


Abstract
We examined the effects of the calcium channel blockers nitrendipine, diltiazem, verapamil, bepridil, and mibefradil on the cloned HERG and KvLQT1/IsK K+ channels. These channels generate the rapid and slow components of the cardiac delayed rectifier K+current, and mutations can affect them, which leads to long QT syndromes. When expressed in transfected COS cells, HERG is blocked in a concentration-dependent manner by bepridil (EC50 = 0.55 μm), verapamil (EC50 = 0.83 μm), and mibefradil (EC50 = 1.43 μm), whereas nitrendipine and diltiazem have negligible effects. Steady state activation and inactivation parameters are shifted to more negative values in the presence of the blockers. Similarly, KvLQT1/IsK is inhibited by bepridil (EC50 = 10.0 μm) and mibefradil (EC50 = 11.8 μm), while being insensitive to nitrendipine, diltiazem, or verapamil. These results demonstrate that both cloned K+channels HERG and KvLQT1/IsK, which represent together the cardiac delayed rectifier K+ current, are sensitive targets to calcium channel blockers. This work may help in understanding the mechanisms of action of verapamil in certain ventricular tachycardia, as well as some of the deleterious adverse cardiac events associated with bepridil.
CCBs constitute an heterogeneous class of compounds with different chemical structures and varying potencies for blocking voltage-dependent Ca2+ channels (Hosey and Lazdunski, 1988). Their primary sites of action include cardiac muscle, systemic, and coronary arterial smooth muscle cells. Their largest domain of prescription is the management of hypertension, angina, and supraventricular arrhythmias. According to the type of voltage-dependent Ca2+ channel they block, one can distinguish T- or L-type calcium antagonists. Mibefradil is the only T-type calcium antagonist approved by the Food and Drug Administration (Clozel et al., 1997). L-type calcium antagonists include the 1,4-dihydropyridines such as nitrendipine and isradipine, the phenylalkylamines such as verapamil, and the benzothiazepines as typified by diltiazem. Many other drugs have effects on L-type Ca2+ channels, like the diarylaminopropylamine bepridil. Verapamil, diltiazem, bepridil, and mibefradil block Ca2+ channels in cardiac cells at clinically relevant concentrations, resulting in a decrease in heart rate and atrioventricular nodal conduction velocity. This forms the basis of their antiarrhythmic efficacy in reentrant arrhythmias or in slowing the ventricular rate in the case of atrial flutter or fibrillation (at least for phenylalkylamines and benzothiazepines) (Roden, 1996). With few exceptions, calcium antagonists do shorten the cell action potential duration; this is why they do not prolong the refractory period and all except bepridil (Campbell et al., 1990) are considered ineffective as ventricular antiarrhythmic agents. Hence, verapamil, diltiazem, and mibefradil can increase the rate and duration of experimentally induced ventricular tachycardia (Billman and Hamlin, 1996), and inappropriate use of verapamil has been shown to jeopardize the outcome of Wolff-Parkinson-White syndrome by further shortening the refractory period of the accessory pathway and increasing the ventricular rate in the case of atrial fibrillation (Gulamhusein et al., 1983). Conversely, verapamil also has been shown to be of use in certain forms of ventricular tachycardia known as “verapamil sensitive” that seem to be triggered by delayed afterdepolarizations (Lauer et al., 1992; Gill et al., 1993; Lee et al., 1996). Bepridil,because of its ability to prolong the refractory period of ventricular myocytes and the QT interval on the electrocardiogram, has been successfully used in ventricular arrhythmias (Roden, 1996). This classic feature of class III antiarrhythmic drugs results from a blockade of IK, which is considered to be an important modulator of the cardiac action potential repolarization. In most mammalian species, including humans, IK is recognized as being composed of at least two currents: IKr and IKs (Sanguinetti and Jurkiewicz, 1990; Li et al., 1996). IKr (rapid component) activates quickly and exhibits inward rectification. IKs (slow component) has much slower kinetics and shows no inactivation. Regarding class III antiarrhythmic drugs, with excessive prolongation of the QT interval, bepridil also can induce polymorphic ventricular tachycardias known as torsades de pointes, especially in the setting of hypokaliemia, bradycardia, or both (Manouvrier et al., 1986). The possibility for verapamil to share, with bepridil, some class III type of activity gives those calcium antagonists a role in modulating the action potential duration (i.e., shortening or lengthening the action potential duration according to the pathophysiological settings). This also raises concerns about their use in populations at risk, such as patients with long QT syndrome (Napolitano et al., 1994).
Because previous studies have suggested that CCBs can interact with cardiac voltage-dependent K+ channels (Hume, 1985), we aimed to extensively explore the potency to block IK of the most prescribed CCBs available on the market. The two types of K+ channels involved in the generation of IKr (Sanguinetti et al., 1995) and IKs (Attali, 1996; Barhaninet al., 1996; Sanguinetti et al., 1996) have been cloned, and mutations in the corresponding genes have been associated with different manifestations of the long QT syndrome (Roden et al., 1996; Chouabe et al., 1997). The IKr and IKs currents are generated by, respectively, the human ether-a-go go-related gene HERG product (Sanguinetti et al., 1995) and a K+ channel resulting from the assembly of two different proteins, KvLQT1 and IsK (Attali, 1996; Barhanin et al., 1996; Sanguinetti et al., 1996). To evaluate separately the effects of calcium antagonists on the two components of IK, we used an in vitro mammalian cell model (COS-7 cells) transfected with either HERG or KvLQT1 and IsK coding sequences.
