for activation by nicotine alone; Hex, concentration-response curve for activation by nicotine in the presence of 300 μmhexamethonium. Mec, concentration-response curve for activation by nic- otine in the presence of 2 μm mecamylamine. B, Effect of DHβE. Control, concentration-response curve for activation by nicotine alone. DHβE, concentration-response curve for activation by nicotine in the presence of 250 μm DHβE. C, Effect ofd-tubocurarine. Control, concentration-response curve for activation by nicotine alone.d-TC, concentration-response curve for activation by nicotine in the presence of d-tubocurarine at the indicated concentrations.
Block of nicotine-stimulated 86Rb+efflux.
The blockade of nicotine-stimulated86Rb+ efflux by several antagonists was investigated. As shown in Fig. 5D, despite the inability of mecamylamine to compete for the agonist binding site (Table 1), it blocked nicotine-stimulated86Rb+ efflux from KXα3β4R2 cells quite potently, with an IC50value of ≈1 μm and a nearly complete blockade of efflux at a concentration of 10 μm. Similarly,d-tubocurarine, DHβE, and hexamethonium blocked nicotine-stimulated ion efflux in a concentration-dependent manner, with IC50 values of ≈10, 100, and 210 μm, respectively (Fig. 5D). In contrast to these drugs, α-bungarotoxin (1.5 μm) had no effect on nicotine-stimulated 86Rb+efflux from KXα3β4R2 cells (data not shown).
Although Fig. 5D indicates the relative potency of these antagonists in blocking the α3/β4 receptor, it does not provide information about the type of block exerted by these drugs. Therefore, to investigate the mechanism of the block produced by these antagonists, concentration-response measurements for nicotine-stimulated86Rb+ efflux were made in the absence and presence of each of the drugs. As shown in Fig. 6A, in the presence of mecamylamine or hexamethonium, the maximum response elicited by nicotine was decreased, but the nicotine EC50 value was not significantly altered, indicating that both of these drugs block these nicotinic receptors by a noncompetitive mechanism. In contrast, the presence of DHβE shifted the nicotine concentration-response curve to the right (Fig. 6B), resulting in a higher EC50 value but no decrease in the maximum response that could be elicited, indicating that DHβE blocks these receptors by a competitive mechanism.
Tubocurarine is a competitive antagonist at nicotinic receptors in muscle, whereas at ganglionic nicotinic receptors, it has been reported to block the ion channel of the receptor (Ascher et al., 1979), which would be consistent with a noncompetitive mechanism. Therefore, because d-tubocurarine might help to distinguish among nicotinic receptor subtypes, we investigated the type of mechanism by which it blocks α3/β4 receptor function. As shown in Fig. 6C, in the presence of increasing concentrations ofd-tubocurarine, the maximum86Rb+ efflux stimulated by nicotine was progressively decreased without noticeably affecting the nicotine EC50 value. Furthermore, this blockade by d-tubocurarine is reversible; therefore, when cells were exposed to 200 μm d-tubocurarine for 2 min (as in the 86Rb+ efflux assay) and then washed in fresh buffer, the response to nicotine was nearly completely restored (data not shown). Thus, d-tubocurarine blocks α3/β4 receptor function by a noncompetitive mechanism consistent with channel blockade.
Tubocurarine thus is unusual in that it competes with [3H]EB for the α3/β4 receptor’s agonist binding site with an apparent Ki of 23 μm (Fig. 4, Table 1), but at similar concentrations it blocks the receptor’s function by a noncompetitive mechanism, suggesting that it might be a channel blocker. Therefore, we investigated the mechanism of the inhibition byd-tubocurarine of [3H]EB binding to α3/β4 receptors in more detail by examining its effect on [3H]EB binding saturation curves and comparing it with the competitive antagonist DHβE. As shown in Fig.7, the block of [3H]EB binding by 250 μm DHβE fit a model of competitive inhibition, shifting the apparent Kd value of [3H]EB by a factor of 4 with no change in its B max (Fig. 7B). The block of [3H]EB binding by 30 μm d-tubocurarine, however, seemed to fit a model of a mixed mechanism. Competitive inhibition was indicated by the ≈3-fold shift in apparent Kd for [3H]EB, but there also was a 25% decrease in the apparent B max value for [3H]EB binding in the presence ofd-tubocurarine (Fig. 7B).
