


Abstract
The effects of cannabinoids on metabolic pathways and signal transduction systems were studied in primary cultures of rat astrocytes. Δ9-Tetrahydrocannabinol (THC), the major active component of marijuana, increased the rate of glucose oxidation to CO2 as well as the rate of glucose incorporation into phospholipids and glycogen. These effects of THC were mimicked by the synthetic cannabinoid HU-210, and prevented by forskolin, pertussis toxin, and the CB1 receptor antagonist SR 141716. THC did not affect basal cAMP levels but partially antagonized the forskolin-induced elevation of intracellular cAMP concentration. THC stimulated p42/p44 mitogen-activated protein kinase (MAPK) activity, Raf-1 phosphorylation, and Raf-1 translocation to the particulate cell fraction. In addition, the MAPK inhibitor PD 098095 and the phosphoinositide 3-kinase inhibitors wortmannin and LY 294002 were able to antagonize the THC-induced stimulation of glucose oxidation to CO2, phospholipid synthesis and glycogen synthesis. The possible involvement of sphingomyelin breakdown in the metabolic effects of THC was studied subsequently. THC produced a rapid stimulation of sphingomyelin hydrolysis that was concomitant to an elevation of intracellular ceramide levels. This effect was prevented by SR 141716. Moreover, the cell-permeable ceramide analogd-erythro-N-octanoylsphingosine, as well as exogenous sphingomyelinase, were able in turn to stimulate MAPK activity, to increase the amount of Raf-1 bound to the particulate cell fraction, and to stimulate glucose metabolism. The latter effect was prevented by PD 098059 and was not additive to that exerted by THC. Results thus indicate that THC produces a cannabinoid receptor-mediated stimulation of astrocyte metabolism that seems to rely on sphingomyelin hydrolysis and MAPK stimulation.
Cannabinoids, the active components of marijuana, exert a wide spectrum of effects such as alterations in cognition and memory, analgesia, anticonvulsing, anti-inflammation, and alleviation of both intraocular pressure and emesis (Abood and Martin, 1992). It is currently well established that cannabinoids exert their effects by binding to specific plasma-membrane receptors (Howlett, 1995). To date, two different cannabinoid receptors have been characterized and cloned from mammalian tissues: CB1 (Matsudaet al., 1990) and CB2 (Munro et al., 1993). The CB1 receptor is mainly distributed in the central nervous system, whereas the CB2 receptor is expressed in cells of the immune system but not in brain (Matsuda et al., 1990; Munro et al., 1993; Howlett, 1995). Experiments conducted in transformed cells have shown that signaling through the CB1 and the CB2 receptor induces inhibition of adenylyl cyclase (Felder et al., 1995; Slipetzet al., 1995) as well as stimulation of the MAPK cascade (Bouaboula et al., 1995a, 1995b, 1996). The CB1 receptor is also coupled to other signal transduction systems, such as inhibition of ion channels (Howlett, 1995; Pan et al., 1997) and mobilization of arachidonic acid (Hunter et al., 1997).
Astrocytes, the major class of glial cells in the mammalian brain, play an important role in the homeostasis of the neuronal microenvironment, the formation of the blood-brain barrier, the guidance of neuron migration in the developing embryo, and the secretion of neurotrophic factors for neuron healing in several neuropathological situations (Fedoroff et al., 1993). A major homeostatic function of astrocytes is the regulation of brain energy metabolism (Magistretti and Pellerin, 1996; Wiesinger et al., 1997). Thus, astrocytes provide neurons with anaplerotic metabolites and substrates for generation of energy (Magistretti and Pellerin, 1996; Wiesingeret al., 1997). It is thus conceivable that neuropathological processes, such as those induced by cannabinoid intoxication (Abood and Martin, 1992), may perturb the homeostatic functions of the astroglia, including those related to energy metabolism. This could in turn lead to an impairment of neuronal functionality. As a matter of fact, THC has been shown to affect glucose metabolism in rat-glioma cellsin vitro (Sánchez et al., 1997) and in rat (Megulies and Hammer, 1991) and human brain (Volkow et al., 1996) in vivo.
A potential direct and specific action of cannabinoids on astrocytes is supported by some recent observations. Thus, for example, the CB1 receptor mRNA is expressed in astrocytes and astrocytoma cells (Bouaboula et al., 1995a). Astrocytes in culture have also been shown to bind and take up anandamide, a putative endogenous ligand of the CB1 receptor (Di Marzo et al., 1994). In astrocytoma cells, cannabinoids lead to the stimulation of the MAPK cascade and to the induction of the immediate-early gene krox-24 (Bouaboulaet al., 1995a, 1995b). However, the molecular events underlying the cannabinoid-induced activation of the MAPK cascade are as yet unknown. In addition, the potential implications of the cannabinoid-induced stimulation of MAPK on astroglial physiology (e.g., on metabolic-regulation systems) are also unknown. Hence, the present work was undertaken to study the mechanism by which cannabinoids may lead to the stimulation of the MAPK cascade and the consequences this may have on glucose metabolism in primary cultures of rat astrocytes.
Methods
Reagents.
THC, neutral sphingomyelinase (fromStaphylococcus aureus) and 1-methyl-3-isobutylxantine were from Sigma Chemical (St. Louis, MO). SR 141716 was a generous donation of Sanofi Recherche (Montpellier, France). HU-210 was kindly given by Prof. R. Mechoulam (Hebrew University, Jerusalem, Israel). Forskolin, pertussis toxin, wortmannin, LY 294002, PD 098059 and C8-ceramide were from Calbiochem (San Diego, CA, USA). d-[U-14C]glucose, [methyl-14C]choline, [9,10-3H]palmitic acid, [32P]Pi, [γ-32P]ATP, the p42/p44 MAPK assay components, the [3H]cAMP assay kit and the electrochemiluminescence detection kit were from Amersham International (Amersham, Bucks, UK). The anti-Raf-1 polyclonal antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-phosphotyrosine polyclonal antibody was from Zymed (San Francisco, CA). FCS and all plastic material for cell cultures were from Nunc (Roskilde, Denmark).
Cell cultures.
