


Abstract
The stress-activated protein kinase (SAPK) and mitogen-activated protein kinase (MAPK) cascades mediate cytotoxic and cytoprotective functions, respectively, in the regulation of leukemic cell survival. Involvement of these signaling systems in the cytotoxicity of 1-β-d-arabinofuranosylcytosine (ara-C) and modulation of ara-C lethality by protein kinase C PKC inhibition/down-regulation was examined in HL-60 promyelocytic leukemia cells. Exposure to ara-C (10 μm) for 6 hr promoted extensive apoptotic DNA damage and cell death, as well as activation of PKC. This response was accompanied by downstream activation of the SAPK and MAPK cascades. PKC-dependent MAPK activity seemed to limit ara-C action in that the toxicity of ara-C was enhanced by pharmacological reductions of PKC, MAPK, or both. Thus, ara-C action was (1) partially attenuated by diradylglycerols, which stimulated PKC and MAPK, but (2) dramatically amplified by sphingoid bases, which inhibited PKC and MAPK. The cytotoxicity of ara-C also was substantially increased by pharmacological reductions of PKC, including down-regulation of PKC by chronic preexposure to the macrocyclic lactone bryostatin 1 or inhibition of PKC by acute coexposure to the dihydrosphingosine analog safingol. Significantly, both of these manipulations prevented activation of MAPK by ara-C. Moreover, acute disruption of the MAPK module by AMF, a selective inhibitor of MEK1, suppressed both basal and drug-stimulated MAPK activity and sharply increased the cytotoxicity of ara-C, suggesting the direct involvement of MAPK as a downstream antiapoptotic effector for PKC. None of these chemopotentiating agents enhanced ara-CTP formation. Ceramide-driven SAPK activity did not seem to mediate drug-induced apoptosis, given that (1) neutralization of endogenous tumor necrosis factor-α with monoclonal antibodies or soluble tumor necrosis factor receptor substantially reduced ceramide generation and SAPK activation by ara-C, whereas the induction of apoptosis was unaffected; (2) pharmacological inhibition of sphingomyelinase by 3-O-methoxysphingomyelin reduced ceramide generation and SAPK activation without limiting the drug’s cytotoxicity; and (3) potentiation of ara-C action by bryostatin 1 or safingol was not associated with further stimulation of SAPK. These observations collectively suggest a primary role for decreased MAPK, rather than increased SAPK, in the potentiation of ara-C cytotoxicity by interference with PKC-dependent signaling.
Multiple lipid messengers contribute to the physiological regulation of leukemic cell survival. We recently reported that the cytotoxic lipid messenger ceramide initiates apoptosis in myeloid leukemia cells through coordinate regulation of the SAPK and MAPK cascades (Jarvis et al., 1997). Lethal signaling by ceramide entails simultaneous recruitment of SAPK activity (Verheij et al., 1996) and suppression of MAPK activity (Jarvis et al., 1997), thereby redirecting the net balance between the two systems (i.e., away from cytoprotective and toward cytotoxic signaling). Moreover, ceramide action is subject to transmodulation by other lipid messengers that regulate conventional and novel isoforms of PKC (cPKC, nPKC), inasmuch as the proapoptotic actions of ceramide are attenuated by diglyceride (Jarvis et al., 1994a, 1994b) and amplified by sphingosine (Jarvis et al., 1996). The divergent influences of these lipids over ceramide action are mediated through reciprocal modulation of one or more cPKC/nPKC-dependent signaling elements that antagonize apoptosis. Given recent evidence of a critical involvement of MAPK in cell survival (Xia et al., 1995), the MAPK cascade represents a plausible downstream effector for the antiapoptotic effects of PKC. Apart from an antiapoptotic role in leukemic cell survival, it is conceivable that MAPK has a significant impact on the lethal actions of genotoxic stressors such as antineoplastic agents. Validation of this hypothesis could have important implications for the design of novel chemomodulatory paradigms.
