


Abstract
The cholecystokinin (CCK) receptor types A and B (CCKAR and CCKBR) are G protein-coupled receptors with approximately 50% amino acid identity; both have high affinity for the sulfated CCK octapeptide (CCK-8), whereas only the CCKBR has high affinity for gastrin. Previously, we identified five amino acids in the second extracellular loop (ECL) of the CCKBR that were essential for gastrin selectivity. Subsequent mutagenesis of one of these five amino acids (H207F) resulted in the loss of radiolabeled CCK-8 binding. CCK-8 stimulated total inositol phosphate accumulation in COS-1 cells transiently expressing the CCKBR-H207F with full efficacy and a 3044-fold reduced potency, which suggests that the loss of radioligand binding was caused by a loss in affinity. Alanine scanning mutagenesis was performed on the amino terminus near the top of transmembrane domain I (TMI) and on ECL1, two extracellular domains implicated in ligand binding by previous mutagenesis studies.125I-Bolton-Hunter-CCK-8 binding to mutant receptors transiently expressed in COS-1 identified one nonconserved amino acid, R57A, at the top of TMI that caused a 21-fold reduction in CCK-8 affinity and four conserved amino acids, N115A, L116A, F120A and F122A, in the ECL1 that caused a 15.6-, 6-, 440-, and 8-fold reduction in affinity or efficacy. Alanine substitution of the equivalent amino acids in the CCKAR corresponding to each of the five amino acids in ECL1 and ECL2 affecting CCK-8 affinity for the CCKBR revealed only two mutations, L103A and F107A, that decreased CCK-8 affinity (68- and 2885-fold, respectively). These data suggest that CCK-8 interacts at multiple contact points in the extracellular domains of CCK receptors and that the CCKAR and CCKBR have distinct binding sites despite their shared high affinity for CCK-8.
CCK is a regulatory peptide hormone found predominately in localized endocrine cells of the gastrointestinal tract and neurotransmitter present throughout the nervous system. CCK is processed from a 115-amino-acid preprohormone to multiple molecular fragments. CCK-8 and larger forms (CCK-58, CCK-39 and CCK-33, CCK-22) have similar high affinity for the two pharmacological receptor subtypes, CCKA and CCKB. The shorter CCK forms (CCK-4 to CCK-7) have a lower affinity and are able to discriminate CCKA and CCKB receptors displaying a higher affinity for the CCKBR (Poirot et al., 1993; Wank, 1995). The CCKBR can also be highly discriminated by the selective affinity for the related gastrin family of peptides that share the same carboxyl-terminal pentapeptide-amide but differ in sulfation at the sixth (gastrin) versus the seventh (CCK) tyrosyl residue. The full biological activity of CCK peptides reside in the carboxyl-terminal sulfated, amidated heptapeptide. CCK acting through the CCKAR and the CCKBR has well-characterized physiological and pharmacological functions. In the gastrointestinal system, CCK regulates motility, pancreatic enzyme secretion and growth, gastric emptying, and inhibition of gastric acid secretion. In the nervous system, CCK is involved in anxiogenesis, satiety, analgesia, and regulation of dopamine release (Crawley and Corwin, 1994).
Although the pharmacology and actions of CCK receptors have been well studied and their possible implication in disorders related to their functions reported (Crawley and Corwin, 1994), much less is known about the structural basis for the interaction of CCK agonists with both the CCKAR and the CCKBR subtypes. The cloning of CCKAR and CCKBR has shown that CCK receptors are seven transmembrane G protein-coupled receptors that have ∼ 50% amino acid homology (Wank et al., 1992a, 1992b). Despite the relatively high degree of homology and shared affinity for CCK-8, a study of the human CCKBR undertaken to evaluate the role of the nonconserved transmembrane domain amino acids for agonist and antagonist selectivity identified three amino acid residues, R57Q, S219H, and H376L, that decrease CCK affinity 6-, 5-, and 29-fold, respectively, when mutated to the corresponding CCKAR amino acids (Kopin et al., 1995). However, mutation of H381 in the rat CCKBR, (equivalent to H376 in the human CCKBR) to Leu did not change the affinity for CCK-8 (Jagerschmidt et al., 1996). For the CCKAR, it has been reported that the amino-terminal domain of the CCKAR in rat and human is important for CCK-9 binding (Poirot et al., 1994; Kennedy et al., 1995). In this region, two nonconserved residues in the human CCKAR (W39 and Q40) caused a decrease of 12.9- and 20.9-fold in CCK-9 affinity, respectively, after conservative mutations (W39F, Q40N). Furthermore, these residues were shown to affect CCK-8 binding by interacting with the amino terminus of CCK-8 (Kennedy et al., 1997). In the course of a previous study that permitted us to identify a segment of five amino acids (residues 204–208; Fig.1, bottom) in the ECL2 of the rat CCKBR that was essential for the high affinity and selectivity of gastrin binding (Poirot and Wank, 1996), we observed that the binding of radiolabeled CCK-8 was lost when we substituted a block of amino acids (amino acid residues 204, 206–209; Fig. 1, bottom) in the ECL2 with the corresponding amino acids of the CCKAR (Fig. 1,top). Further mutations in the ECL2 revealed that the substitution of H207F alone could account for this loss of affinity. The importance of H207 in ECL2 for CCK-8 binding indicated that the extracellular domains of the CCKBR were involved in CCK-8 binding, which was consistent with our previous studies aimed at determining the importance of this region for gastrin selectivity. Previously, no studies have evaluated the role of the extracellular domains of the CCKBR for CCK-8 binding affinity. Therefore, based on our previous data that demonstrates the importance of this single amino acid substitution H207F in the ECL2, the present work was undertaken to evaluate the previously undetermined role of the first ECL1 and amino-terminal region in CCK-8 binding to the CCKBR and compare these findings with those for the corresponding positions in the CCKAR. These regions were chosen because they are likely to be proximal to H207 in the ECL2 of the CCKBR by virtue of a putative disulfide bridge shown for other G protein-coupled receptors (Karnik et al., 1988) and the demonstrated importance of amino-terminal region for CCK-8 binding to the CCKAR (Poirot et al., 1994; Kennedy et al., 1995, 1997).