Cortical or hippocampal astrocytes were derived from 1–2-day-old rats and cultured as described previously (Galve-Roperh et al., 1997b). Briefly, cells were seeded on plastic plates previously coated with 5 μg/mll-polyornithine in water. Cells were cultured for 3 weeks in a mixture of DMEM medium and Ham’s F12 medium (1:1, v/v) supplemented with 0.66% (w/v) glucose, 5 μg/ml streptomycin, 5 units/ml penicillin and 10% FCS. Cell cultures consisted of 95% astrocytes as judged by immunocytochemical staining of glial fibrillary acidic protein (Galve-Roperh et al., 1997b).
For all the experimental determinations performed (see below), 48 hr before the experiment, the FCS-containing medium was removed and cells were transferred to a chemically-defined, serum-free medium consisting of DMEM/Ham’s F12 (1:1, v/v) supplemented with 5 μg/ml insulin, 50 μg/ml human transferrin, 20 nm progesterone, 50 μm putrescine, 30 nm sodium selenite, and 0.1% (w/v) defatted and dialyzed bovine serum albumin. Except for pertussis toxin, which was prepared in water, 1000 × stock solutions of the different cellular modulators used in this study were prepared in Me2SO. Then, control incubations had the corresponding Me2SO content. No significant influence of Me2SO on any of the parameters determined was observed at the final concentration used (0.1%, v/v). Experiments were performed at a cell density of ca. 6 × 104 cells (7 μg protein)/cm2.
Determination of rates of [14C]glucose metabolism.
Rates of [14C]glucose metabolism were monitored essentially as described before (Sánchez et al., 1997). Briefly, astrocytes were cultured as indicated above in 25-cm2 flasks. Reactions were started by the addition of 2 μCi ofd-[U-14C]glucose and different effectors to the cell cultures, and stopped with 0.3 ml of 2m HClO4 after 6 hr (pilot experiments had shown that glucose utilization was linear at least up to 8 hr). At the same time, 0.15 ml of benzethonium hydroxide (1 m in methanol) was injected in a center well containing filter paper. Samples were allowed to equilibrate for 12 additional hr, and the center well (with the 14CO2fixed as bicarbonate) was transferred to vials for radioactive counting. The cell precipitates were neutralized with K2CO3 and used to quantify phospholipids after lipid extraction and thin layer chromatography on silica-gel G60 plates, using hexane/diethyl ether/acetic acid (70:30:1, v/v/v) as developing system (Sánchez et al., 1997).
For glycogen determination, after the 6-hr incubation period, the medium was removed, cells were washed three times with NaCl/Pi, and reactions were stopped with 2 ml of 2 m NaOH. Five milligrams of unlabeled glycogen was added as carrier, cells were scraped and transferred to tubes, and glycogen was precipitated four times with 70% ethanol. Ethanol precipitates were digested with amyloglucosidase and glycogen was quantified as described previously (Sánchez et al., 1997).
Determination of cAMP concentration.
Astrocytes were cultured as indicated above in multiwell plates (9.4 cm2 in diameter) in the presence of 0.5 mm 1-methyl-3-isobutylxantine, a phosphodiesterase inhibitor. After 15 min, cells were further exposed to different effectors. Then, reactions were stopped by the addition of 1 ml of acidic ethanol (1 ml of concentrated HCl/100 ml ethanol). Cell lysates were scraped off, transferred to tubes and then centrifuged (2000 × g, 10 min) to remove any remaining cell fragments. Supernatants were subsequently collected, lyophilized, and quantified for cAMP using a cAMP assay kit.
MAPK assay.
Astrocytes were cultured as indicated above in multiwell plates (9.4 cm2 in diameter) and exposed to different agents. Reactions were terminated by washing with ice-cold PBS (1× = 10 mm NaPi, 150 mm NaCl, pH 7.4) and addition of 500 μl of ice-cold lysis buffer consisting of 10 mm Tris·HCl, pH 7.4, 150 mm NaCl, 2 mm EGTA, 2 mm dithiothreitol, 40 μg/ml digitonin, 1 mm orthovanadate, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin. Cell debris were precipitated at 25,000 × g for 20 min and MAPK activity was measured in the supernatant. p42/p44 MAPK activity was then assayed by mixing 15 μl of the cell extract with 10 μl of substrate buffer (6 mm substrate peptide, 75 mmHEPES, pH 7.4, 0.3 mm sodium orthovanadate, and 0.05% sodium azide) and 5 μl of ATP medium (0.3 mm[γ-32P]ATP (0.3 μC/μl) and 90 mm MgCl2) according to Galve-Roperhet al. (1997b).
Raf-1 immunoprecipitation.
Raf-1 was immunoprecipitated from astrocytes cultured in 57-cm2 dishes essentially as described by Galve-Roperh et al. (1997a, 1997b). In the case of 32P-labeling experiments, the chemically defined medium was replaced with Pi-free DMEM and cells were loaded with [32P]Pi (100 μCi/dish) for 4 hr. Cells were stimulated as described in the text and lysates were further obtained by treating cells with a buffer containing 50 mm Tris·HCl, pH 7.5, 2 mm EDTA, 1 mm EGTA, 1% Triton X-100, 1 mg/ml bovine serum albumin, 150 mm NaCl, 10 mm 2-mercaptoethanol, 1 mm PMSF, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, and 10 μg/ml benzamidine. Samples were treated with 7.5 μg/ml anti-Raf-1 antibody bound to anti-rabbit agarose-linked IgG. Phosphorylation was determined in the immunoprecipitates after sodium dodecyl sulfate-polyacrylamide gel electrophoresis and either autoradiography of the gels (32P-labeled Raf-1) or luminography. In the latter case, samples were transferred onto nitrocellulose membranes. The blots were then blocked with 5% fat-free dried milk in PBS supplemented with 0.1% Tween 20. They were subsequently incubated with the anti-phosphotyrosine antibody (1 μg/ml) in PBS/Tween 20 for 1 hr at room temperature, and washed thoroughly. The blots were then incubated with anti-rabbit peroxidase-conjugated secondary antibody (1:5000) for 1 hr at room temperature, and finally subjected to luminography with an electrochemiluminescence detection kit.
Western blot analysis of Raf-1.