Materials and Methods
Expression in COS cells.
The pSI HERG expressing vector was kindly provided by D. J. Snyders (Snyders and Chaudhary, 1996). The human KvLQT1 and IsK coding sequences were amplified by polymerase chain reaction and respectively subcloned into the pCI and pCMV expression vector as described previously (Barhanin et al., 1996).
The African green monkey kidney-derived cell line COS-7 was obtained from the American Type Culture Collection (Rockville, MD) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated fetal calf serum (GIBCO, Paisley, Scotland) at 37° in a humidified incubator. They were subcultured regularly by enzymatic treatment. COS-7 cells were transfected using the DEAE-Dextran method (diethylaminoethyl dextran, chloride form) according to the manufacturer’s instructions (Sigma Chemical, St. Louis, MO). Cells were seeded at a density of 20,000 cells/35-mm-diameter Petri dish 24 hr before transfection. They then were transfected with 0.05 μg of pSI-HERG/dish or 0.5 μg of pCI-KvLQT1 and 0.5 μg of pCMV-hIsK/dish. A CD8-expressing plasmid (one fifth of the required K+ channel-expressing plasmid) was added in all transfection experiments to visualize transfected cells using anti-CD8 antibody-coated beads (Jurman et al., 1994). Cells were tested electrophysiologically 48 hr after transfection. In those conditions, 30–40% of the cells expressed CD8 receptors and >90% of the bead-coated cells expressed K+ channels.
Electrophysiological methods.
Electrophysiological recordings were carried out at 22 ± 2° (room temperature) in the whole-cell configuration of the patch-clamp technique (Hamillet al., 1981). A Petri dish containing cells was placed on the stage of an inverted microscope and superfused continuously with the standard extracellular solution. Patch pipettes with a resistance of 2–5 MΩ were used routinely and connected electrically to the head stage of a RK300 patch-clamp amplifier (Biologic, Grenoble, France) with a 100-MΩ feedback resistor. Junction potentials were zeroed with the pipette in the standard extracellular solution. After the whole-cell configuration was established, the capacitative transients elicited by symmetrical 10-mV voltage-clamp steps from −80 mV were recorded at 50 kHz (filtered at 10 kHz) for calculation of capacitative surface area and series resistance, which were 34.2 ± 0.8 pF and 4.8 ± 0.2 MΩ, respectively (82 experiments). Membrane capacitance and series resistance were not compensated. Voltage commands and simultaneous signal recording were performed with pClamp software (Axon Instruments, Foster City, CA). A microperfusion system allowed local application and rapid change of the different experimental solutions. Superfusion flow rate was 50–100 μl/min.
Solutions and drugs.
The intracellular pipette filling solution contained 150 mm KCl, 0.5 mmMgCl2, 5 mm EGTA, and 10 mm HEPES/KOH, pH 7.2, and the standard extracellular solution contained 150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 1 mmMgCl2 and 10 mm HEPES/NaOH, pH 7.4. Nitrendipine (Bayer AG, Wuppertal, Germany), bepridil (Sigma), and mibefradil (F. Hoffmann-La Roche, Basel, Switzerland) were dissolved in dimethylsulfoxide (0.1 m). Diltiazem and verapamil (Sigma) were dissolved in distilled water (10 mm). The stock solutions were kept in the dark at 4°. The drug solutions were prepared fresh from these stock solutions and vortexed immediately before each use. The solvent concentration never exceeded 1:10,000.
Pulse protocols and analysis.
The holding potential in all experiments was −80 mV. Current traces were uncorrected for the leak. In all experiments, recordings were started after a 5-min dialysis of the cell. The rundown of the current was negligible during the course of the experiments (generally <10% within 20 min for KvLQT1/IsK current, whereas HERG current was stable during this period). HERG currents were evoked every 7 sec by 2-sec depolarizing voltage steps ranging from −80 mV to +80 mV in 10-mV increments and then by repolarizing steps to −40 mV for 2 sec (sampling rate, 250 Hz; cutoff frequency, 80 Hz). KvLQT1/IsK currents were evoked every 10 sec by 4-sec depolarizing voltage steps ranging from −80 mV to +120 mV in 20-mV increments and then by repolarizing steps to −40 mV for 1 sec (sampling rate, 200 Hz; cutoff frequency, 60 Hz). For both HERG and KvLQT1/IsK currents, the current-voltage relationships were obtained by measuring the current amplitude at the end of the depolarizing voltage steps and at the peak of tail currents with respect to the zero of the A/D converter. The HERG and KvLQT1/IsK activation curves were determined by fitting peak values of tail currents (Itail) versus test potential (Vt) to a Boltzmann function: Itail = Itail-max/(1 + exp[(V0.5 − Vt)/k], where Itail-max is the maximum tail current. The voltage at which the current was half-activated (V0.5) and the slope factor (k) were calculated from these data. The HERG inactivation curves were assessed by brief steps (20 msec) to various hyperpolarized potentials (ranging from −120 mV to +80 mV in 10-mV increments) from +50 mV. After allowing inactivation to relax to steady state at various test potentials, the membrane voltage was stepped to +50 mV to assess the relative number of channels available to activate. The HERG inactivation curves were determined by fitting the initial current measured on return to +50 mV versus the previous test potential to a Boltzmann function.