Model of the primary structure of the rat CCKA and CCKB receptors. Shown are the models of the deduced amino acid sequences of the rat CCKAR and CCKBR illustrating putative transmembrane helices (boxed amino acids), N-linked glycosylation sites (tridents), serine and threonine phosphorylation sites (-PO3), conserved cysteines in the first and second extracellular loops possibly forming a disulfide bridge (—S—S—) and a palmitoylated cysteine in the carboxy-terminus (jagged line extending into the membrane). Filled circles, amino acid residues conserved between CCKAR and CCKBR. Amino acid residues mutated to alanine are denoted by enlarged heptagons, except A55 in the CCKBR, which was mutated to leucine.
Using an alanine scanning mutagenesis approach (Hjorth et al., 1994; Leong et al., 1994; Huang et al., 1995; Kim et al., 1995; Mouillac et al., 1995), we report that a mutation at the top of TMI (R57A), four mutations in the first ECL (N115A, L116A, F120A, and F122A), and a mutation in the second ECL (H207A) resulted in decreased CCKBR affinity for the agonist, CCK-8. When these mutations were made in the equivalent positions of the CCKAR, only two, L103A and F107A in the first ECL of the CCKAR, affected CCK-8 affinity. Unlike the CCKAR, no mutations in the distal amino terminus (proximal to the top of TMI) of the CCKBR had an affect on CCK-8 affinity. These results indicate that although the extracellular domains of both the CCKBR and CCKAR significantly contribute to CCK-8 binding, CCK-8 does not interact with all equivalently positioned amino acids. Consistent with the observed selectivity of shorter CCK forms for the CCKBR, these results suggest that the CCK-8 binding site differs in the two receptor subtypes despite their shared high affinity for CCK-8.
Materials and Methods
Construction of mutant receptor.
cDNAs-mutant receptor cDNAs were constructed by oligonucleotide-directed mutagenesis (Muta-Gene Phagemid in vitro Mutagenesis Kit, Bio-Rad, Hercules, CA), using the CCKBR or the CCKAR cDNAs as a single strand template as described previously (Poirot and Wank, 1996). Oligonucleotides were designed to include a silent restriction site to facilitate analysis of mutant constructs by restriction endonuclease digestion. Mutations were confirmed by DNA sequencing using the dsDNA Cycle Sequencing System (Gibco BRL, Gaithersburg, MD) or using the Dye Terminator kit and an automated 377 DNA Sequencer (Applied Biosystems, Norwalk, CT).
Transfection of wild-type and mutant receptor cDNAs into mammalian cells.
COS-1 cells were grown in DMEM supplemented with 10% fetal calf serum. Two micrograms of the CCKBR, CCKAR and mutant receptor cDNAs subcloned in pCDL-SRα was transiently transfected into COS-1 cells using the DEAE/Dextran method as described previously (Poirot and Wank, 1996). Twenty-four hours after transfection, the transfected cells were transferred to 24-wells culture plates and seeded at a density of approximately 1 × 105 cells/well and assayed for radioligand binding or IP hydrolysis.
Radioligand binding to transfected COS-1 cells.
Twenty-four hours after the transfer of transfected cells to 24-well plates, the cells were washed once with cold PBS, pH 7.4, containing 0.1% BSA and incubated in DMEM containing 0.1% BSA for 60 min at 37° with either 50 pm of 125I-BH-CCK-8 (2200 Ci/mmol) or 1.8 nm [3H]SR27897 (31 Ci/mmol) (Gully et al., 1993) or for 30 min with 0.4 nm[3H]PD140,376 (51 Ci/mmol) with and without the indicated concentrations of unlabeled ligand. The cells were washed two times with PBS containing 2% BSA and were collected with 0.5 ml of 0.1n NaOH added to each well and radioactivity detected in either a γ or β counter (Packard, Downers Grove, IL). Nonspecific binding (determined in presence of 1 μm CCK-8) was always less than 10% of total binding. Binding assays were performed in duplicate in at least three separate experiments.Kd andB max values were determined using the nonlinear least-squares curve-fitting computer program, Ligand (Munson and Rodbard, 1980).
Measurement of total IP accumulation.
Twenty-four hours after COS-1 cell transfection, the transfected cells were transferred to 24-well culture plates and incubated overnight in DMEM with 3 μCi/well of myo-2-[3H]inositol (18.6 Ci/mmol)(Dupont-New England Nuclear, Boston, MA). After the aspiration of the medium containing themyo-[3H]inositol, the cells were incubated at 37° for 20 min with 2 ml of PBS, pH 7.45, containing 20 mm LiCl, incubated 1 hr at 37° with IP buffer (20 mm HEPES, 135 mm NaCl, 2 mm CaCl2, 1.2 mm MgSO4, 1 mm EGTA) containing the indicated concentrations of CCK-8. The reaction was stopped with 1 ml of methanol/HCl added to each well and the content transferred to a Dowex AG 1-X8 (formate form) column (Bio-Rad, Hercules, CA). Each column was washed with 5 ml of water followed by 2 ml of 5 mm sodium tetraborate/60 mm sodium formate. Total IP were eluted from the columns with 2 ml of 1m ammonium formate/100 mm formic acid. The total [3H]IP β-radioactivity was detected in a liquid scintillation counter (Packard).