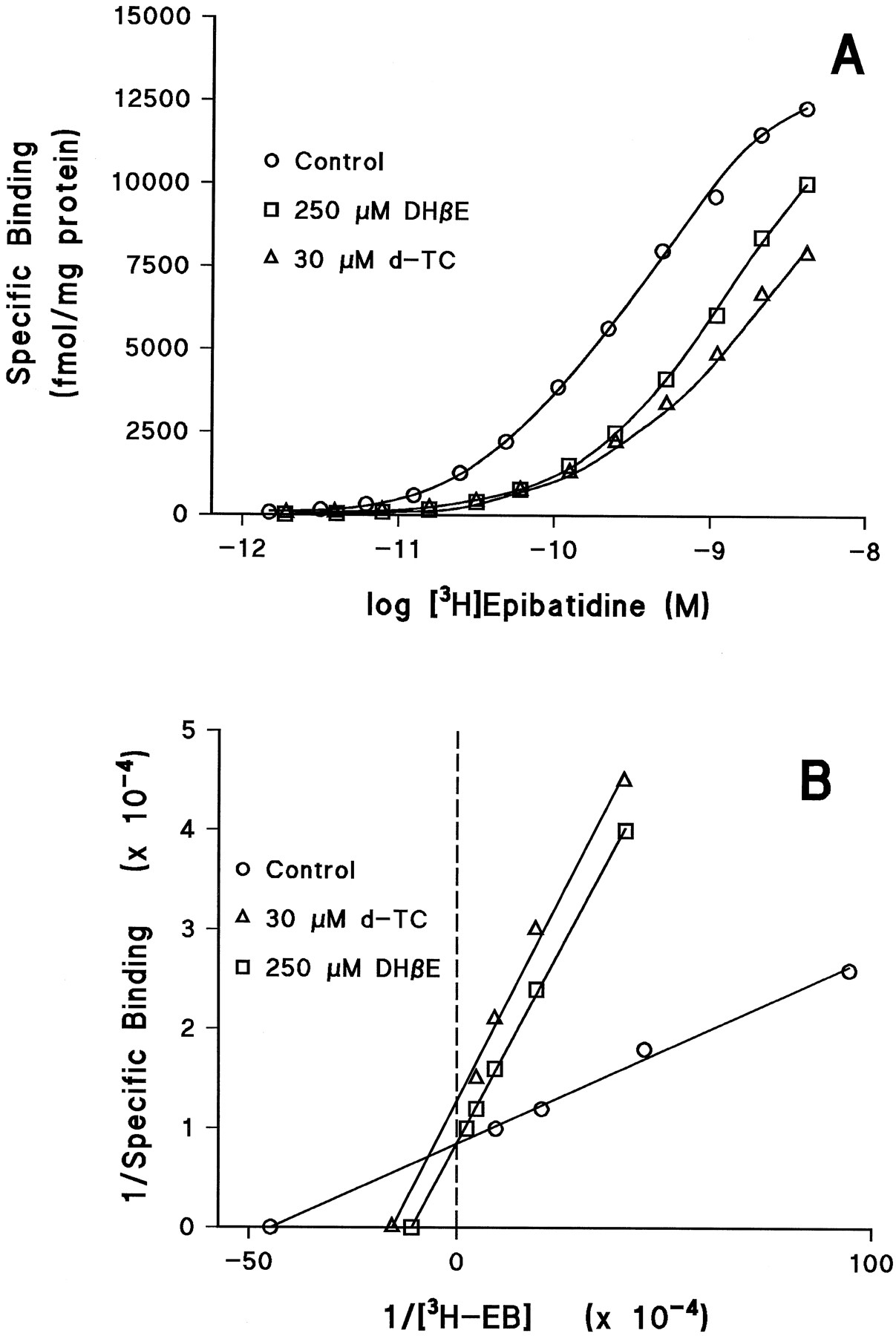
Effect of DHβE and d-tubocurarine on the saturation binding of [3H]EB to KXα3β4R2 membrane homogenates. Saturation binding assays were carried out as described in Experimental Procedures. A, Semilogarithmic plots showing specific binding of [3H]EB alone (control) and in the presence of 250 μm DHβE or 30 μm d-tubocurarine. B, Lineweaver-Burk plots of the specific binding shown in A. Data from saturation binding experiments were analyzed by nonlinear least-squares regression (Accufit Saturation Two-Site Program) for curve-fitting andK d andB max estimation. The apparentK d andB max values were, respectively, 276 pm and 12,960 fmol/mg of protein for control, 1,076 pm and 12,700 fmol/mg of protein in the presence of 250 μm DHβE, and 954 pm and 9,685 fmol/mg of protein in the presence of 30 μm d-tubocurarine.