The antileukemic actions of the deoxycytidine analog ara-C are well characterized (reviewed in Grant, 1997). The ara-C-related cytotoxicity arises from its conversion to the lethal derivative ara-CTP and incorporation of this metabolite into template-specific sites within elongating DNA strands, thereby interfering with normal DNA synthesis. ara-CTP incorporation subsequently leads to extensive DNA fragmentation and endonucleolytic chromatinolysis associated with apoptotic cell death. In addition to effects on DNA synthesis, ara-C engages an array of signaling elements in myeloid leukemia cells, including generation of the lipid messengers diglyceride (Kucera and Capizzi, 1992) and ceramide (Strum et al., 1994), activation of cPKC (Kharbandaet al., 1991) and the MAPK (Kharbanda et al., 1994) and SAPK (Saleem et al., 1995) cascades, and up-regulation of various transregulatory factors (e.g., activator protein-1, nuclear factor-κB) (Kharbanda et al., 1990;Brach et al., 1992a, 1992b)). Although the functional contributions of these systems to the cytotoxicity of ara-C remain to be established, simultaneous recruitment of two distinct pathways could plausibly participate in ara-C action: (1) ceramide-dependent activation of the SAPK cascade (possibly via ceramide-activated protein kinase) and (2) diglyceride-dependent activation of the MAPK cascade (via cPKC/nPKC). ara-C action may therefore depend on the extent to which the proapoptotic effect of SAPK is engaged relative to the antiapoptotic influence of MAPK.
We previously demonstrated that ara-C action is substantially augmented by pharmacological reductions in PKC activity, through either (1) down-regulation after chronic exposure to the macrocyclic lactone bryostatin 1 (Jarvis et al., 1994c) or (2) inhibition on acute exposure to the fungal metabolite staurosporine (Grant et al., 1994). We also examined the effects of the nonphysiological sphingoid base analog l-threo-dihydrosphingosine (SPC-100270; safingol) (Kedderis et al., 1995) based on evidence of a similar potentiation of mitomycin C cytotoxicity by this agent in gastric carcinoma cells (Schwartz et al., 1995). It is presently unknown whether the influence of PKC-directed chemopotentiation is manifested at the level of downstream signaling elements such as the MAPK and SAPK cascades. To address this question directly, we monitored MAPK and SAPK activity in HL-60 human promyelocytic leukemia cells accompanying potentiation of ara-C-related apoptosis by bryostatin 1 and safingol. In addition, we compared those responses with the effects of the selective MEK1 inhibitor AMF (Alessiet al., 1995; Dudley et al., 1995). Our results demonstrate that potentiation of ara-C lethality by inhibition or down-regulation of PKC correlates closely with suppression of the MAPK cascade, rather than recruitment of the SAPK cascade.
Experimental Procedures
Drugs and reagents.