Immunofluorescence assay.
A peptide epitope, YPYDVPDYA, from the hemagglutinin of the influenza virus (HA1) was fused to the amino terminus of the wild-type CCKBR and the mutant CCKBRs, C127A-CCKBR and C205A-CCKBR, using the polymerase chain reaction (Field et al., 1988) The day after the transient transfection into COS-1 cells, 1.25 × 105 cells were transferred to Lab-Tek chambered coverglass slides for tissue culture (Nunc, Naperville, IL). Two days after transfection, cells were washed two times with PBS and fixed with 2% formaldehyde in PBS for 15 min at room temperature. To permeabilize the cell membranes, cells were treated with 0.1% saponin in PBS (w/v) for 10 min. After washing with PBS three times and blocking with DMEM containing 10% fetal calf serum, cells were incubated with 12CA5 monoclonal antibody at 10 mg/ml (BAbCo, Richmond, CA) for 3 hr at 37°. The cells were subsequently washed with PBS and incubated with a 1/100 dilution of a fluorescein isothiocyanate-conjugated anti-mouse IgG antibody (Sigma, St. Louis, MO) in DMEM containing 10% fetal calf serum for 1 hr at 37°. After washing with PBS, the cells were analyzed on a confocal laser scanning microscope (LSM 410; Carl Zeiss, Thornburg, NY).
Results
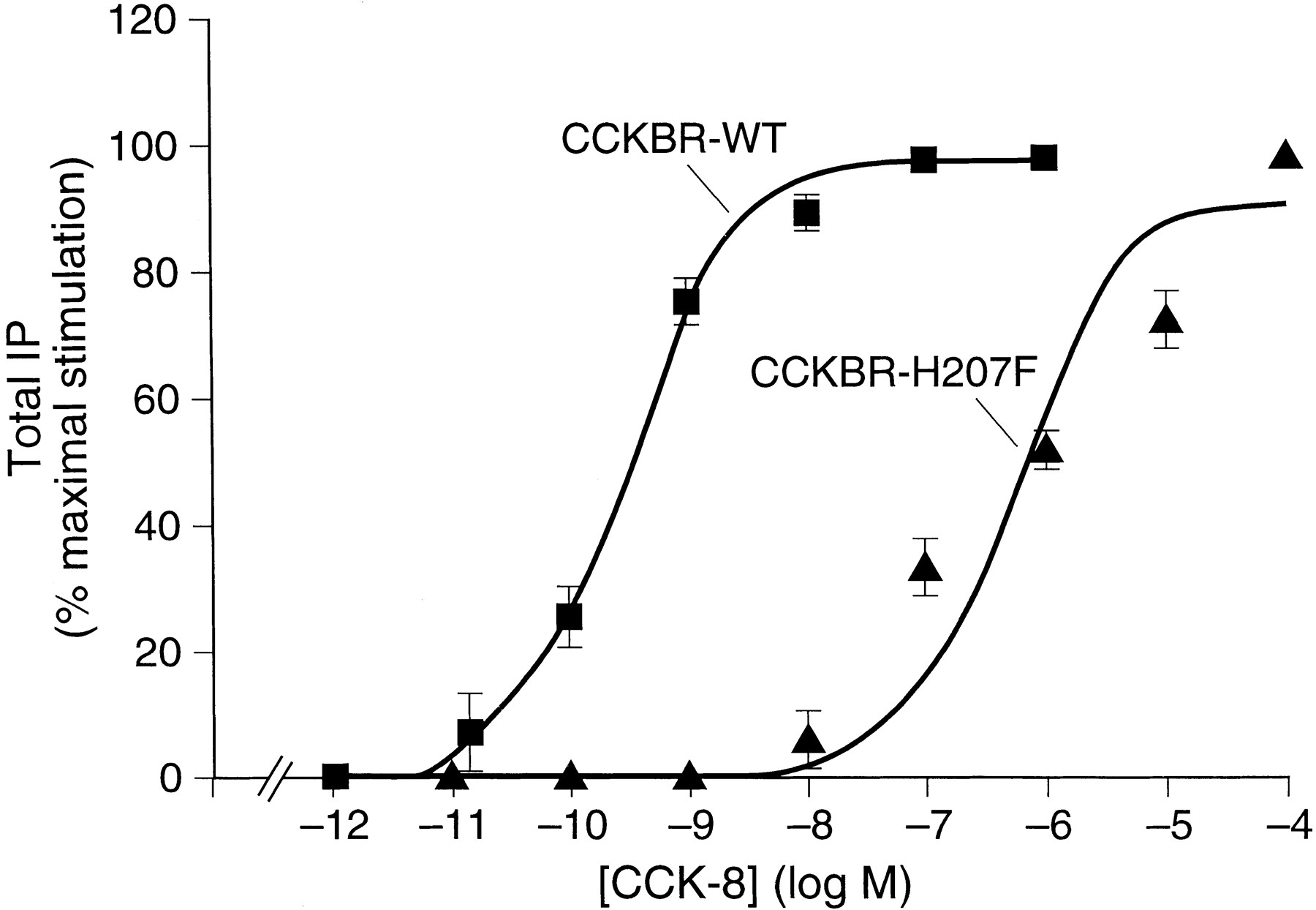
In a previous study of chimeric CCKA and CCKB receptors, we determined that a segment of five amino acids (amino acid residues 204, 206–209; Fig. 1, bottom) in the ECL2 was responsible for conferring gastrin-17-I selectivity (Poirot and Wank, 1996). In the course of our studies, it was observed that the binding of radiolabeled CCK-8 was lost when a block of amino acids (amino acid residues 204, 206, and 207; Fig. 1, bottom) in the ECL2 was substituted with the corresponding amino acids of the CCKAR, whereas other segment substitutions elsewhere in the ECL2 had no effect on CCK-8 affinity. To determine which of the three amino acids in this segment of the ECL2 could account for this loss of affinity for CCK-8, each was individually mutated to the corresponding amino acid in the CCKAR. These mutants and wild-type CCKBR were transiently expressed in COS-1 cells and their affinity for CCK-8 was determined by radioligand binding inhibition studies using 125I-BH-CCK-8. These studies revealed that the substitution H207F alone could account for this loss of affinity and that reciprocal mutations of amino acids Q204 and M206 with the corresponding CCKAR amino acids did not significantly change CCK-8 affinity. TheKd obtained was 1.03 (± 0.05) nm for the mutant CCKBR-Q204m and 0.65 (± 0.12) nm for the mutant CCKBR-M206R compared with 0.46 (± 0.08) nm for the wild-type CCKBR. In the absence of detectable binding of a 50 pm concentration of125I-BH-CCK-8 by the mutant CCKBR-H207F, we measured CCK-8 stimulated total IP production to determine if this mutant was functionally expressed. The mutant receptor, CCKBR-H207F, increased total IP with full efficacy relative to the wild-type CCKBR (11-fold increase over basal), although the dose-response curve was displaced 3044-fold to the right compared with the wild-type CCKBR (Fig. 2) with EC50values of 1016 (±406) nm and 0.33 (±0.07) nm, respectively. Consistent with the fully efficacious stimulation of total IP by the CCKBR-H207F mutant, Scatchard analysis of the CCKBR specific nonpeptide antagonist, [3H]PD 140376, binding revealed a similar level of expression of the CCKBR-H207F mutant (B max = 3.95 ± 0.34 pmol/106 cells) compared with wild-type CCKBR (B max = 4.33 ± 0.26 pmol/106 cells). To determine whether the loss of CCK binding for the mutant CCKBR-H207F was caused by an unfavorable interaction peculiar to the side chain of the substituted Phe residue, we mutated the His residue to an Ala. Similar to the mutant CCKBR-H207F, the mutant CCKBR-H207A displayed no detectable binding of125I-BH-CCK8, indicating that the side chain of the Phe residue was not uniquely responsible for the loss of CCK binding. These data suggest that the loss of125I-BH-CCK8 binding was caused by a substantial reduction in CCK-8 affinity rather than a loss in receptor expression and that H207 is critical for conferring high affinity for CCK-8.