Astrocytes were cultured as indicated above in 57-cm2 dishes. After stimulation with different effectors as indicated in the text, the medium was aspirated, cells were washed with ice-cold PBS, and 500 μl of ice-cold lysis medium was added onto the plates. This medium contained 50 mm Tris·HCl, pH 7.5, 5 mm EDTA, 1 mm EGTA, 10 mm 2-mercaptoethanol, 1 mm PMSF, 5 μg/ml leupeptin, 2 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, and 10 μg/ml benzamidine. Cells were scraped from the plates, sonicated (2 × 10 sec) on ice, and the particulate fraction was obtained after centrifugation at 40,000 × g for 60 min. Samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and proteins were further transferred onto nitrocellulose membranes. Luminographic analysis was performed as described above after incubation of the blots with the anti-Raf-1 antibody (1:250).
Determination of sphingomyelin hydrolysis.
Astrocytes were cultured in multiwell plates (9.4 cm2 in diameter). Two days before the experiment, cells were transferred to chemically defined medium (see above) supplemented with 1 μCi of [methyl-14C]choline per well. Reactions were started by the addition of the different agonists and terminated by aspiration of the medium and addition of 1 ml methanol. Samples were further treated as described by Mathias et al. (1993). Briefly, lipids were extracted with chloroform/methanol/HCl (100:100:1, v/v/v) containing 15 mm EDTA and saponified in 0.1 m methanolic KOH. Sphingomyelin was resolved by thin layer chromatography on silica-gel G60 plates with chloroform/methanol/acetic acid/water (60:30:8:5, v/v/v/v) as developing system.
Determination of ceramide levels.
Astrocytes were cultured as described above for determination of sphingomyelin hydrolysis but 1 μCi of [9,10-3H]palmitate was used instead of radiolabeled choline. Lipids were extracted and saponified (Mathiaset al., 1993), and ceramide was resolved by thin layer chromatography on silica-gel G60 plates with chloroform/methanol/water (100:42:6, v/v/v) as developing system until the front reached two thirds of the plate. The solvent was then evaporated and plates were subsequently run with chloroform/methanol/acetic acid (94:1:5, v/v/v) until the front reached the top of the plate.
Statistical analysis.
Results shown represent the mean ± standard deviation of the number of experiments indicated in every case. In the case of metabolic parameters, five or six different replicates of the various conditions included in each experiment were routinely performed. In the rest of the parameters, every experimental condition was assayed in triplicate. Statistical analysis was performed by ANOVA. A post hoc analysis was made by the Student-Neuman-Keuls test.
Results
Effect of THC on glucose metabolism.
The effect of THC on glucose metabolism was studied in primary cultures of newborn-rat astrocytes. At physiologically relevant concentrations (Abood and Martin, 1992), THC induced a significant dose-dependent stimulation of glucose oxidation to CO2 in primary cultures of rat cortical astrocytes. Half-maximal stimulation of CO2 production was observed at ∼ 8 nm THC (Fig. 1), which is in the range of the binding affinity of THC for cannabinoid receptors in whole-cell systems (Howlett, 1995). Because the maximal effect of THC occurred at 100 nm (Fig. 1), further experiments were conducted with that standard dose of THC.

Dose-dependent stimulation of glucose oxidation by THC and HU-210 in primary cultures of cortical astrocytes. Cells were incubated with different concentrations of THC (○) or HU-210 (•). Results are expressed as percentage of incubations with no additions and correspond to four different experiments for each condition.
As shown in Table 1, not only glucose oxidation to CO2, but also glucose utilization for phospholipid and glycogen syntheses were activated by THC. The effects exerted by THC on these three metabolic pathways were quantitatively similar (Table 1). In addition, although it has been reported that the CB1 cannabinoid receptor may display an heterogeneous distribution within the newborn-rat brain (Romero et al., 1997), no significant differences were observed between cortical and hippocampal astrocytes either in the basal rates of glucose metabolism or in the response of glucose-metabolizing pathways to THC (Table 1).
Effect of THC and other cellular modulators on glucose metabolism in primary cultures of cortical and hippocampal astrocytes
Bouaboula et al. (1995a) have shown that rat astrocytes and astrocytoma cells express the mRNA for the CB1 cannabinoid receptor. Therefore, several experiments were performed to test whether the THC-induced stimulation of glucose metabolism in astrocytes was a cannabinoid receptor-mediated process. The maximal stimulatory effect elicited by THC on glucose oxidation was similar to that produced by HU-210, a very potent and specific synthetic cannabinoid (Fig. 1). HU-210 has been reported to bind to cannabinoid receptors with much higher affinity than THC (Howlett, 1995). Thus, half-maximal stimulation of CO2 production (Fig. 1) occurred at ∼ 0.8 nm HU-210 [i.e., in the range of the binding affinity of HU-210 for cannabinoid receptors in whole-cell systems (Howlett, 1995)]. As shown in Table 1, HU-210 also induced a ∼50% stimulation of phospholipid and glycogen syntheses from glucose.
Because both pertussis toxin and forskolin are widely used to demonstrate receptor-dependent effects of cannabinoids (Howlett, 1995), the effect of these two compounds on the metabolic effects of THC was tested. As shown in Table 1, the stimulatory effect of THC on glucose utilization was abolished by treatment of cells with pertussis toxin, which prevents the dissociation of Gi/Go proteins, and with forskolin, which stimulates adenylyl cyclase. Furthermore, the stimulatory effect of THC on glucose metabolism was antagonized by SR 141716 (Table 1), a selective CB1 receptor antagonist. Forskolinper se produced a significant inhibition of phospholipid and glycogen syntheses, whereas neither pertussis toxin nor SR141716 at the concentrations used had any significant effect on the three metabolic pathways studied (Table 1).
Effect of THC on cAMP concentration.
In a first attempt to elucidate the mechanism of the cannabinoid-induced stimulation of glucose metabolism, the intracellular concentration of cAMP was determined. THC and HU-210 partially antagonized the forskolin-induced elevation of intracellular cAMP concentration (Fig.2), pointing to a Gi protein-mediated inhibition of adenylyl cyclase by cannabinoids in primary astrocytes. As also shown for glucose oxidation (Fig. 1), HU-210 was much more potent than THC in preventing forskolin-stimulated cAMP accumulation (Fig. 2). The effect of cannabinoids on the forskolin-induced elevation of cAMP levels was not evident when SR 141716 was present in the culture medium (Table2), pointing to an involvement of the CB1 cannabinoid receptor in this cannabinoid action. However, neither THC nor HU-210 were able per se to affect basal cAMP levels (Table 2), indicating that factors in addition to modulation of adenylyl cyclase should be involved in the cannabinoid-induced stimulation of glucose utilization.