For drug effect screening, currents were evoked by depolarizing voltage steps to +50 and +30 mV for HERG and KvLQT1/IsK, respectively, and then by repolarizing steps to −40 mV before, during, and after application of the compounds. The effects of drugs were determined from the reduction of peak tail currents that were recorded after achieving steady state block. The percent block of HERG and KvLQT1/IsK currents by different concentrations of drugs were best fitted to the Hill equation: relative tail current = 1/{([drug]/EC50)n + 1} to determine the concentration required for half-block (EC50) and the Hill coefficient (n).
Results are expressed as mean ± standard error. Statistical significance of the observed effects was assessed by mean of a Student’s t test. A value of p < 0.05 was considered statistically significant.
Results
HERG and KvLQT1/IsK expression in COS cells.
Cells were clamped at a holding potential of −80 mV. For HERG, depolarizing steps applied for 2 sec to voltages between −80 mV and +80 mV in 10-mV increments activated a time-dependent outward current that increased in amplitude from a threshold of −50 mV to peak at 0 mV (Fig.1A). Depolarizing steps to more positive voltages resulted in an inward rectification because of a fast C-type inactivation (Sanguinetti et al., 1995). Deactivating outward tail currents elicited on repolarization to −40 mV were large as a result of the instantaneous removal of the inactivation process combined with a slow deactivation. HERG outward and tail currents were totally suppressed by the benzenesulfonamide antiarrhythmic agent E-4031 (5 μm, four cells; data not shown) as already described for IKr (Sanguinetti and Jurkiewicz, 1990). For KvLQT1/IsK, 4-sec depolarizing steps were applied to voltages ranging from −80 mV to +120 mV in 20-mV increments. A slowly time-dependent outward current developed from a threshold of −40 mV, and the amplitude increased linearly with more positive voltage steps (Barhanin et al., 1996; Chouabe et al., 1997) (Fig. 1B). Deactivating outward tail current recorded on repolarization to −40 mV increased in amplitude with depolarizing steps from −40 mV up to +100 mV, where they reached a maximum. The KvLQT1/IsK currents were E-4031 insensitive (5 μm, three cells; data not shown). The current-voltage relationships were evaluated for both currents at the end of the depolarizing steps and at the peak of tail currents. For HERG, the maximal amplitude of the outward current present at the end of the depolarizing step was of 1.1 ± 0.1 nA at 0 mV and the maximal amplitude of the tail current was of 2.0 ± 0.2 nA (31 cells; Fig. 1C). The amplitudes of KvLQT1/IsK at the end of the depolarizing steps to 0 mV and +60 mV were 1.0 ± 0.1 and 4.7 ± 0.3 nA, respectively, and the maximal amplitude of the tail current was of 1.9 ± 0.1 nA (14 cells; Fig. 1D). Because inactivation of the HERG channel was rapidly removed on repolarization, the plot of the normalized peak tail currents measured on repolarization at −40 mV versus the test potentials yielded the steady state activation curves (Fig. 1E). The mean control values of half-point activation and the corresponding slope factor were −13.0 ± 0.9 and 8.3 ± 0.2 mV, respectively (31 cells). The steady state HERG inactivation curves were assessed by brief steps to various hyperpolarized potentials from +50 mV. Normalized peak currents released from inactivation at +50 mV were plotted as a function of prepulse test potentials. The mean control values of half-point inactivation and the corresponding slope factor were −31.1 ± 3.1 and 16.9 ± 0.5 mV (29 cells). For KvLQT1/IsK, which does not inactivate, the relative activation curves were determined at the beginning of the deactivating tail currents after return to −40 mV (Fig. 1F). The mean control values of half-point activation and the corresponding slope factor were 26.9 ± 3.0 and 24.9 ± 1.3 mV (14 cells). Nontransfected COS cells or cells transfected with pCI plasmid alone exhibited no time-dependent current, as described (Barhanin et al., 1996; Chouabe et al., 1997) (Fig. 1B, inset).

HERG and KvLQT1/IsK expression in transfected COS cells. A, Typical HERG currents elicited by depolarizing voltage steps. Currents are activated by 2-sec pulses applied in 20 mV from −60 mV to +60 mV. Current magnitude progressively increased during the pulse and then decreased with voltage according to a voltage-dependent inactivation. Tail currents elicited on repolarization to −40 mV (voltage protocol inset) increased progressively with voltage and saturated at +20 mV. B, Typical KvLQT1/IsK currents elicited by depolarizing voltage steps. Currents are activated by 4-sec pulses applied in 20 mV from −60 mV to +100 mV. Time-dependent outward currents slowly increased during the pulse according to the voltage. Tail currents recorded on repolarization to −40 mV (voltage protocolinset) increased progressively with voltage and saturated at +100 mV. Top traces, superimposed current traces recorded in a nontransfected COS cell showing the absence of K+ currents on depolarization. C and D, K+currents measured at the end of the test pulses (▵) and at the peak of the deactivating tail currents after return to −40 mV (■) plotted against test potentials. Data points are mean ± standard error for 31 cells for HERG (C) and 14 cells for KvLQT1/IsK (D). The maximal amplitude of the HERG current occurs at ∼0 mV (∼1 nA), whereas the tail currents saturate at ∼2 nA from +20 mV on. The amplitude of the KvLQT1/IsK current linearly augments whereas the tail currents saturate at ∼ 2 nA from +100 mV on. E and F, Gating properties of HERG (E) and KvLQT1/IsK (F) channels in control condition. Steady state activation curves were determined for both channels at the peak of the deactivating tail current on return to −40 mV. The voltage dependence of HERG channel inactivation were assessed by 20-msec steps to test potentials ranging from −120 mV to +80 mV in 10-mV increments from +50 mV. After allowing inactivation to relax to steady state at these various test potentials, the membrane voltage was stepped to +50 mV. Steady state inactivation curves were determined by fitting the initial current measured on return to +50 mV versus previous test potentials. Data points were fitted to a Boltzmann distribution. Averaged half-point activation values (corresponding slope factors) were −13.0 ± 0.9 mV (8.3 ± 0.2 mV) for HERG (31 cells) and 26.9 ± 3.0 mV (24.9 ± 1.3 mV) for KvLQT1/IsK (14 cells). The mean value of HERG half-point inactivation and the corresponding slope factor were −31.1 ± 3.1 and 16.9 ± 0.5 mV (29 cells).