CCK-8 stimulated increase in total IP in COS cells expressing the wild-type CCKBR (CCKBR-WT) and mutant CCKBR-H207F receptors. Transfected COS-1 cells expresssing the wild-type CCKBR or the mutant CCKBR-H207F were incubated with increasing concentration of CCK-8 for 1 hr at 37° and the production of IP was determined. The IP accumulation is expressed as the percent of the maximal increase obtained using 1 × 10−6 m CCK-8. Results are the mean ± standard error from three experiments performed in duplicate.
Conserved cysteines in the first extracellular loop at the top of TM III and in the ECL2 (C127 and C205; Fig. 1, bottom) are likely to form a disulfide bridge in the CCKBR similar to other G protein-coupled receptors (Karnik et al., 1988). This would then bring ECL1 in proximity to H207 in the ECL2, shown above to be important for high affinity CCK-8 binding. To assess this hypothetical role for C127 and C205, the two cysteines were individually mutated to alanine and the effect on CCK-8 affinity was determined. No binding of CCK-8 was detected for both mutants at a 50 pmconcentration of 125I-BH-CCK-8. Immunofluorescence studies using an antibody directed against the hemagglutinin HA1 epitope fused to the amino terminus of the wild-type and the C127A and C205A CCKB receptors were used to determine if the respective receptors were expressed at the cell surface of transiently transfected COS-1 cells. Unlike the wild-type receptor-expressing cells that exhibited a fluorescent signal at the cell surface, the mutant C127A-CCKBR and C205A-CCKBR transfected cells exhibited a fluorescent signal in the intracellular compartment only when the cells were permeabilized with saponin (data not shown). In addition, CCK-8 was unable to stimulate an increase in total IP in cells expressing the mutant C127A-CCKBR and C205A-CCKBR. These results indicate that the CCKBR mutants C127A and C205A are important for the proper folding and surface expression of the CCKB receptor and are consistent with the formation of a disulfide bridge that would bring the ECL1 and the ECL2 closer together.
Subsequently, the role of the first extracellular loop in CCK binding was investigated. To evaluate the possible influence of the ECL1 on CCK-8 affinity, alanine scanning mutagenesis was applied to conserved and nonconserved amino acids in the first extracellular loop (except G118, which was mutated to Lys). All amino acids were mutated except P114 and G123 that are conserved in both the CCKAR and the CCKBR in all species and are felt to have at best an indirect structural affect on ligand binding on the basis of studies with other G protein-coupled receptors. Nonconserved amino acids were included having found that a nonconserved amino acid residue, H207, in the ECL2 was important for the high affinity CCK-8 binding. Nonetheless, mutating each of these nonconserved amino acid residues in the ECL1 did not significantly affect the affinity of CCK-8 relative to the wild-type CCKBR with each mutation exhibiting less than a 3-fold decrease in CCK-8 affinity (Table 1). In contrast, mutation of four conserved amino acids, N115A, L116A, F120A, and F122A, resulted in a significant decrease in CCK-8 affinity. Substitution of L116A and F122A led to a moderate reduction in CCK affinity of 6.2-fold (Kd = 2.94 (±0.84) nm) and 8-fold (Kd = 3.74 (±0.25) nm), respectively, whereas the mutation of N115A caused a more pronounced 15.6-fold (Kd = 7.33 (±1.46) nm) decrease in CCK-8 affinity (Table 1). For the mutant F120A, there was no detectable binding of CCK-8 using a 50 pm concentration of125I-BH-CCK-8. However, this mutant retained full efficacy (11-fold increase over basal) for signaling CCK-8 stimulated increase in total IP, although the EC50 was reduced 440-fold (EC50 = 147 (±38) nm for the F120A mutant compared with 0.33 (±0.07) nm for the wild-type CCKBR)(Fig.3). Consistent with the fully efficacious stimulation of total IP by the CCKBR-F120A mutant, Scatchard analysis of the CCKBR specific nonpeptide antagonist, [3H]PD 140376, binding revealed a similar level of expression of the CCKBR-F120A mutant (B max = 2.04 ± 1.37 pmol/106 cells) compared with wild-type CCKBR (B max = 4.33 ± 0.26 pmol/106 cells). These results indicate that the mutation F120A in the CCKBR results in a major loss in CCK-8 affinity that cannot be attributed to a nonspecific effect resulting in either a loss in receptor expression or a major disruption in receptor conformation.