Dose-dependent inhibition by THC and HU-210 of forskolin-stimulated cAMP accumulation in primary cultures of cortical astrocytes. Cells were exposed to different concentrations of THC (○) or HU-210 (•) for 10 min and incubations were continued for an additional 10-min period in the presence of 20 μmforskolin. Results are expressed as percentage of incubations with forskolin and correspond to four different experiments for each condition.
Effect of THC and other cellular modulators on intracellular cAMP concentration in primary cultures of cortical astrocytes
Effect of THC on MAPK activity, Raf-1 phosphorylation, and Raf-1 intracellular localization.
Different studies have demonstrated an activation of the MAPK cascade in astrocytes in response to growth factors and regulatory peptides that act via G protein-coupled receptors (Bhat, 1995). Likewise, the activity of p42/p44 MAPK was stimulated by THC, and this effect was prevented by SR141716 and pertussis toxin (Table 3). In line with data obtained by Bouaboula et al. (1995a, 1995b) in astrocytoma cells, forskolin per se produced a strikingly strong stimulation of p42/p44 MAPK in astrocytes (Table 3).
Effect of THC and other cellular modulators on p42/p44 MAPK activity in primary cultures of cortical astrocytes
Raf-1 represents a pivotal element in the MAPK cascade (Morrison and Cutler, 1997). It is generally believed that Raf-1 kinase activity is tightly controlled by multisite phosphorylation, although the molecular details underlying this process are as yet unknown (Morrison and Cutler, 1997; Wartmann et al., 1997). As shown in Fig.3A, THC was able to stimulate the phosphorylation of Raf-1 in primary cultures of astrocytes. As previously reported (Wartmann et al., 1997), forskolin also induced Raf-1 phosphorylation (Fig. 3A).
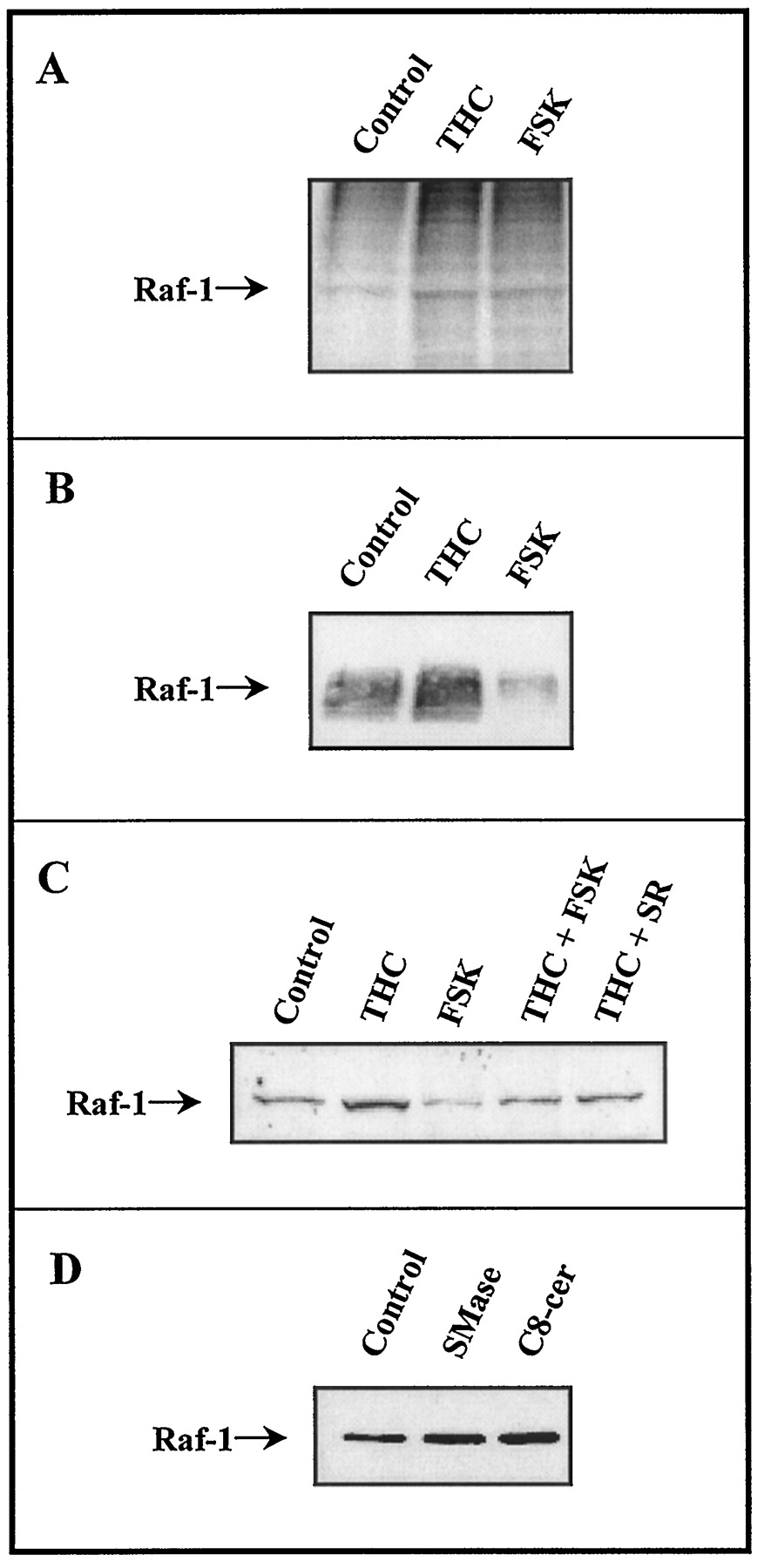
Effect of THC and other cellular modulators on the phosphorylation state of Raf-1 and the recovery of Raf-1 in the particulate fraction in primary cultures of cortical astrocytes. Cells were incubated as described in Tables 2 and 3. One representative experiment is shown in every case. Essentially identical results were obtained in two other experiments of each condition. A, Raf-1 phosphorylation as determined by immunoprecipitation from32P-labeled cells. B, Raf-1 phosphorylation as determined by Western blotting with an anti-phosphotyrosine antibody. C and D, Amount of Raf-1 bound to the particulate fraction as determined by Western blotting with an anti-Raf-1 antibody. FSK, forskolin; SR, SR 141716; SMase, sphingomyelinase; C 8 -cer, C8-ceramide.