Ca2+ ion imaging.
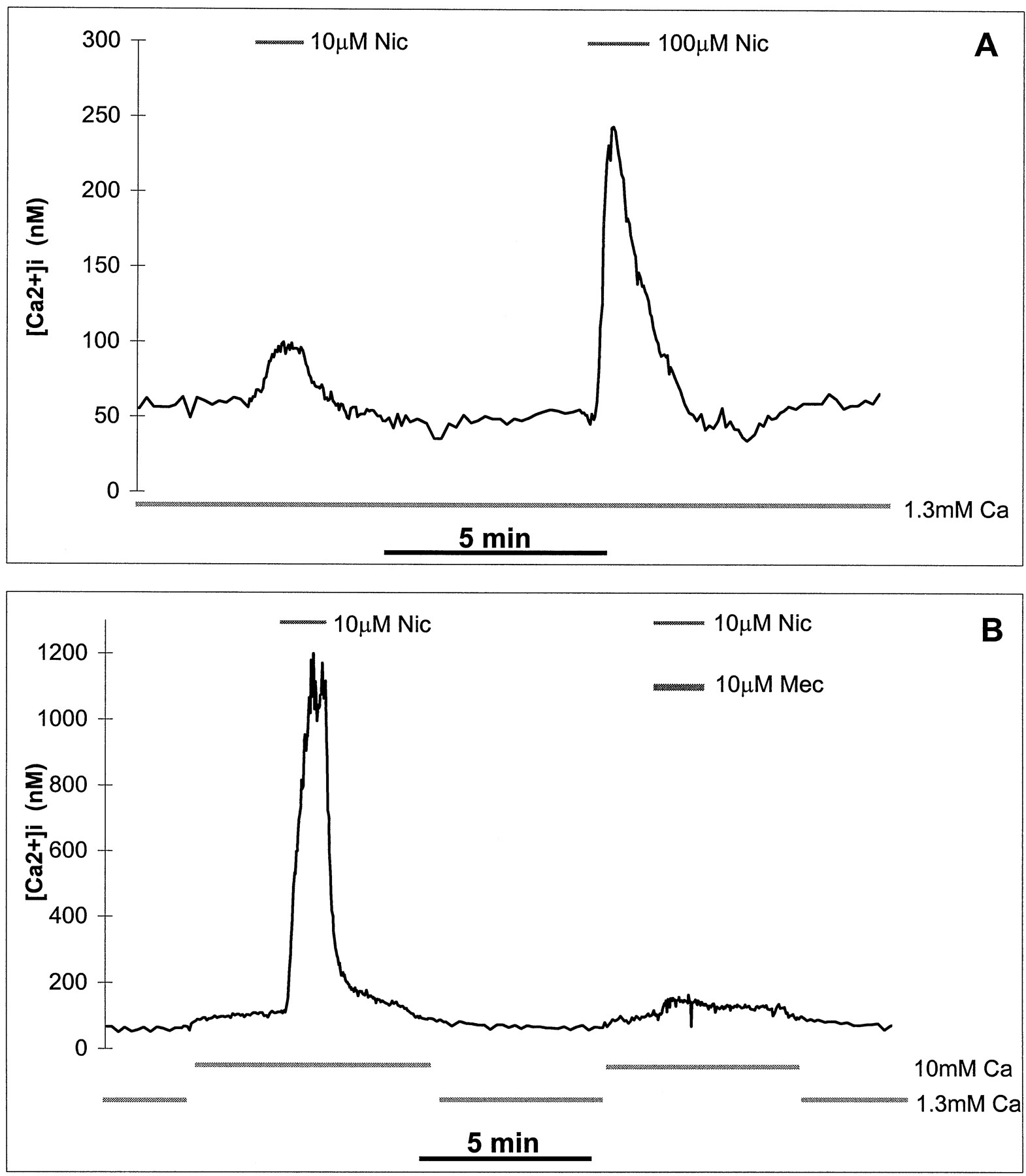
As shown in Fig.8A, nicotine stimulated an increase in [Ca2+]i concentration in KXα3β4R2 cells in a concentration-dependent manner. When the extracellular Ca2+ concentration was maintained at 1.3 mm, nicotine at concentrations of 10 and 100 μm increased [Ca2+]i levels to 2-fold and 5-fold of base-line, respectively. When the extracellular Ca2+ was raised from 1.3 to 10 mm(Fig. 8B), the basal level of [Ca2+]i increased slightly, but the increase in [Ca2+]i stimulated by 10 μm nicotine was markedly enhanced to >10-fold of base-line. This nicotine stimulated increase in [Ca2+]i was almost completely blocked by 10 μm mecamylamine (Fig. 8B).

Effects of nicotine on [Ca2+]i in KXα3β4R2 cells. Changes in [Ca2+]i were measured and calculated as described in Experimental Procedures. Cells were loaded with the Ca2+ fluorescent indicator Fura-2 and then exposed to buffer alone or buffer containing nicotine with or without mecamylamine. Traces, [Ca2+]ichanges. Concentrations of nicotinic drugs during the period indicated (horizontal bars) are marked (above traces). Extracellular Ca2+ concentration and time scales are shown (under each trace). Nic, (−)-nicotine. Mec, mecamylamine. Ca, extracellular Ca2+concentration. A, [Ca2+]i increase is stimulated by nicotine at 1.3 mm extracellular Ca2+. Data shown are representative of three independent experiments, and the trace shown is the average [Ca2+]i of 58 cells. B, [Ca2+]i increase is stimulated by nicotine and blocked by mecamylamine at an extracellular Ca2+concentration of 10 mm. Data shown are representative of three independent experiments, and the trace shown is the average [Ca2+]i of 37 cells.