CCK-8 binding affinities of the wild-type CCKB (CCKBR-WT) and ECL1 mutant CCKB receptors

CCK-8 stimulated increase of total IP in COS cells expressing the wild-type CCKBR (CCKBR-WT) and mutant CCKBR-F120A receptors. Transfected COS-1 cells expressing either the CCKBR-WT or the mutant CCKBR-F120A were incubated with increasing concentrations of CCK-8 for 1 hr at 37° and the production of IP was determined. The IP accumulation is expressed as the percent of the maximal increase obtained using 1 × 10−6 m CCK-8. Results are the mean ± standard error from three experiments performed in duplicate.
A recent report has shown that two nonconserved amino acids, W39 and Q40, in the amino-terminal region of the CCKAR interact with the amino terminus of CCK-8 to affect high affinity binding (Kennedy et al., 1997). Therefore, to determine the potential role of the corresponding area of the CCKBR, we first mutated the equivalent amino acids in the CCKB receptor (amino acid residues L52 and E53) and the amino acids adjacent to these two positions. Each amino acid was individually substituted to alanine with the exception of A55, which was mutated to Leu. For the wild-type CCKBR, theKd value obtained for CCK-8 affinity was 0.46 (±0.08) nm (Table2). For the mutants CCKBR-L52A and CCKBR-E53A, the Kd value was 0.50 (±0.22) nm and 0.94 (±0.05) nm, respectively, indicating that CCK affinity was not significantly affected (Table 2). To determine if this region of the CCKBR was important for CCK-8 binding as shown previously for the CCKAR, we evaluated the role of the amino acid residues neighboring these two positions. Similarly, the mutations of residues E51A and M54A did not affect the affinity for CCK-8, theKd values were 0.57 (±0.17) nm and 0.74 (±0.21) nm for the respective mutants (Table 2). A report of a small decrease of 6-fold in CCK-8 affinity when R57 in the CCKBR was substituted to Gln, the equivalent amino acid in CCKAR (6), prompted the mutation of amino acids further down TMI of the CCKBR. Mutations of A55L, I56A, and I58A did not affect CCK-8 affinity, the Kd values obtained were 0.30 (±0.17) nm, 0.99 (±0.20) nm, and 0.60 (±0.33) nm, respectively (Table 2). However, when we mutated R57 to Ala, we observed a decrease on CCK affinity of 21.3-fold compared with the wild-type CCKBR. TheKd value was 9.96 (±0.48) nm for the mutant CCKBR-R57A compared with 0.46 (±0.08) nm for the wild-type CCKBR (Table 1). These results indicate that the points of interaction of CCK-8 in this region of the CCKBR are distinct from the equivalent points of interaction reported for the CCKAR despite their shared high affinity for CCK-8.
CCK-8 binding affinities of the wild type CCKB (CCKBR-WT) and amino-terminal/TMI mutant CCKB receptors
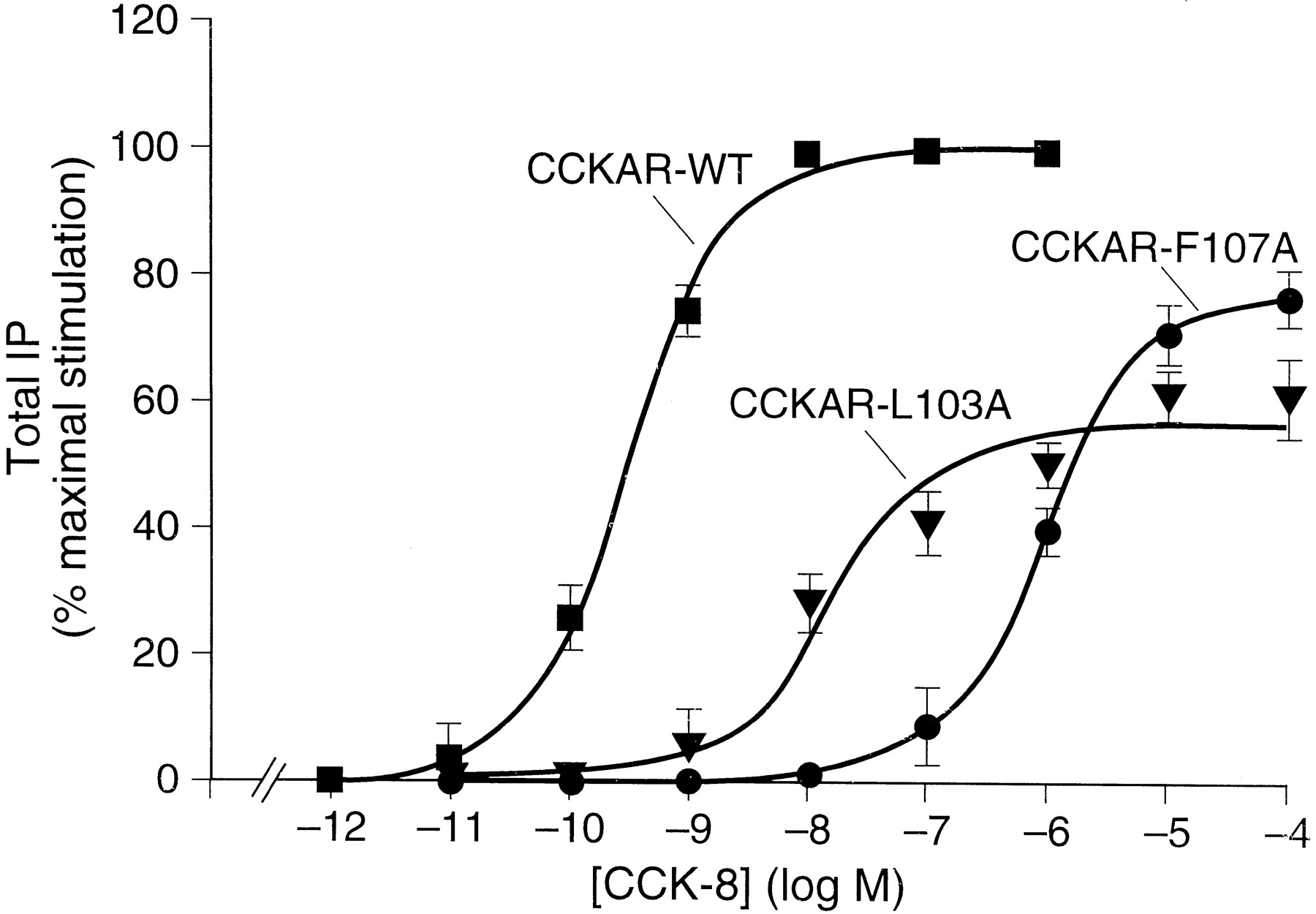