Effects of nitrendipine, diltiazem, verapamil, bepridil, and mibefradil on HERG and KvLQT1/IsK currents.
A 10 μmconcentration of the different CCBs first was tested on HERG and KvLQT1/IsK currents to evaluate the range of efficacy of the different drugs. Verapamil, bepridil, and mibefradil turned out to be potent HERG blockers (Fig. 2A), inducing a sharp decrease in both the outward and tail currents. A representative effect of bepridil on HERG current is shown on Fig. 2B. Diltiazem had a much smaller effect (14.6 ± 2.7% block of tail current; four cells), whereas nitrendipine (8.7 ± 2.9%; three cells) and isradipine effects (3.1 ± 1.8%; two cells, data not shown) were merely negligible. KvLQT1/IsK outward and tail currents were effectively blocked only by bepridil (Fig. 2C) and mibefradil with little or no effect from nitrendipine (5.2 ± 2.4%; three cells), verapamil (9.6 ± 3.5%; four cells), and diltiazem (8.0 ± 1.8%; three cells) (Fig. 2D).

Effects of nitrendipine, diltiazem, verapamil, bepridil, and mibefradil on HERG and KvLQT1/IsK currents. A and D, Relative blockade of HERG (A) and KvLQT1/IsK (D) tail currents by an equimolar 10 μm concentration of calcium antagonists. Bepridil and mibefradil share an equivalent potency to block both currents. Verapamil inhibits only HERG current significantly. Comparatively, diltiazem does not exert a relevant effect on HERG, whereas nitrendipine seldom blocks any of the current. B and C, Representative HERG (B) and KvLQT1/IsK (C) currents elicited in transfected COS cells according to the voltage protocol under control conditions (○) and after bepridil (10 μm) (•). Bepridil decreased both the outward currents elicited on depolarization and the tails resulting from return to −40 mV.
Concentration dependence of HERG blockade by verapamil.
Verapamil reversibly blocked HERG currents in a dose-dependent manner (Fig. 3A). Both the outward and tail currents were suppressed in the presence of verapamil. Each given concentration (0.1–10 μm) was maintained until steady state block was attained (Fig. 3B). Blockade occurred rapidly and was completely reversible within 2 min of washout. The concentration dependence of the blockade of HERG tail currents was best fitted to the Hill equation that yielded an EC50 value of 0.83 μm (Fig. 3C).

Concentration-dependent blockade of HERG current by verapamil. A, HERG current was evoked according to the voltage protocol (inset). Top trace, recording of tail current on repolarization to −40 mV in control conditions. Increasing concentrations of verapamil (0.1–10 μm) dose-dependently decreased the current and its corresponding tail. B, Plot of the maximal amplitude of the tail current against time from the same cell as above during control condition and superfusion with increasing concentrations of verapamil. The blockade produced by verapamil was totally reversible within 2 min of washout (WO) with standard solution. C, Verapamil induced a concentration-dependent blockade of HERG tail currents (mean ± standard error, six cells). Data were best fitted with the Hill equation (inset, chemical structure of verapamil). The EC50 value is 0.83 μm (Hill coefficient, 1.19).
Verapamil influences HERG voltage-dependent gating.
To evaluate the voltage-dependence characteristics of verapamil blockade, a concentration close to verapamil EC50 (1 μm) was applied on HERG current. Verapamil significantly blocked both the outward and tail currents (Fig.4, A and B). Verapamil shifted HERG steady state activation curves by ∼8 mV to more negative values (Fig.4C and Table 1). Half-point activation value was −13.9 ± 1.1 mV (control) and −21.9 ± 2.1 mV with verapamil (p < 0.05; 11 cells), with no significant change in slope factors [8.4 ± 0.3 and 7.9 ± 0.3 mV for control and verapamil, respectively; p > 0.1 (NS)]. Steady-state inactivation curves also were shifted to more negative values by ∼22 mV. Half-point inactivation value was −26.1 ± 4.6 mV (control) and −48.4 ± 4.5 mV with verapamil (p < 0.05, 11 cells), again with no significant change in slope factors [17.8 ± 0.6 and 20.2 ± 0.7 mV for control and verapamil, respectively; p > 0.1 (NS)].