Crystalline preparations of ara-C (Sigma Chemical, St. Louis, MO) were stored desiccated at 4° and dissolved in sterile PBS immediately before use. Synthetic preparations of diglyceride (1,2-sn-dioctanoylglycerol; 2,3-sn-dioctanoylglycerol; 1,3-sn-dioctanoylglycerol) were obtained from Sigma Chemical. Synthetic preparations of sphingosine (d-erythro-sphingosine,l-erythro-sphingosine,l-threo-sphingosine, andd-threo-sphingosine) were obtained from BIOMOL Research Laboratories (Plymouth Meeting, PA) or were synthesized and purified as described previously (Jarvis et al., 1996). All lipids were initially dissolved in 100% ethanol and stored at −70°; for experimental use, glycerolipids were used directly as organic stocks, whereas sphingolipids were used complexed with delipidated albumin. Lipid preparations were prewarmed to 37° and added directly to complete medium as indicated below. Bacterial phospholipase C (type XI from B. cereus; Sigma) in a vehicle of 3.25m(NH4)2SO4, pH 6.0, was stored at 4°. Both bryostatin 1 (provided by Dr. A. J. Murgo, Cancer Treatment Evaluation Program, National Cancer Institute) and UCN-01 (provided by Dr. E. A. Sausville, Developmental Therapeutics Program, National Cancer Institute) were dissolved in dimethylsulfoxide and delivered as concentrated organic stocks. Safingol (SPC-100270; obtained variously from Sphinx (Durham, NC) Pharmaceuticals (Darmstadt, Germany) or Calbiochem (San Diego, CA) was prepared and delivered as noted above for sphingoid bases. AMF (Calbiochem) was dissolved in dimethylsulfoxide and delivered directly to complete medium. Lyophilized preparations of rhTNFα, rhsTNF-RI, and murine anti-hTNFα mAbs were obtained from R and D Systems (Minneapolis, MN); rhsTNF-RI, anti-hTNFα, and rhTNFα mAbs were dissolved in sterile PBS (138 mm NaCl, 3 mm KCl, 10 mm Na2PO4, 2 mm NaH2PO4) containing 0.2% bovine serum albumin. The neutral/acidic sphingomyelinase inhibitor 3-O-methoxy-N-octanoyl-sphingosyl-1-phosphorylcholine (methoxysphingomyelin, d-erythro isomer) was synthesized as described previously (Lister et al., 1995); the ceramide synthase inhibitor fumonisin B1 was obtained from Sigma Chemical. Both methoxysphingomyelin and fumonisin B1 were dissolved in 100% methanol and diluted in serum-free medium. All test reagents were presented at final concentrations in complete medium at 37°; the vehicles used were without effect in HL-60 cells.
Cell culture.
The human promyelocytic leukemia cell line was grown in complete RPMI-1640 medium (phenol red-free formulation, supplemented with 1.0% sodium pyruvate, nonessential amino acids,l-glutamine, penicillin, and streptomycin; all from Life Technologies) and 10% heat-inactivated fetal bovine serum (Hyclone). HL-60 cell cultures were passed twice weekly and exhibited a doubling time of ∼24 hr. Cultures were maintained under a humidified atmosphere of 95% room air, 5% CO2, at 37°. Cell densities were determined with a Coulter Products (Buffalo, NY) counter, and basal cell viability was assessed by vital dye exclusion.
Test exposures.
All experimental incubations were performed as described previously (Jarvis et al., 1994a). Cells in log-phase growth were pelleted, rinsed twice in complete medium, resuspended at a density of 4 × 105cells/ml, and maintained as indicated above. Cells were exposed to test agents for appropriate intervals in complete medium; loss of cells under these conditions caused by either washing or cell adherence was negligible (≤5%). Test incubations were terminated with gentle pelleting of the cells by centrifugation at 400 × gfor 10 min at 4°; in some instances, aliquots of the medium were retained for direct assay of released DNA. After the determination of cell density, the cells were pelleted and prepared as outlined below for agarose gel electrophoresis; spectrofluorophotometric assays of DNA damage; assay of clonogenicity; SAPK, MAPK, and PKC activities; quantification of cellular ceramide levels; or examination of cellular morphology.
Quantitative analyses of DNA damage.
The formation and release of DNA fragments and the corresponding breakage of bulk DNA were assessed by bisbenzimide spectrofluorophotometry as described previously (Jarvis et al., 1997). To measure DNA fragments, pelleted cells (4 × 106 cells/pellet) and medium aliquots were mixed with 5 mm Tris·HCl, 30 mm EGTA, 30 mm EDTA, and 0.1% Triton X-100, pH 8.0. Lysate and medium preparations were centrifuged at 30,000 ×g at 4° for 40 min; nonsedimenting DNA fragments in the extracts were quantified by spectrofluorophotometry in the presence of Hoechst-33258 (1 μg/ml; λex = 365, λem = 460). Values are expressed as ng/μg DNA recovered or released from 106 cells. To measure corresponding loss of integrity of bulk DNA, pelleted cells (8.25 × 106 cells/pellet were resuspended in cold PBS and subjected to timed alkaline denaturation in 0.1 n NaOH; denaturation was terminated by neutralization in 0.1 n HCl. Cells then were lysed by the addition of 200 mmK2HPO4, 50 mmEDTA, and 0.16% N-lauroylsarcosine. Bulk DNA breakage was quantified by spectrofluorophotometry in the presence of Hoechst-33258 (λex = 350, λem = 450). Values are expressed as rad-equivalents.