Together, these data show that CCK-8 binding to the CCKBR involve amino acid residues that are both conserved and divergent from the corresponding amino acids located in the extracellular regions of the CCKAR. Having demonstrated that mutation of amino acid residues at equivalent positions in the amino-terminal domain of the CCKAR and the CCKBR do not have the same effect on CCK-8 affinity, mutations at divergent residues shown above to be important for CCK-8 affinity for the CCKBR were made in the equivalent positions in ECL1 and ECL2 of the CCKAR to determine their effect on CCK-8 affinity. Residues N102, L103, F107, and F109 (equivalent to amino acid residues N115, L116, F120, and F122 in the ECL1 of CCKBR) and amino acid residue F198 (equivalent to residue H207 in the ECL1 of CCKBR) were mutated to alanine (Fig. 1,top). Similar to the above studies on the CCKBR, the CCKAR mutants and the wild-type CCKAR were transiently expressed in COS-1 cells and their affinity for CCK-8 assessed by125I-BH-CCK-8 radioligand displacement by CCK-8. Scatchard analysis of 125I-BH-CCK-8 binding on the wild-type CCKAR results in a single class of high affinity binding sites as described previously for the CCKAR in COS-1 cells with aKd of 0.18 (±0.10) nm Huang et al., 1994). The mutants CCKAR-N102A and CCKAR-F109A displayed a similar affinity for CCK-8 compared with the wild-type CCKAR with aKd value of 0.45 (±0.35) nm and 0.14 (±0.15) nm, respectively (Table 3). For the mutants CCKAR-L103A and CCKAR-F107A there was no detectable binding of125I-BH-CCK-8 using a 50 pmconcentration of radioligand. Using higher ligand concentrations, CCK-8 was able to stimulate these mutant receptors to increase total IP with an EC50 value that was increased 2885-fold for the mutant CCKAR-F107A [EC50 = 939 (±133) nm] and 68.4-fold for the mutant CCKAR-L103A [EC50 = 22.2 (±6.2) nm] compared with the wild-type CCKAR [EC50 = 0.325 (±0.034) nm] (Fig.4). However, both mutants were unable to induce the same maximum in the IP response compared with the wild-type CCKAR. The maximum stimulation attained was 77% for the mutant CCKAR-F107A and 61% for the mutant CCKAR-L103A of the wild-type CCKAR (18-fold increase over basal) (Fig. 4). To determine whether the lower efficacy observed for the mutants could be attributable to reduction in receptor expression, Scatchard analysis was performed using the CCKAR-specific nonpeptide antagonist [3H]SR27897 (Gully et al., 1993). These studies revealed a similar affinity and maximal binding capacity for the two mutants compared with the wild-type CCKAR withKd andB max values of 1.52 (±0.23) nm and 0.88 (±0.10) pmol/106 cells, respectively, for the mutant CCKAR-F107A, 1.88 (±0.56) nm and 1.65 (±0.22) pmol/106 cells, respectively, for the mutant CCKAR-L103A versus 3.96 (±0.02) nm and 1.92 (±0.6) pmol/106 cells, respectively, for the wild-type CCKAR. These results suggest that the residues F107 and L103 in the ECL1 of the CCKAR play a substantial role in CCK-8 affinity and, in addition, may participate in the conformational change necessary for G protein-coupling as indicated by the loss in CCK-8 affinity as well as the decrease in potency and efficacy for stimulating total IP observed for the two mutants. In contrast, the affinity of CCK-8 was not significantly affected for the ECL2 mutant, CCKAR-F198A, having aKd value of 0.53 (±0.22) nm compared with 0.18 (±0.10) nm for the wild-type CCKAR (Table 3). These results suggest that F198 is not involved in CCK-8 binding in the CCKAR although this position seems critical for CCK-8 binding in the CCKBR.