It has been reported that depending on the amino acid residues in which it is phosphorylated, Raf-1 may become activated or inhibited (Morrison and Cutler, 1997). It is generally considered that phosphorylation of Raf-1 tyrosine residues produces an activation of Raf-1 protein kinasein vivo (Morrison and Cutler, 1997). Therefore, we determined the effect of THC and forskolin on Raf-1 tyrosine phosphorylation in cultured astrocytes. As shown in Fig. 3B, THC enhanced and forskolin inhibited the phosphorylation of Raf-1 tyrosine residues, indicating that the former activates Raf-1 and the latter inhibits Raf-1. To support this notion, and because it is well established that Raf-1 activation occurs in concert with its translocation from the soluble to the particulate cell fraction (Morrison and Cutler, 1997; Wartmann et al., 1997), the recovery of Raf-1 protein in the particulate fraction of astrocytes was determined. As shown in Fig. 3C, treatment of astrocytes with THC increased the recovery of Raf-1 in the particulate fraction of the cell, and this effect was prevented by SR141716. In contrast, forskolin decreased the recovery of Raf-1 in the membrane fraction and prevented the THC-induced translocation of Raf-1. Data therefore indicate that THC and forskolin seem to have opposite effects on Raf-1 activity, the former activating Raf-1 and the latter inhibiting Raf-1.
Effect of MAPK inhibitors on the THC-induced stimulation of glucose metabolism.
To test whether the MAPK cascade may be involved in the THC-induced stimulation of glucose metabolism in primary astrocytes, experiments with PD 098059 were conducted. PD 098059 is a cell-permeable, specific inhibitor of MAPK kinase which is widely used to demonstrate MAPK-dependent processes in intact cells (Cohen et al., 1997). As shown in Fig. 4, PD 098059 significantly antagonized the THC-induced stimulation of glucose oxidation to CO2 as well as of phospholipid and glycogen syntheses. Interestingly, PD 098059 was unable to prevent the inhibitory effects of forskolin on glucose metabolism in cultured astrocytes (Fig. 4).

Effect of PD 098059, wortmannin, LY 294002, exogenous sphingomyelinase, and C8-ceramide on glucose metabolism in primary cultures of cortical astrocytes. Final concentrations of the different additions were: THC, 100 nm; PD 098059 (PD), 50 μm; wortmannin (WM), 100 nm; LY 294002 (LY), 50 μm; sphingomyelinase (SMase), 0.2 U/ml; C8-ceramide (C 8 -cer), 25 μm; and forskolin (FSK), 20 μm. Results are expressed as percentage of incubations with no additions and correspond to six different experiments for each condition. ∗, Significantly different (p < 0.01) versus incubations with no additions.
The cannabinoid-induced stimulation of the MAPK cascade has been shown to be prevented by wortmannin, an inhibitor of PI3K (Bouaboula et al., 1997). Hence the effect of this compound was also tested on the THC-induced stimulation of glucose metabolism in primary astrocytes. Like PD 098059, wortmannin significantly antagonized the THC-induced stimulation of CO2, phospholipid and glycogen formation from glucose (Fig. 4). The addition of LY 294002 -another PI3K inhibitor- to the astrocyte incubation medium also prevented the effects of THC on glucose metabolism (Fig. 4). Neither PD 098059 nor wortmannin nor LY 294002 added alone had any significant effect on the three metabolic pathways studied (results not shown).
Effect of THC on sphingomyelin hydrolysis and ceramide formation.
Sphingomyelin hydrolysis is a key process in the control of many physiological events related to signal transduction and cellular regulation (Hannun, 1996). Thus, a role of ceramide (the product of sphingomyelin breakdown) in the activation of the MAPK cascade has been suggested (Hannun, 1996). Hence we investigated the possible effect of THC on sphingomyelin hydrolysis. As shown in Fig.5A, the addition of THC to the astrocyte culture medium produced a rapid and significant time-dependent breakdown of cellular sphingomyelin. This was concomitant to a remarkable elevation of intracellular ceramide levels (Fig. 5B). The effect of THC on sphingomyelin breakdown and ceramide generation seemed to rely on a cannabinoid receptor-dependent process because it was prevented by SR141716. Thus, relative values of radioactivity in sphingomyelin were 100 ± 5 for untreated cells, 76 ± 4 for cells treated for 30 min with 100 nm THC (p < 0.01 versus untreated cells), 98 ± 6 for cells treated for 30 min with 1 μm SR141716, and 95 ± 4 for cells pretreated with 1 μm SR141716 for 30 min followed by 30 min with 100 nm THC (n = 4). In addition, relative values of radioactivity in ceramide were 100 ± 7 for untreated cells, 212 ± 21 for cells treated for 10 min with 100 nm THC (p < 0.01 versus untreated cells), 104 ± 18 for cells treated for 30 min with 1 μm SR141716, and 95 ± 38 for cells pretreated with 1 μm SR141716 for 30 min followed by 10 min with 100 nm THC (n = 4).