Determination of clonogenicity.
Pelleted cells were rinsed extensively and prepared for soft-agar cloning as described previously (Jarvis et al., 1994c). Cells were resuspended in cold PBS and seeded onto 35-mm culture plates at a fixed density (400 cells/ml/well) in complete RPMI-1640 medium containing 20% fetal calf serum, 10% 5637-CM, and 0.3% Bacto agar. Cultures were maintained for 10–12 days before the formation of colonies (defined as groups of ≥50 cells) was scored.
Cytological characterization of apoptosis.
Pelleted cells were resuspended in PBS and fixed in cytocentrifuge preparations according to established procedures (Jarvis et al., 1997). For visualization of apoptotic morphological alterations, fixed cells were stained with 20% Wright-Giemsa stain. At least five 100-cell fields were scored for each treatment by conventional light microscopy by assessing the expression of cytoarchitectural characteristics of apoptosis (i.e., condensed nucleoplasm and cytoplasm, formation of membrane blebs, karyolytic degeneration of the nucleus into apoptotic bodies, and overall cell shrinkage). For visualization of apoptotic DNA damage, fixed cells were sequentially (1) treated with ethanol-acetic acid (2:1, v/v) at 20° for 5 min, (2) stained for broken DNA by treatment with terminal deoxynucleotidyl transferase in the presence of fluorescein isothiocyanate-dUTP; Molecular Probes, Eugene, OR) at 37° for 60 min, and (3) counterstained for intact DNA with 0.01% propidium iodide in sodium citrate at 20° for 10 min. At least three 100-cell fields were scored for each treatment by fluorescent microscopy by assessing increased direct fluorescence of end-labeled double-stranded DNA.
Assessment of ara-CTP metabolism.
Pelleted cells were rinsed in cold PBS, repelleted, and then lysed in 0.6 ntrichloroacetic acid. Pyrimidine nucleotide extracts were then prepared as previously explained in detail (Jarvis et al., 1994c). Levels of ara-CTP were separated by high pressure liquid chromatography; values are expressed as pmol of ara-CTP present in 1.5 × 106 cells.
Determination of cPKC/nPKC activity.
Pelleted cells were rinsed in PBS, repelleted, and homogenized in 20 mmTris·HCl, 500 μm EDTA, and 500 μm EGTA, pH 7.5, containing protease inhibitors (40 μg/ml aprotinin, 15 μg/ml leupeptin). After partial purification of homogenates over DEAE-cellulose, particulate (i.e., membrane-associated) and soluble (i.e., cytosolic) enzyme fractions were separated by ultracentrifugation at 100,000 × g at 4° for 2 hr. Membrane fractions were added to reaction mixtures containing lysis buffer and mixed micelles of phosphatidylserine and synthetic dioleoylglycerol (10 μm). Particulate activity was assayed using synthetic acetylated myelin basic proteinN-terminal peptide AcMBP4–14 as described previously (Grant et al., 1996). Reactions were initiated by the addition of 25 μCi of [γ-32P]ATP and 20 μmnonisotopic ATP and allowed to proceed for 5 min at 30°. Reactions were terminated by transfer to nitrocellulose filters and immersion in cold orthophosphoric acid (1% v/v). Filters were rinsed sequentially in orthophosphoric acid and PBS, and radioactivity was determined by liquid scintillation counting. Because this in vitro assay monitors only diglyceride-driven PKC activity, only the activity conventional and novel isoforms are directly measured; the contribution of diglyceride-insensitive (i.e., atypical) species is subtracted on correction for nonspecific and background activity. Therefore, data derived form this assay system are reported throughout the text as cPKC/nPKC activity.