CCK-8 binding affinities of the wild type CCKA (CCKAR-WT) and mutant A receptors

CCK-8 stimulated increase of total IP in COS cells expressing the wild type A (CCKAR-WT) and mutant CCKAR-L103A and CCKAR-F107A receptors. Transfected COS-1 cells expressing either the wild-type CCKBR or one of the mutants CCKAR-L103A and CCKAR-F107A were incubated with increasing concentrations of CCK-8 for 1 hr at 37° and the production of IP was determined. The IP accumulation is expressed as the percent of the maximal increase obtained using 1 × 10−6 m CCK-8. Results are the mean ± standard error from three experiments performed in duplicate.
Discussion
Previously, chimeric studies between CCKA and CCKB receptors initially involving large segments and subsequently smaller site-directed mutations identified five amino acids (Q204, M206, H207, R208, and W209; Fig. 1, bottom) in the CCKBR conferring selective high affinity binding for gastrin, a CCK-8 related peptide sharing the same carboxyl-terminal five amino acids (Poirot and Wank, 1996). During the course of these experiments, labeled CCK-8 affinity was lost when a group of amino acids (Q204, M206, and H207; Fig. 1,bottom) in the ECL2 were mutated. It was subsequently determined that the mutation of only one residue, H207F, could account for this loss of binding of labeled CCK-8. This loss in CCK-8 binding along with the discovery of nearby amino acids affecting gastrin selectivity suggested that the ECL2 region could be involved in CCK-8 binding. To date, no results have been reported evaluating the role of the extracellular domains in CCK binding to the CCKBR. Therefore, we studied further the contribution of H207 to the CCK-8 binding site in the CCKBR as well as the potential role of five amino acids within two other proximal extracellular domains, one amino acid in the amino-terminal extension and top of TMI shown to be important for CCK binding to both the CCKAR and CCKBR and four amino acids in the ECL1 drawn toward the second extracellular loop by virtue of a putative disulfide bridge. The probability for disulfide bond formation between the conserved cysteines in ECL1 at the top of TM III and in the second ECL (C127 and C205; Fig. 1, bottom) is supported by the loss of receptor surface expression demonstrated by the absence of radioligand binding, the loss of CCK-8 stimulated total IP production, the negative immunolocalization studies after mutation of each of these cysteines to alanine reported in this study, the loss of affinity for CCK upon dithiothreitol reduction of CCKB-like receptors from toad retina (Bone and Rosenzweig, 1988), and similar findings reported for other G protein-coupled receptors (Dixon et al., 1987;Karnik et al., 1988; Hulme et al., 1990).
In the absence of detectable binding of the CCKBR-H207F mutant, the CCK-8 stimulated increase in total IP was measured for the CCKBR-H207F mutant expressed transiently in COS-1 cells. The mutant CCKBR-H207F increased total IPs with full efficacy although the potency was reduced 3044-fold compared with the CCKBR-WT. These data suggest that the CCKBR-H207F mutation does not lead to misfolding and subsequent reduced expression and suggests that although a direct affect on ligand binding cannot be concluded for this loss of function point mutation, the loss in CCK-8 affinity is not the result of a gross conformational change. Interestingly, CCKBR-H207 is involved in the sequence of five amino acids that we determined to be essential for the selective binding of gastrin in the CCKBR (Poirot and Wank, 1996). These results suggest that the ECL2 of the CCKBR is important for both selective and nonselective agonist binding.
Although the amino-terminal extension of a number of peptide receptors has been shown to interact with their ligand (Fong et al., 1992; Leong et al., 1994; Hjorth et al., 1994) as well as specifically for the CCKAR where the mutation of two amino-terminal residues (W39 and Q40) were reported to be involved in CCK binding and to interact directly with the amino-terminal moiety of CCK-8 (Kennedy et al., 1997), the reciprocal changes in CCKBR did not affect the affinity of CCK-8. In fact, the closest residue from this locus that significantly affects CCK binding was the residue R57 located at the top of TMI. A 21.3-fold decrease in CCK-8 affinity was observed when R57 was mutated to Ala (Kopin et al., 1995). This value is in the same range as the 12.9 and 20.9-fold decrease obtained for the mutation of the residues W39 and Q40 in the CCKA (Kennedy et al., 1997). It is also in agreement with the 6.1 and 10-fold decrease in affinity observed for the more conservative R57Q CCKB/A chimeric mutant (Kopin et al., 1995) and the naturally occurring amino-terminal truncated isoform, ΔCCKBR, in which the amino-terminal 66 amino acids are absent (Miyake, 1995).
In addition to the residue R57 in the top of TMI and H207 located in ECL2, four residues, N115, L116, F120, and F122, that significantly decreased CCK-8 binding in the CCKBR were identified by alanine scanning mutagenesis in the ECL1. The mutation F120A in ECL1, similar to H207F in ECL2 of the CCKBR, resulted in a complete loss of radiolabeled CCK-8 binding and a concomitant reduction in potency for CCK stimulated IP-response of 440-fold. Also similar to H207A, F120A reached the same maximal IP response compared with the control CCKBR, making it unlikely that an indirect effect of this mutation on CCK affinity was caused by low expression or a gross conformation alteration. This is an important point, because F120 is within a region of ECL1 loosely corresponding to a WPXF motif seen in the AT1 and other 7TM receptors, in which mutations lead to loss of function that is suspected on the basis of alteration in conformation and expression (Hjorth et al., 1994). Therefore, this important shift in the IP response for both mutants along with a relatively small or no change in receptor expression indicates a strong effect of the mutations on CCK affinity and suggests that residues H207 and F120 are important for the high affinity binding of CCK to the CCKBR.