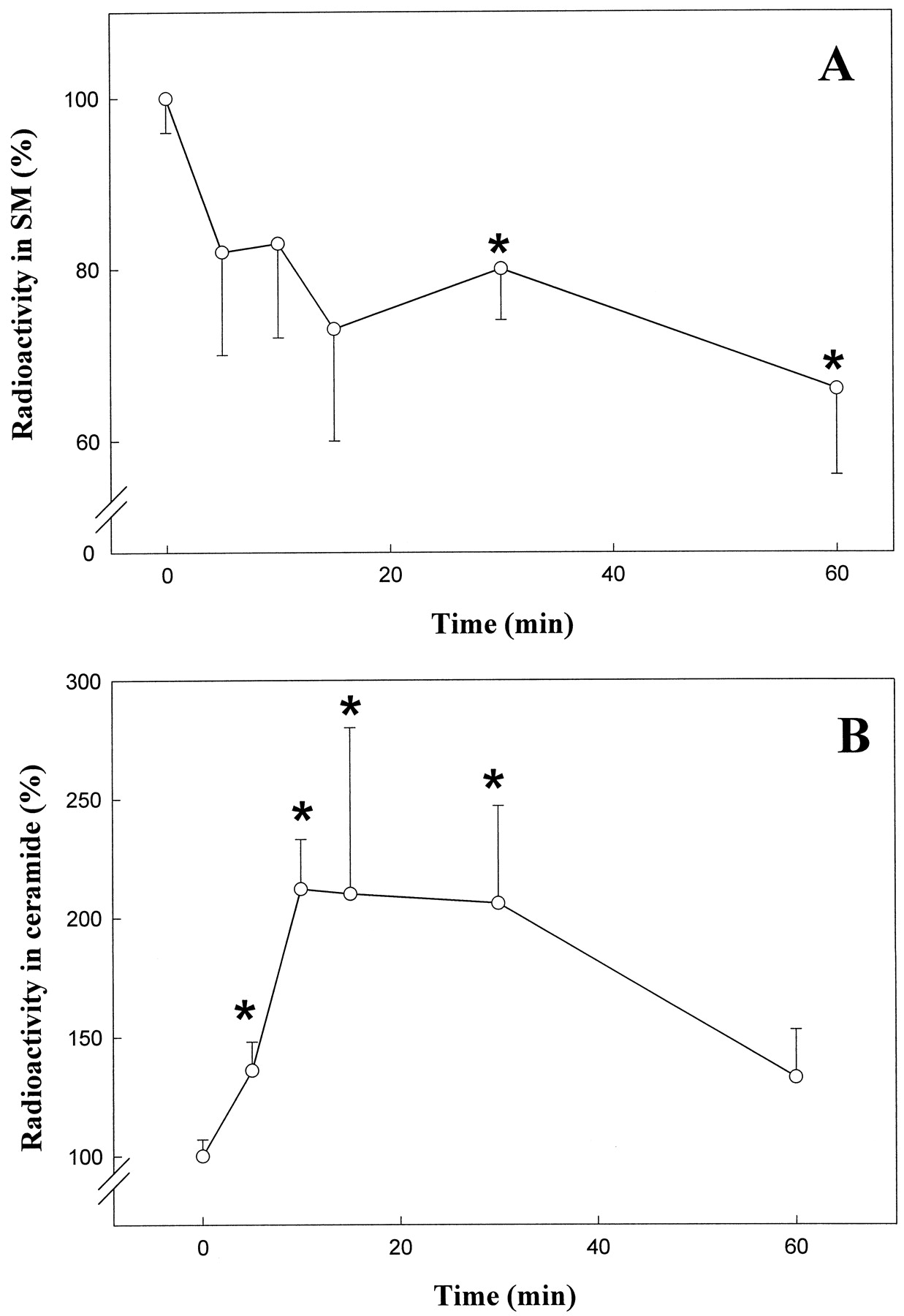
Stimulation of sphingomyelin hydrolysis and ceramide formation by THC in primary cultures of cortical astrocytes. Cells were exposed to 100 nm THC for the indicated times. A, Sphingomyelin (SM) hydrolysis. B, Ceramide formation. Results are expressed as percentage of incubations with no additions and correspond to six (A) or four (B) different experiments. ∗, Significantly different (p < 0.01) versus incubations with no additions.
Effect of C8-ceramide and exogenous sphingomyelinase on MAPK activity, Raf-1 intracellular localization, and glucose metabolism.
Because THC triggers sphingomyelin breakdown and ceramide generation (see above), and ceramide has been shown to activate the MAPK cascade (Hannun, 1996), experiments were conducted subsequently to study whether sphingomyelin breakdown may be linked to the metabolic effects of THC in primary astrocytes. Sphingomyelin breakdown was thus triggered by addition of exogenous neutral sphingomyelinase to the astrocyte incubation medium. In addition, ceramide action was mimicked by addition of C8-ceramide, a cell-permeable ceramide analog. Both exogenous sphingomyelinase and C8-ceramide were able to stimulate MAPK activity (Table 2) and to induce the translocation of Raf-1 to the astrocyte particulate fraction (Fig. 3D). Exogenous sphingomyelinase and C8-ceramide have also been shown to increase Raf-1 phosphorylation in primary cultures of rat astrocytes (Galve-Roperh et al., 1997b). Furthermore, both exogenous sphingomyelinase and C8-ceramide stimulated glucose oxidation to CO2 and glucose incorporation into phospholipids and glycogen in a similar fashion than THC (Fig. 4). Fig. 4 also shows that the metabolic effects of both exogenous sphingomyelinase and C8-ceramide were prevented by PD 098059, indicating that they rely on MAPK activation, and were not additive to those exerted by THC, pointing to a common mechanism of action. All these observations indicate in short that the activation of the sphingomyelin pathway may be linked to the stimulatory effects of THC on MAPK activity and glucose metabolism in cultured astrocytes.
Discussion
Receptor-Dependency of the Effects of THC
Data herein show that in primary astrocytes, THC stimulates glucose metabolism at doses similar to those found in plasma from humans who had smoked marijuana or from laboratory animals that were injected with THC (i.e., up to 0.2–0.3 μm) (Abood and Martin, 1992). In addition, the effects of THC and HU-210 occurred at concentrations ranging the respective binding affinities for cannabinoid receptors in whole-cell systems (Howlett, 1995). All this supports a possible physiological relevance of the metabolic effects of cannabinoids described herein.