Na+ ion imaging.
Nicotine also stimulated an increase in [Na+]i, measured by SBFI/Na+ imaging, and this increase was blocked by mecamylamine (Fig. 9A). Unlike changes in [Ca2+]i, however, the nicotine-stimulated increase in [Na+]i was not affected by increasing the extracellular Ca2+ from 1.3 to 10 mm (Fig. 9B).

Effects of nicotine on [Na+]i of KXα3β4R2 cells. Relative changes in [Na+]i are shown in arbitrary units of fluorescence ratio F334/F380, as described in Experimental Procedures. Cells were loaded with the Na+ fluorescent indicator SBFI and then exposed to buffer containing nicotinic drugs.Traces, [Na+]i changes. Concentrations of nicotinic drugs during the period indicated (horizontal bars) are marked (above traces). Extracellular Ca2+concentration and time scales were shown (under each trace).Nic, (−)-nicotine. Mec, mecamylamine.Ca, extracellular Ca2+concentration. A, [Na+]i increase is stimulated by nicotine and blocked by mecamylamine at 10 mm extracellular Ca2+. Data shown are representative of two independent experiments, and the trace shown is the average [Na+]i of 27 cells. B, [Na+]i increase is stimulated by nicotine at different extracellular Ca2+ concentrations. Data shown are representative of three independent experiments, and the trace shown is the average [Na+]i of 77 cells. Dotted horizontal lines, [Na+]i in cells that were permeabilized with 5 μm Gramicidine A when extracellular [Na+] was 154 mm (top line) and 0 mm (bottom line), respectively.