Effects of verapamil on HERG currents. A, Current traces of HERG were recorded by using depolarizing pulses to seven different potentials with 20-mV steps between −60 mV and +60 mV from a holding potential of −80 mV (voltage protocol inset).Left traces, control conditions. Right traces, recorded in the presence of verapamil (1 μm), which decreased significantly the outward and tail currents. B, Current-voltage relationships for the currents measured at the end of the 2-sec depolarizing pulse (▵) and at the peak of the deactivating tail currents (■) on return to −40 mV in control conditions (mean ± standard error). Closed symbols, currents measured after verapamil (11 cells). Verapamil significantly decreased the tail currents according to the voltage and shifted the peak of the outward current by 10 mV to hyperpolarized values. Inset, normalized HERG tail currents (○) and relative tail current blockade magnitude (•) according to the test pulse potential. The intensity of verapamil blockade parallels the steady state activation of the current. C, Steady state activation and inactivation curves determined by Boltzmann equation in control conditions (○) and after verapamil (•, 11 cells) characterized a shift toward more hyperpolarized values. Data are mean ± standard error (vertical bars). D, HERG current profile during a cardiac action potential. The cardiac action potential was simulated as a brief pulse (10 msec) from −90 mV to +50 mV followed by a ramp (400 msec) from +50 mV to −90 mV. This waveform (gray trace) was applied as voltage command. HERG currents (black traces) were recorded in control condition, in steady state condition during application of 1 μm verapamil, and after washing out with control solution.
Steady-state activation and inactivation parameters of HERG and KvLQT1/IsK currents in the absence and presence of verapamil, bepridil, and mibefradil
Current-voltage relationships of HERG outward and tail currents showed a voltage dependence of HERG blockade by verapamil. Verapamil decreased the tail current amplitude by 33.7 ± 5.4% at −20 mV and by 64.0 ± 2.7% at +20 mV (11 cells, p < 0.05 in both cases). For voltages >−40 mV, both the channel availability and the level of blockade by verapamil increased accordingly, suggesting that verapamil blockade requires HERG channel activation (Fig. 4B,inset). In the presence of verapamil, the time course of HERG-activating current displayed a slow inactivation-like behavior (Fig. 4A), again suggesting an open channel block mechanism.
The magnitude and the time course of the HERG current during a cardiac action potential and the effects of verapamil on this current were evaluated using a simplified cardiac action potential-like waveform as voltage command (Fig. 4D). As expected, the HERG current was small at the end of the step depolarization from −90 mV to +50 mV. The ramp repolarization (400 msec in duration) from +50 mV to −90 mV first induced an increase of the outward current likely due to the inactivation relief and then induced a linear decline of the current to zero, corresponding to the reduction in driving force while HERG channels are still open. Verapamil (1 μm) decreased the HERG response by ∼50% and delayed its peak value compared with control.
Bepridil and mibefradil blockade of HERG current.
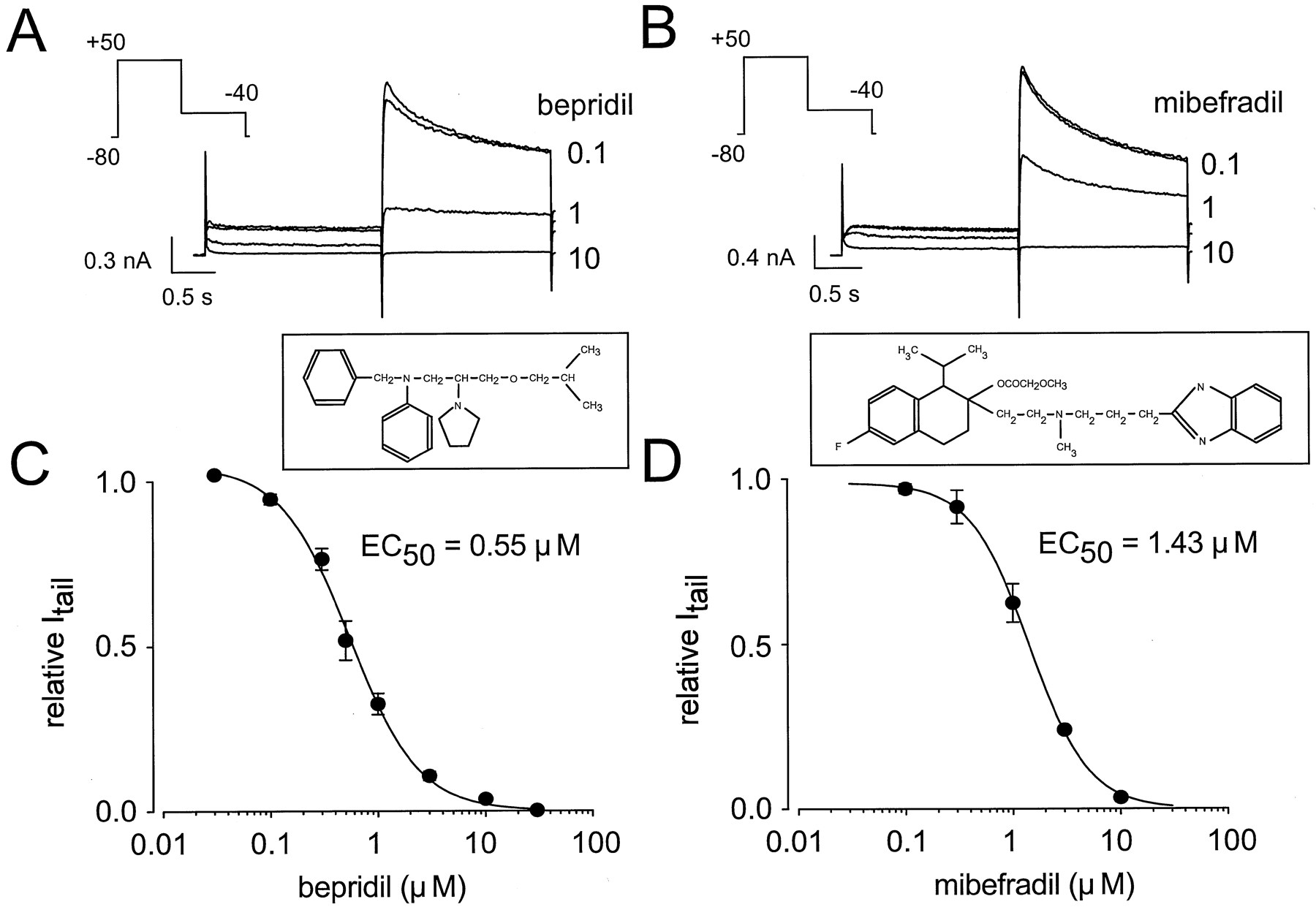
Regarding verapamil, bepridil and mibefradil also decreased dose-dependently and reversibly both the HERG outward and tail currents. Similar analysis of the data obtained from each drug (six cells in both cases) resulted EC50 values of 0.55 μm for bepridil and 1.43 μm for mibefradil (Fig. 5, A–D). Again, bepridil and mibefradil blockade of HERG tail current seemed to be voltage dependent (Fig. 6, A–D). Bepridil and mibefradil decreased the tail current amplitude by 4.4 ± 1.2% and 20.6 ± 6.0% at −20 mV and by 46.6 ± 8.0% and 52.8 ± 2.0% at +20 mV, respectively (10 cells, p < 0.05 in each case). Similar modifications of the HERG gating properties were observed with both drugs (Fig. 6, E and F, and Table 1).