On the basis of the antagonism exerted by pertussis toxin and especially SR 141716, the THC-induced metabolic stimulation of astrocytes seems to be a CB1 receptor-mediated process. This is in line with the observation that primary rat astrocytes and human astrocytoma cells express the CB1 receptor mRNA (Bouaboula et al., 1995a). However, these observations must be interpreted with caution, because other investigators have been unable to detect the CB1 receptor mRNA in primary cultures of rat astrocytes (Fernández-Ruiz JJ, Ramos JA, Hernández ML, Garcìa-Gil L, and Barrendero F, unpublished observations). The reason for this discrepancy is not obvious, because similar protocols for mRNA extraction and amplification were used in the two studies. In addition, cannabinoids produce a quantitatively similar stimulation of glucose metabolism in cortical and hippocampal astrocytes, despite the severalfold higher abundance of the CB1 receptor mRNA in the former than in the latter (Casellas P and Bouaboula M, unpublished observations). Furthermore, the possible existence of two different isoforms of the central cannabinoid receptor (i.e., CB1 and CB1A) in rat brain should be kept in mind. Both CB1 and CB1A bind SR141716 and are coupled to activation of the MAPK cascade through a pertussis toxin-sensitive G protein (Rinaldi-Carmonaet al., 1996).
Possible Mechanism of THC Action
In the context of the data presented in this report, several potential mechanisms may be considered to explain the effects of cannabinoids on the MAPK cascade:
Involvement of a Gi/Go protein.
The involvement of a Gi/Goprotein in the cannabinoid-induced stimulation of the MAPK cascade and glucose utilization is supported by the blockade exerted by pertussis toxin (see also Bouaboula et al., 1995b). Cannabinoid receptors are considered to be coupled to Giprotein (Howlett, 1995). Upon occupation of cannabinoid receptors, Gi protein should therefore dissociate into Giα and βγ subunits. Besides Giα’s well known effect on adenylyl cyclase, it may have additional targets (compare Post and Brown, 1996). However, in the context of the cannabinoid-induced stimulation of the MAPK cascade a possible role for Giα is not obvious. More likely, the βγ subunits released from Gi proteins, as suggested by Bouaboula et al. (1995b), may mediate (at least in part) the observed response to THC by acting on a component of the MAPK cascade. The activation of Ras by βγ subunits leads to the binding of Raf-1 to the plasma membrane and the subsequent activation of Raf-1 protein kinase activity (Post and Brown, 1996; Morrison and Cutler, 1997). The observation that THC induces the translocation of Raf-1 to the particulate cell fraction by a SR 141716-sensitive process indicates that the activation of Ras by βγ subunits might play a role in the cannabinoid-induced stimulation of the MAPK cascade.
PI3K: a link between G protein-coupled receptors and Gβγ-mediated MAPK activation?
It is well established that PI3K plays a central role in the control of many events related to cell growth and development as regulated by growth factors such as insulin, platelet-derived growth factor, epidermal growth factor, and interleukin-2 (Toker and Cantley, 1997). Class IA PI3Ks are activated upon occupation of growth factor receptors with tyrosine kinase activity through adaptor subunits containing Src homology domains (Vanhaesebroeck et al., 1997). Interestingly, class IB PI3Ks (e.g., PI3K γ) are stimulated by G protein βγ subunits and do not interact with Src homology-domain-containing adaptors (Vanhaesebroeck et al., 1997). Thus, PI3K has been suggested to be a link between G protein-coupled receptors and Gβγ-mediated MAPK activation (e.g., López-Ilasaca et al., 1997). Although the PI3K isoform pattern expressed by rat astrocytes has not been reported to date, a potential role for PI3K in cannabinoid action in astrocytes might be suggested on the basis of the antagonistic effect of PI3K inhibitors on the cannabinoid-induced stimulation of glucose metabolism (discussed herein) and MAPK activity (Bouaboulaet al., 1997). Nevertheless, we are aware that some of the antagonistic effects of wortmannin have been claimed to be independent of PI3K inhibition, though wortmannin has been repeatedly used as a tool to infer PI3K-dependent processes in intact cells (compare Cohenet al., 1997; Vanhaesebroeck et al., 1997). For this reason we also made use of LY 294002, an inhibitor of PI3K that is much more specific than wortmannin (compare Cohen et al., 1997; Vanhaesebroeck et al., 1997).
The lipid products of PI3K catalytic action mediate the activation of at least two protein kinases (termed PDK1 and PDK2) that in turn activate protein kinase B, which seems to mediate many of the metabolic effects of insulin (Cohen et al., 1997). A similarity exists between cannabinoid- and insulin-triggered metabolic events, because (a) cannabinoids produce similar metabolic effects than insulin (i.e., stimulation of glucose utilization for lipid and glycogen syntheses), (b) both cannabinoids and insulin stimulate the MAPK cascade, and (c) the metabolic effects of both cannabinoids and insulin seem to be dependent on the activation of PI3K (Cohen et al., 1997;Wiesinger et al., 1997). Interestingly, SR 141716 blocks the stimulation of MAPK induced by insulin (Bouaboula et al., 1997).
Paradigm for Raf-1 activation not applicable to cannabinoid receptors.
The effect of cannabinoids on the sphingomyelin cycle, as well as the activation of Raf-1 and MAPK induced by cannabinoids, exogenous sphingomyelinase, and C8-ceramide, suggest a model for Raf-1 activation different from that triggered by growth factor receptors with tyrosine kinase activity. Cannabinoid receptors do not possess tyrosine kinase activity, and therefore the well established paradigm for Raf-1 activation through receptors with tyrosine kinase activity, involving adaptor proteins that contain Src homology domains and the activation of Ras (Post and Brown, 1996;Morrison and Cutler, 1997), does not seem to be applicable to cannabinoid receptors. Thus, as previously shown for the 55 kDa TNF receptor (Zhang et al., 1997), our data indicate that cannabinoid receptors might activate Raf-1 through a ceramide-dependent mechanism. This activation may involve a ceramide-activated protein kinase (Zhang et al., 1997) or the direct binding of ceramide to Raf-1 (Huwiler et al., 1996). We are nevertheless aware that the possible link between Gi protein βγ subunits, PI3K and sphingomyelinase is as yet unknown. Our research is currently focused on the mechanism by which cannabinoid receptors stimulate neutral and/or acid sphingomyelinase.
Is cAMP Involved in the Metabolic Effects of THC?