Binding of [3H]EB to cell surface receptors.
The KXα3β4R2 cells produce a very high density of α3/β4 receptors, and the ligand binding studies indicate that these receptors have characteristics of a single homogenous population. However, ligand binding measurements in membrane homogenates do not distinguish between receptors on the cell surface membrane and receptors on intracellular membranes. Therefore, to estimate the fraction of the total α3/β4 receptor population that is located on the cell surface of KXα3β4R2 cells, [3H]EB binding was measured in intact cells attached to 24-well tissue culture plates, and nonspecific binding was determined with either nicotine (which easily crosses cell membranes) or carbachol (which does not readily cross cell membranes). As shown in Fig. 10, nicotine blocked >95% of the [3H]EB binding sites in intact cells, whereas in contrast, the nonpermeant carbachol blocked a maximum of 40% of the [3H]EB binding sites even at a concentration 25,000 times higher than its affinity constant (Ki ) for the receptor, as determined in membrane binding assays (see Table 1). Thus, we estimate that 40% of the total number of α3/β4 receptors measured in KXα3β4R2 cell homogenates are located on the cell surface.
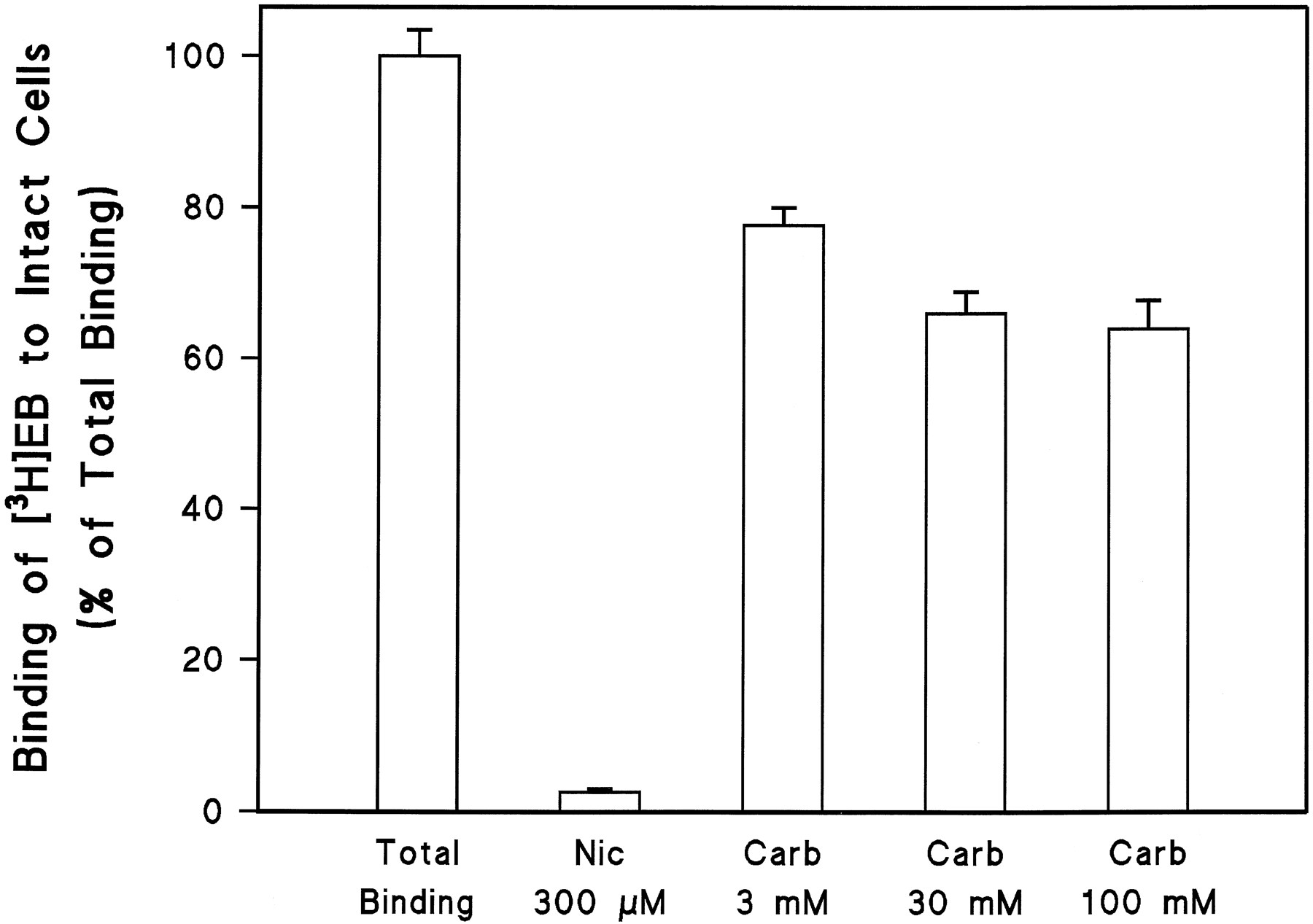
Binding of [3H]EB to intact KXα3β4R2 cells. Intact cell binding assays were carried out as described in Experimental Procedures. Total binding of [3H]EB (3 nm) was determined in the absence of any competing ligand. Nic, (−)-nicotine.Carb, carbachol. Values are the mean ± standard error from six independent experiments.
Discussion
The KXα3β4R2 cells described here stably express α3/β4 nAChRs that bind [3H]EB with high affinity and function to gate cations through their channels in response to nicotinic agonists. In contrast, cells expressing the mRNA for either the α3 or β4 subunit alone do not express [3H]EB binding sites or cation channels that respond to nicotine. Both [3H]EB saturation binding and drug competition studies indicate that KXα3β4R2 cells express a single class of nicotinic receptor