Concentration-dependent blockade of HERG current by bepridil and mibefradil. A and B, HERG current was evoked with 2-sec depolarizing pulses to +50 mV from a holding potential of −80 mV (voltage protocol inset). Tail current was recorded on repolarization to −40 mV. Increasing concentrations (0.1–10 μm) of bepridil (A) and mibefradil (B) dose-dependently decreased the current and its corresponding tail. In each case, the top trace corresponds to control conditions. C and D, Concentration-dependent blockade by bepridil (C, six cells) and mibefradil (D, six cells) of HERG tail currents (insets, chemical structures of bepridil and mibefradil). The Hill equation was fitted to the data with 100% blockade taken as the fixed maximal effect. EC50 values are 0.55 and 1.43 μm for bepridil and mibefradil, respectively. Hill coefficients are 1.35 (bepridil) and 1.57 (mibefradil). Data are expressed as mean ± standard error (vertical bars).

Effects of bepridil and mibefradil on HERG currents. A, Current traces of HERG were recorded by using depolarizing pulses to five different potentials with 20-mV steps between −40 mV and +40 mV from a holding potential of −80 mV (voltage protocolinset). Top traces, control conditions.Bottom traces, recorded in the presence of bepridil (0.5 μm). B, HERG traces in control conditions (top) and after mibefradil (1 μm) (bottom). The same voltage protocol is used as in A. C and D, Current-voltage relationships for the currents measured at the end of the 2-sec depolarizing pulse (▵) and at the peak of the deactivating tail currents (■) on return to −40 mV in control conditions (mean ± standard error). Closed symbols, the currents measured after bepridil (C, 10 cells) and mibefradil (D, 10 cells). E and F, Steady state activation and inactivation curves determined by Boltzmann equation in control conditions (○) and after drug (•) for bepridil (E, 10 cells) and mibefradil (F, 8–10 cells) show a shift toward more negative value of both curves. Data are expressed as mean ± standard error (vertical bars).
Bepridil and mibefradil blockade of KvLQT1/IsK current.
Increasing concentrations of bepridil and mibefradil (1–100 μm) were tested on KvLQT1/IsK currents. Bepridil and mibefradil dose-dependently blocked both the outward and tail currents (Fig. 7A). At each given concentration, the drug was maintained until steady state block was attained (Fig.7B). Blockade occurred rapidly and was fully reversible when the drug was washed out. The concentration-effect relationships were fitted to the Hill equation and yielded EC50 values of 10.0 and 11.8 μm for bepridil and mibefradil, respectively (Fig. 7, C and D).

Concentration-dependent blockade of KvLQT1/IsK current by bepridil and mibefradil. A, KvLQT1/IsK current was evoked with 4-sec depolarizing pulses to +30 mV from a holding potential of −80 mV (voltage protocol inset). Tail current was recorded on repolarization to −40 mV. Top trace, control conditions. Increasing concentrations of bepridil (1–30 μm) dose-dependently decreased the current and its corresponding tail. B, Plot of the tail current amplitude against time from cell (A) before, during, and after superfusion with increasing concentrations of bepridil.WO, washout. C and D, Concentration-dependent blockade by bepridil (C, seven cells) and mibefradil (D, eight cells) of KvLQT1/IsK tail currents. The Hill equation was fitted to the data with 100% blockade taken as the fixed maximal effect. EC50values are 10.0 and 11.8 μm for bepridil and mibefradil, respectively. Hill coefficients are 1.42 (bepridil) and 0.97 (mibefradil). Data are expressed as mean ± standard error (vertical bars).