Further supporting the involvement of ECL1 in the ligand binding site for CCK-8 was the 15.6-, 8-, and 6-fold reduction in affinity as a result of the N115A, L116A, and F122A mutations in the ECL1, respectively. Similar to these findings for the CCKBR, point mutations involving multiple amino acids clustered within a single extracellular domain have been shown to affect ligand binding for a number of G protein-coupled receptors (Leong et al., 1994; Hjorthet al., 1994; Huang et al., 1995; Kennedyet al., 1997) and suggest either multiple points of contact or an indirect effect by multiple amino acids on a single contact point within a limited domain.
The lack of effect of reciprocal mutations in the ECL1 (N102A and F109A) and ECL2 (F198A) in the CCKAR suggest that all the amino acids determined to affect CCK-8 affinity in the CCKBR are not necessarily important for CCK-8 binding in the CCKAR. In fact, of the four positions that were determined to be important for CCK-8 binding in the CCKBR, only two positions seem to affect CCK-8 affinity in both the CCKBR and CCKAR. Only the mutation of residues L103A and F107A both localized in the ECL1 of the CCKAR induced a strong effect on CCK-8 binding as demonstrated by the loss of radiolabeled CCK-8 binding and the decrease in potency to stimulate IP of 68.4-fold for the mutant CCKAR-L103A and 2885-fold for the mutant CCKAR-F107A. In addition, there was a reduction in the efficacy to stimulate IP production observed for the mutants CCKAR-L103A and CCKAR-F107A of 39% and 23%, respectively. In the absence of a reduction in receptor expression at the cell surface as determined by [3H]SR27897 binding, these results suggest that the mutations L103A and F107A, unlike their equivalents in the CCKBR, can influence the coupling of CCKAR to its G protein. Similarly, the lack of effect of the reciprocal mutations in the proximal amino-terminal extension of the CCKBR suggest that the two amino acids (W39 and Q40; Fig. 1, top) previously shown to affect CCK-8 binding to the CCKAR (Kennedy et al., 1997) are not necessarily important for CCK-8 binding to the CCKBR. Although both the CCKAR and the CCKBR have high affinity for the shared ligand, CCK-8, and therefore might be expected to have similar binding sites, these data suggest that this is not entirely the case. As demonstrated in this study, only four of the seven amino acids that have been shown to affect ligand affinity for either one or both of the CCK receptors are conserved and only one of these four conserved amino acids influence binding of CCK to both receptors. In fact, this is not unusual, because there are several similar examples of G protein-coupled receptor subtypes with high affinity for the same ligand in which the binding pocket has not been completely conserved, such as the neurokinin, angiotensin, dopamine, serotonin, and somatostatin receptor families (Gingrich and Caron, 1993; Humphreyet al., 1993; Reisine and Bell, 1993; Hjorth et al., 1994).
Initial detailed studies of ligand binding sites in G protein-coupled receptors pointed to the importance of multiple contact points within the transmembrane domains as best exemplified by the monoamine ligands (Schwartz and Rosenkilde, 1996). Subsequent studies of protein and peptide receptors increasingly has drawn attention to the important contribution of the extracellular domains either alone or in combination with the transmembrane domains toward the interaction with the naturally occurring agonist (Strader et al., 1994). Although it is difficult to interpret whether point mutations that decrease agonist affinity do so on the basis of direct ligand interaction versus indirect changes in local conformation, the results of this and previous studies of CCKAR and CCKBR interaction with their structurally related agonists, CCK and gastrin, together suggest that both ligands interact predominantly with multiple sites within several extracellular domains and to a lesser extent with the outer third of TMI and TMIII (Kopin, et al., 1995; Poirot and Wank, 1996;Schmitz et al., 1996; Kennedy et al., 1997; Wuet al., 1997). In addition to the proposed tight helical packing of the transmembrane domains (Baldwin, 1993; Schertler et al., 1993), data in this study suggest that the apparently distant sites of interaction of CCK and gastrin with the amino-terminal, first and second ECLs, and transmembrane domains of CCK receptors may actually be reasonably close by virtue of a disulfide bridge between two highly conserved cysteines located in the middle of ECL2 and ECL1 just at the top of TMIII (Fig. 1). The present study also supports the hypothesis that the ligand binding site for CCK differs for the two CCK receptor subtypes as well as between CCK and gastrin within the CCKBR.
Acknowledgments
We thank Michelle Bouisson (INSERM, Toulouse, France) for DNA sequencing.
Footnotes
- Received December 2, 1997.
- Accepted April 15, 1998.
-
Send reprint requests to: Dr. Stephen A. Wank, National Institutes of Health, Building 10, Room 9C-103, Bethesda, MD 20892-1804. E-mail: stevew{at}bdg10.niddk.nih.gov
-
This work was supported in part by a grant from the Association pour la Recherche sur le Cancer (ARC 6234) and a grant from the Region Midi-Pyrénées. S.S.-P. is in charge of research for Centre National de la Recherche Scientifique.
Abbreviations
- CCK
- cholecystokinin
- CCK-8
- cholecystokinin octapeptide
- CCKAR
- cholecystokinin receptor type A
- CCKBR
- cholecystokinin receptor type B
- ECL
- extracellular loop
- TM
- transmembrane
- BH
- Bolton-Hunter
- DMEM
- Dulbecco’s modified Eagle’s medium
- PBS
- phosphate-buffered saline
- BSA
- bovine serum albumin
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IP
- inositol phosphate(s)
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}