binding sites; similarly, the studies of the receptor function are consistent with a single class of receptors. These cells produce a very high density of α3/β4 receptors, and binding studies in intact cells indicate that ∼40% of the total number of receptors are located on the cell surface. Only the receptors on the cell surface would be expected to mediate functional responses, whereas receptors on intracellular membranes might be in various stages of their receptor cycle, either before insertion into the cell surface membrane or after removal from it. Furthermore, we cannot exclude the possibility that these cells express receptors with more than one stoichiometric combination of α3 and β4 subunits but very similar pharmacological and functional properties. Nevertheless, these cells provide a model system in which the pharmacology, function, and regulation of the α3/β4 nAChR can be examined in detail.
Drug binding competition studies indicated that the affinity of every agonist examined is lower at the α3/β4 receptor binding site in these cells than at the α4/β2 nicotinic receptor site in rat forebrain, with affinity ratios ranging from 7 for EB to >750 for A85380. One consequence of the lower affinity of agonists for α3/β4 receptors is that with the exception of [3H]EB, radioligands such as [3H]cytisine, [3H]nicotine, and [3H]acetylcholine, which have been very useful for labeling the α4/β2 nAChR, the predominant receptor subtype in brain, probably would not be useful for labeling α3/β4 receptors. This is because their lower affinities would require ligand concentrations (200–880 nm) at which nonspecific binding would obscure specific binding. This probably explains why these radioligands have not been useful for labeling nicotinic receptors in autonomic ganglia or adrenal gland, where a receptor subtype containing α3 subunits seems to predominate. Although the affinity of EB also is lower at α3/β4 receptors than at α4/β2 receptors in brain, its affinity at these α3/β4 receptors (300 pm) is still very high, making it an excellent radioligand for these receptors.