Regarding HERG, to evaluate the voltage-dependence characteristics of bepridil and mibefradil blockade, a drug concentration of 10 μm, which is close to the EC50 values of bepridil and mibefradil, was applied on KvLQT1/IsK current. Both drugs significantly blocked the KvLQT1/IsK outward and tail currents (Fig.8, A and B). The current-voltage relationships as measured at the end of the depolarizing steps and at the peak of tail currents in presence of both drugs (Fig. 8, C and D) showed little if any voltage-dependency at −20, + 20, and +60 mV; the resulting block of KvLQT1/IsK current was 61.3 ± 7.9%, 55.6 ± 7.8%, and 52.6 ± 6.4% for bepridil (eight cells) and 46.4 ± 6.6%, 43.4 ± 8.0%, and 39.0 ± 7.7% for mibefradil (six cells). The voltage dependence of KvLQT1/IsK activation was unaffected by either bepridil or mibefradil (Fig. 8, E and F, and Table 1).

Effects of bepridil and mibefradil on KvLQT1/IsK currents. A, Current traces of KvLQT1/IsK were recorded by using depolarizing pulses to five different potentials with 40-mV steps between −60 mV and +100 mV from a holding potential of −80 mV (voltage protocol inset). Top traces, control conditions. Bottom traces, recorded in the presence of bepridil (10 μm). B, KvLQT1/IsK traces in control conditions (top) and after mibefradil (10 μm) (bottom). The same voltage protocol is used as in A. C and D, Current-voltage relationships for the currents measured at the end of the 4-sec depolarizing pulse (▵) and at the beginning of the deactivating tail currents (■) on return to −40 mV in control conditions (mean ± standard error). Closed symbols, currents measured after bepridil (C, eight cells) and mibefradil (D, six cells). E and F, Steady state activation curves determined in control conditions (○) and after drug (•) at the beginning of the deactivating tail currents on return to −40 mV (mean ± standard error) for bepridil (E, eight cells) and mibefradil (F, six cells).
Discussion
The cardinal feature of this study is that calcium antagonists have various potentialities to block IK. Bepridil and mibefradil block both HERG and KvLQT1/IsK currents, verapamil only blocks HERG current, and nitrendipine and diltiazem seldom block any of these currents.
Among the five CCBs that were tested, only verapamil, bepridil, and mibefradil blocked HERG current with an EC50value relevant to therapeutic concentrations. Open channel block by verapamil of native K+ currents different from IKr has been reported previously in humans (Pancrazio et al., 1991) and animal cell models (DeCoursey, 1995). Similar results also have been observed with an expression model of cloned K+ channels (Kv1.5 and Kv1.3) other than HERG (Rampe et al., 1993; Rauer and Grissmer, 1996). In this study, we have shown the effect of verapamil on IKr in a transfected COS cell model and found similar results with bepridil and mibefradil. These drugs, at their respective EC50 values, produced similar changes in steady state activation and inactivation curves. The midpoint potential (V0.5) of the activation curves was shifted ∼8 mV in the hyperpolarizing direction with no significant change in the slope (k). For the inactivation curves, there was a greater hyperpolarization shift of V0.5(∼22 mV) without any change in k. The greater shift of inactivation compared with activation only partially (∼30%) accounts for the reduction in HERG currents during depolarizing voltage steps. Hence, in tail currents reflecting mainly the activation process, their blockade cannot be explained solely by the shift of inactivation. In fact, the degree of channel block directly correlates with channel activation (Fig. 4B, inset). The blockade of activation and the voltage shifts can be separate mechanisms of action as more clearly shown by using the simulated action potential protocol (Fig. 4D). Besides the blocking effect exerted by verapamil, the delay in the peak current during the ramp repolarization reflects the shift of the inactivation curve to more hyperpolarized voltages. In addition, verapamil seems to be an open-channel blocker of HERG channel (Fig.4A).
Only bepridil and mibefradil block KvLQT1/IsK current with an EC50 value close to 10 μm, which is comparable to that observed with the IKs blocker chromanol 293B (Loussouarn et al., 1997) or the antiarrhythmic drug amiodarone (Balser et al., 1991) and greater than that with azimilide, a new IKsblocker currently under development (Salata and Brooks, 1997). The blockade exerted by bepridil or mibefradil did not seem to be either voltage dependent or accompanied by a significant change in the channel gating. Nitrendipine, diltiazem, and verapamil did not have any effect on this current.
Ca2+ and K+ currents, flowing through different time- and voltage-dependent channels, are major determinants of the plateau and repolarization phases of the cardiac action potential. In most mammalian species, the IK is composed of two main currents: a rapidly activating component, IKr, and a slowly activating component, IKs (Sanguinetti and Jurkiewicz, 1990). Similarly, IKr and IKs have been found in human cardiac cells (Liet al., 1996). Numerous attempts have been made to discriminate between these two components of the cardiac delayed rectifier, including the use of specific IKrblockers, Ca2+ channel blockers, β-adrenoreceptor agonists, and low external Ca2+ and K+ (Sanguinetti and Jurkiewicz, 1990; Salata et al., 1996). Most of these conditions interfere with interpretation of the results. In our model, mammalian COS cells are transfected with either HERG or KvLQT1/IsK human coding sequences, therefore expressing separately each component of the IK and avoiding any unneeded interfering condition.