It is well established that signaling through the CB1 receptor is coupled to the inhibition of adenylyl cyclase through Gi protein (Howlett, 1995). Data presented herein indicate that although cannabinoid receptors in astrocytes may be coupled to adenylyl cyclase inhibition through Giprotein, inhibition of adenylyl cyclase does not seem to be the mechanism underlying the metabolic effects of THC in astrocytes because THC per se was able to stimulate glucose metabolism but was unable to change intracellular cAMP content. Nevertheless, two questions remain to be answered:
How does forskolin inhibit Raf-1 and concomitantly enhance MAPK activity?
This points to a Raf-1-independent activation of MAPK by cAMP. In line with this notion, Vossler et al. (1997) have shown that in PC12 cells cAMP activates MAPK through a pathway that is independent of Ras and Raf-1 but dependent on B-Raf and Rap1. Anyway, cAMP-elevating agents have been shown to stimulate (Bouaboula et al., 1995a, 1995b; data herein) or to inhibit (Kurino et al., 1996; Willis and Nisen, 1996) the MAPK cascade in astrocytes. The reason for this discrepancy is not obvious, but may rely on the fact that Raf-1 exhibits multisite phosphorylation, and depending on both the amino acid positions phosphorylated and the extent of Raf-1 overall phosphorylation either activation or inhibition of Raf-1 kinase activity may ensue (Morrison and Cutler, 1997; Wartmann et al., 1997). In this respect, it is noteworthy that forskolin inhibited the phosphorylation of Raf-1 tyrosine residues, and those phosphorylated sites seem to be linked to Raf-1 activation in vivo (Morrison and Cutler, 1997).
How does forskolin enhance MAPK activity and concomitantly inhibit glucose utilization?
Because MAPK stimulation seems to be linked to the stimulation of glucose utilization induced by, for example, growth factors (Cohen et al., 1997), cytokines (Yu et al., 1995), and cannabinoids (data herein), factors in addition to MAPK stimulation should be responsible for the forskolin-induced inhibition of glucose utilization by astrocytes. The lack of antagonistic effect of PD 098059 on the metabolic effects of forskolin is in line with this notion. It is well known that cAMP-dependent protein kinase directly mediates the inhibition of glucose metabolism in a number of cell types (including astrocytes) by modulating the activity of regulatory enzymes of glycolysis/gluconeogenesis and glycogen and lipid metabolism (compare Cohen et al., 1997;Wiesinger et al., 1997). Thus, our data indicate, in short, that MAPK (and not cAMP) seems to be responsible for the metabolic effects of THC, whereas cAMP (and not MAPK) is very much involved in the metabolic effects of forskolin. The cross-talk between the two pathways could occur at the level of the cAMP-dependent inhibition of Raf-1 (compare Post and Brown, 1996).
Metabolic Considerations
Astrocytes play an active role in the regulation of brain energy metabolism (Fedoroff et al., 1993; Magistretti and Pellerin, 1996). Accruing evidence shows that glucose utilization by astrocytes is sensitive to mediators such as neurotransmitters (e.g., vasoactive intestinal peptide, norepinephrine) (Pellerin et al., 1997;Wiesinger et al., 1997) and cytokines (e.g. TNFα, interleukin-1α) (Yu et al., 1995). Our data indicate that cannabinoids may alter brain glucose metabolism by stimulating glucose utilization by astroglial cells. As a matter of fact, low doses of THC (0.2 mg/kg body weight) have been shown to stimulate glucose uptake by rat brain cortical and limbic areas in vivo (Megulies and Hammer, 1991). A stimulation of glucose metabolism induced by a single dose of THC has also been described in the brain cortex of chronic marijuana smokers (Volkow et al., 1996). The enzymatic steps potentially affected by cannabinoids and responsible for the metabolic effects described in this study have not been identified. However, the observation that the three metabolic pathways studied are quantitatively affected in a similar fashion may indicate either that the primary site of cannabinoid action is one of the first steps of glucose metabolism (e.g. glucose uptake, glucose phosphorylation) or that the control of metabolic pathways exerted by cannabinoids occurs at multiple sites. In the latter case, glycogen synthase, the key regulatory enzyme of glycogen synthesis, might be an example of a potential target for cannabinoid action. Activation of glycogen synthase underlies the stimulation of glycogen synthesis induced by insulin (Cohen et al., 1997). This process, like the THC-induced stimulation of glycogen synthesis, is sensitive to wortmannin and relies on the PI3K-dependent stimulation of protein kinase B, which phosphorylates and inactivates glycogen synthase kinase 3, thereby leading to a de-inhibition of glycogen synthase (Cohenet al., 1997).
In conclusion, the present report indicates that activation of sphingomyelin breakdown and the MAPK cascade by cannabinoids leads to remarkable metabolic changes in primary astrocytes. Although the physiological consequences of these changes are not obvious, as far as we know this is the first time in which a connection between stimulation of a signal transduction system and a cellular response is established for the action of cannabinoids on astroglial cells. Anyway, it is clear that further characterization of the cellular localization and signaling properties of brain cannabinoid receptors is required to understand the role of cannabinoid receptors in the modulation of astroglial functions.
Acknowledgments
We are indebted to Mr. Andrés Daza for expert technical assistance; to Dr. Thierry Levade for kind advice in the determination of sphingomyelin and ceramide levels; and to Dr. J. J. Fernández-Ruiz and Dr. P. Casellas for providing access to unpublished data.
Footnotes
- Received March 9, 1998.
- Accepted August 20, 1998.
-
Send reprint requests to: Dr. Manuel Guzmán, Department of Biochemistry and Molecular Biology I, School of Biology, Complutense University, 28040-Madrid, Spain. E-mail:mgp{at}solea.quim.ucm.es
-
This study was supported by grants from Comisión Interministerial de Ciencia y Tecnologı́a (SAF 96/0113), Fondo de Investigación Sanitaria (FIS 97/0039) and Comunidad Autónoma de Madrid (CAM-6648).
Abbreviations
- MAPK
- mitogen-activated protein kinase
- C8-ceramide
- d-erythro-N-octanoylsphingosine
- PI3K
- phosphoinositide 3-kinase
- THC
- Δ9-tetrahydrocannabinol
- FCS
- fetal calf serum
- DMEM
- Dulbecco’s modified Eagle’s medium
- PMSF
- phenylmethylsulfonyl fluoride
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}