The affinities of d-tubocurarine and DHβE, the two antagonists examined that competed for the binding site, like those of the agonists, are lower at the α3/β4 receptor binding site than at the α4/β2 binding site in brain. In fact, DHβE displays the largest difference in affinity for these two receptor subtypes of any drug examined. Thus, DHβE, with a receptor binding site affinity ratio of >7500, and A85380, with an affinity ratio of >750, could be useful drugs for distinguishing between these two subtypes of nicotinic receptors in native tissues. In addition to the absolute difference in affinity of DHβE, a second way to distinguish between these two receptor subtypes could be to take advantage of the difference in the rank orders of affinities of d-tubocurarine and DHβE. Thus, at α3/β4 receptor binding sites, d-tubocurarine has >9 times higher affinity than DHβE, whereas in contrast, at α4/β2 binding sites, DHβE has >75 times higher affinity thand-tubocurarine.
Consistent with a previous report (Houghtling et al., 1995), both d-tubocurarine and DHβE show low Hill coefficients in competing for brain [3H]EB binding sites, which are predominantly α4/β2 receptors. Similarly,d-tubocurarine shows a low Hill coefficient in competing for α3/β4 receptors in these transfected cells. These low Hill coefficients suggest the interesting possibility that some antagonists can distinguish between two states of the same receptor, for example, as the receptor conformation changed from a resting state to a desensitized state (with high affinity for agonists) during prolonged incubation with an agonist, as in the receptor binding assay.
The α3/β4 nAChR function and its pharmacology were assessed by measuring agonist-stimulated86Rb+ efflux. Nicotine and acetylcholine both stimulated86Rb+ efflux in a concentration-dependent manner, with EC50 values of ≈28 and 114 μm, respectively. The difference between the affinity of the receptor for these agonists as assessed by this measurement of receptor function compared with binding studies (Table1) presumably reflects the shift of the receptor to a state with high affinity for agonists under equilibrium binding conditions. In the cases of nicotine and acetylcholine, the EC50values for functional activation were 59- and 129-fold higher, respectively, than the Ki values measured in binding studies.
Among the antagonists tested, mecamylamine, with an IC50 value of 1 μm, was the most potent in blocking nicotine-stimulated86Rb+ efflux in KXα3β4R2 cells. It was ∼10 times more potent thand-tubocurarine, 100 times more potent than DHβE, and 200 times more potent than hexamethonium. Both mecamylamine and hexamethonium blocked receptor function by a noncompetitive mechanism, suggesting that they block the receptor ion channel. This would explain their effective block of receptor function in the face of their near-total inability to compete for the agonist binding site of the α3/β4 receptor.
Tubocurarine, on the other hand, presents a more interesting case. In the binding assay, it competes for the α3/β4 receptor agonist recognition site with a Ki value of ≈23 μm (Table 1), but at similar concentrations, its blockade of receptor function seems to be entirely by a noncompetitive mechanism. In fact, there was no indication thatd-tubocurarine competes for the agonist binding site in the86Rb+ efflux assay; that is, the concentration-response curves for nicotine were not shifted to the right in the presence of any concentration of tubocurarine tested (Fig. 6C). One explanation for this apparent contradiction between the binding assay and the 86Rb+efflux assay is that in the receptor’s functional state, as measured in the 86Rb+ efflux assay, tubocurarine has virtually no affinity for the agonist recognition site of the receptor but nevertheless very effectively blocks receptor function noncompetitively by blocking the ion channel. However, when the receptor conformation shifts to a desensitized state with its higher affinity for nicotinic agonists, as occurs in the binding assay, its affinity for tubocurarine also increases, enabling this antagonist to then compete with micromolar affinity for the agonist recognition site. This explanation also can account for the observation that tubocurarine competes for the agonist recognition sites of both the α3/β4 receptor in these cells and the α4/β2 receptor in rat brain with a low Hill coefficient because this could reflect the different affinities of the drug for the different conformations of the receptors. In fact, the [3H]EB saturation binding studies shown in Fig. 7 suggest that in the presence of 30 μm tubocurarine, ∼25% of the receptors may remain in a conformation with an affinity for [3H]EB too low to be measured in a typical binding assay.