One must consider the relevance of the concentrations that were used in this study. The therapeutic plasma levels of calcium antagonists range from 1 to 10 nm for dihydropyridines and up to several micromolar concentrations for verapamil (Rampe et al., 1993) and mibefradil (Clozel et al., 1997), which is well within the range of concentrations inducing a blockade of HERG and KvLQT1/IsK currents in our study.
It is recognized that agents prolonging cardiac repolarization, such asd-sotalol or amiodarone, might be antiarrhythmic because they inhibit the delayed rectifier IK (Balseret al., 1991) and do prolong the refractory period of the ventricular cardiomyocytes. This mechanism can represent a relevant alternative to the suppression of the delayed afterdepolarization in the verapamil sensitivity of certain ventricular tachycardias (Laueret al., 1992).
Conversely, the prolongation of the action potential has the ability to induce early afterdepolarizations that may trigger a complex form of ventricular arrhythmias known as torsades de pointes (Napolitanoet al., 1994). This is the case for bepridil, which prolongs the action potential duration (Campbell et al., 1990) and was shown to be, apart from a potent antiarrhythmic agent, promoting polymorphic ventricular arrhythmias (Manouvrier et al., 1986). Verapamil does not seem per se to trigger such cardiac adverse events, and the slight differences observed in the EC50 values of mibefradil and bepridil for HERG and KvLQT1/IsK currents hardly explain the widely different clinical profiles of these compounds (Massie, 1997), especially concerning the bepridil arrhythmogenicity. Several reasons might explain such a discrepancy. (1) Verapamil targets only the rapid component of the cardiac delayed rectifier, which renders bepridil and mibefradil likely to be more potent blockers of IK by blocking both of its components. (2) The ratio of potassium and calcium channel blockade potency may be of importance in favoring prolongation over shortening of the action potential duration; in this study, bepridil at therapeutic concentrations has the same propensity to block Ca2+ channels (Yatani et al., 1986) than HERG channel. (3) Other ancillary properties, such as Na+ channel blockade, can modify the myocardial cell refractory period. Unlike many of the other CCBs, bepridil also inhibits fast Na+ channels (Yatani et al., 1986), thus imparting with a class I antiarrhythmic activity the same class proarrhythmogenicity (CAST, 1989). This may explain why verapamil decreases the occurrence of early afterdepolarizations (Singh, 1989; Cosio et al., 1991), whereas bepridil does not (Campbell et al., 1990).
Drug ancillary properties also are of importance in the therapeutic goal to achieve in patients. Amiodarone, for example, is a multifaceted drug that bears class I–IV antiarrhythmic efficacy (Nattel et al., 1992). It also has been shown to better preserve patients with heart failure from death (Pinto et al., 1997) than its more specific “pure” class I (CAST, 1989) or class III (Waldoet al., 1996) counterparts. This could be the case also for calcium antagonists bearing K+ channel blockade properties (DAVIT, 1990).
Study limitations should be considered. Obviously, species differences concerning the respective prominence of the delayed rectifier and the inward calcium currents in the process of repolarization should be considered. Nevertheless, mutations of HERG, KvLQT1, or IsK in the setting of the congenital long QT syndromes have emphasized the importance of the delayed rectifier in human cardiac repolarization (Roden et al., 1996; Chouabe et al., 1997). Regarding bepridil, it is possible that some individuals may be more susceptible to torsades de pointes than others. They may possess a mutation in a channel or regulatory element responsible for repolarization that renders them more susceptible to K+ channel blockade (Roden et al., 1996).
In conclusion, the dual approach of the IKprovided by our model permitted us to differentially quantify the potassium channel blockade exerted by calcium antagonists. The fact that drugs have the ability to block both K+ and Ca2+ currents renders them potentially useful “action potential modulators,” for example, with arrhythmias complicating essential hypertension or stable angina.
Acknowledgments
We are grateful to D. J. Snyders and S. Kupershmidt (Vanderbilt University School of Medicine, Nashville, TN) for the gift of the HERG-expressing plasmid. Mibefradil was kindly provided by Hoffmann-La Roche (Basel, Switzerland). We thank M. Jodar for expert technical assistance and Y. Benhamou for secretarial assistance.
Footnotes
-
Send reprint requests to: Prof. Michel Lazdunski, Institut de Pharmacologie Moléculaire et Cellulaire, CNRS, 660 route des Lucioles, Sophia Antipolis, F-06560 Valbonne, France. E-mail:ipmc{at}ipmc.cnrs.fr
-
This work was supported by the Centre National de la Recherche Scientifique. C.C. is a recipient of a Grant from the Association Française contre les Myopathies.
- Abbreviations:
- IK
- cardiac delayed rectifier K+ current
- IKr
- rapidly activating component of cardiac delayed rectifier K+ current
- IKs,slowly activating component of cardiac delayed rectifier K+current
- EGTA, ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- CCB
- calcium channel blocker
- Received April 6, 1998.
- Accepted June 17, 1998.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}