In contrast to these other antagonists, DHβE blocked receptor function in a competitive manner. Its affinity constant for the functional state of the receptor, calculated from its IC50 value in blocking nicotine-stimulated86Rb+ efflux (after correction for the nicotine concentration), is ∼20 μm, which is ∼10 times lower than theKi value derived from the ligand binding competition assays (Table 1). In other words, DHβE is ∼10 times more potent in blocking receptor function than would have been predicted from the binding assay. This suggests that DHβE has higher affinity for the functional conformation of the receptor than for its conformation in the desensitized state, which would occur during the binding assay.
Activation of the α3/β4 receptor in KXα3β4R2 cells resulted in an increase in both [Ca2+]i and [Na+]i. The nicotine-stimulated increase in [Ca2+]i was markedly enhanced when the concentration gradient was increased by raising the extracellular Ca2+ concentration from 1.3 to 10 mm. The higher extracellular Ca2+concentration, however, seemed to have no effect on the nicotine-stimulated increase in [Na+]i. These results suggest that extracellular Ca2+ can actually pass through the α3/β4 receptor channel.
The α3/β4 nAChR has been expressed previously in HEK 293 cells, both transiently (Wong et al., 1995) and stably in conjunction with a voltage-gated Ca2+ channel (Stetzer et al., 1996). Although pharmacological comparisons of these previously expressed α3/β4 nAChRs and those in the KXα3β4R2 cells are very limited, there seems to be agreement that acetylcholine, nicotine, and cytisine are full agonists, confirming studies in frog oocytes (Luetje and Patrick, 1991; Covernton et al., 1994; Papke and Heinemann, 1994). However, nicotine and acetylcholine seemed to be somewhat more potent in stimulating the α3/β4 receptors in these KXα3β4R2 cells than was reported in the other transfected HEK 293 cells.
In conclusion, KXα3β4R2 cells provide a model system in which to study α3/β4 receptors, a subtype that may mediate neurotransmission in autonomic ganglia and brain. These cells have maintained a high level of receptor expression and function for >300 generations and thus have allowed a detailed examination of the pharmacology of the receptor. The pharmacological studies reported here, for both drug binding and functional activation, should help in studying the distribution, physiological role, and pharmacology of α3/β4 nAChRs in brain and peripheral tissues. In addition, this cell line provides a stable and convenient source of highly functional channels for patch-clamp experiments to study in detail the electrophysiological properties of α3/β4 nAChRs (Zhang et al., 1997). Furthermore, the binding site of the receptor and its function in these cells are altered by exposure to nicotinic drugs (Meyer et al., 1997); thus, these stably transfected cells should prove useful in studying the chronic and acute effects of nicotinic drugs on the density, cellular distribution, and function of α3/β4 nAChRs.
Acknowledgments
We thank Susan Hernandez for her participation in intact cell binding assays and Parul Mehta for her assistance with tissue culture.
Footnotes
- Received November 18, 1997.
- Accepted May 11, 1998.
-
Send reprint requests to: Dr. Kenneth J. Kellar, Department of Pharmacology, Georgetown University School of Medicine, Washington, D.C. 20007. E-mail:kellark{at}gunet.georgetown.edu
-
This work was supported by National Institutes of Health Grants DA06486 and AG09973. E.L.M. was supported by National Institutes of Health Predoctoral Fellowship Grant DA05739–01.
-
A preliminary report of this work has been presented previously [Xiao Y, Meyer EL, Thompson JM, and Kellar KJ (1997) Generation and characterization of a stably transfected cell line expressing rat α3β4 neuronal nicotinic acetylcholine receptors. Soc Neurosci Abstr 23:385].
Abbreviations
- nAChR
- nicotinic acetylcholine receptor
- CNS
- central nervous system
- EB
- (±)-epibatidine
- DHβE
- dihydro-β-erythroidine
- GAPDH
- glyceraldehyde-3-phosphate-dehydrogenase
- [Ca2+]i
- intracellular Ca2+concentration
- [Na+]i
- intracellular Na+ concentration
- HEK
- human embryonic kidney
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}