Determination of MAPK and SAPK activities.
Pelleted cells were rinsed in PBS, repelleted, and flash-frozen. Cell pellets were lysed in 25 mm HEPES, pH 7.4, containing 5 mmEGTA, and 5 mm EDTA, supplemented with protease inhibitors (5 mm benzamidine, 1 mm phenylmethylsulfonyl fluoride, 1 mg/ml soybean trypsin inhibitor, 40 μg/ml pepstatin, 40 μg/ml chymotrypsinogen, 40 μg/ml E64, 40 μg/ml aprotinin, 1 μm microcystin LR), phosphatase inhibitors (0.5 mm trisodium orthovanadate, 0.5 mm tetrasodium pyrophosphate), and containing 0.05% sodium deoxycholate (w/v), 1% Triton X-100 (v/v), and 0.1% 2-mercaptoethanol (v/v). Lysates were clarified by centrifugation at 5000 × g at 4° for 5 min. MAPK/SAPK was immunoprecipitated from clarified lysates with Protein A/agarose-conjugated antibody/antisera, and activities were determined as described previously (Jarvis et al., 1997). MAPK activity was assayed after immunoprecipitation of p42-ERK1/p44-ERK2 using MBP as substrate; alternatively, SAPK activities were assayed after immunoprecipitation of p54-JNK1/p46-JNK2 using GST-c-Jun 1–169 as substrate. Preimmune controls were also run to ensure selectivity of substrate phosphorylation. Reaction mixtures consisted of immunoprecipitated enzyme, substrate, and [γ-32P]ATP (5000 Ci/pmol) in 25 mm HEPES, pH 7.4, containing 15 mmMgCl2, 100 mm trisodium orthovanadate, 0.01% (v/v) 2-mercaptoethanol, and 1 μmmicrocystin LR. Reactions were initiated by the addition of substrate. MAPK reactions were terminated by transfer to p81 filter paper; filters were rinsed repeatedly in 185 mm orthophosphoric acid and then dehydrated in acetone. SAPK reactions were terminated by transfer to 10% polyacrylamide gels; phosphorylated products were resolved by electrophoresis, and appropriate substrate bands were excised. Total radioactivity in gels/filters was determined by liquid scintillation counting.
Quantification of cellular ceramide levels.
Pelleted cells were resuspended in 1× PBS containing 100 mm EGTA/EDTA and placed on ice. Cellular lipids were extracted by the addition of CHCl3-MeOH-HCl (100:100:1, v/v/v), and the organic phase was repartitioned by the addition of CHCl3-MeOH-H2O (1:1:1, v/v/v). Glycerolipids were removed by mild alkaline hydrolysis in methanolic KOH followed by reextraction and repartitioning. Final lipid extracts were assayed for ceramide content through phosphorylation by recombinant bacterial diglyceride kinase as described previously (Jarvis et al., 1994a). Assay products were recovered by reextraction and repartitioning as before. The organic phase was dried under gN2, and the residue was dissolved in CHCl3-MeOH (2:1, v/v). Ceramide phosphate in final extracts was resolved by silane high performance thin layer chromatography with development (kieselguhr-G-hp; EM Science, Darmstadt, Germany) in CHCl3-MeOH-HAc (65:15;5, v/v/v); authentic ceramide standards (type III ceramide; Sigma Chemical) were run in parallel. Radiolabeled chromatographic bands in sample and standard lanes were visualized by radioautography and recovered from the silane adsorbent. Radioactivity in each sample was determined by conventional liquid scintillation counting. Values are expressed as pmol of ceramide/106 cells.
Results
Activation of cPKC/nPKC and the SAPK and MAPK cascades by ara-C.
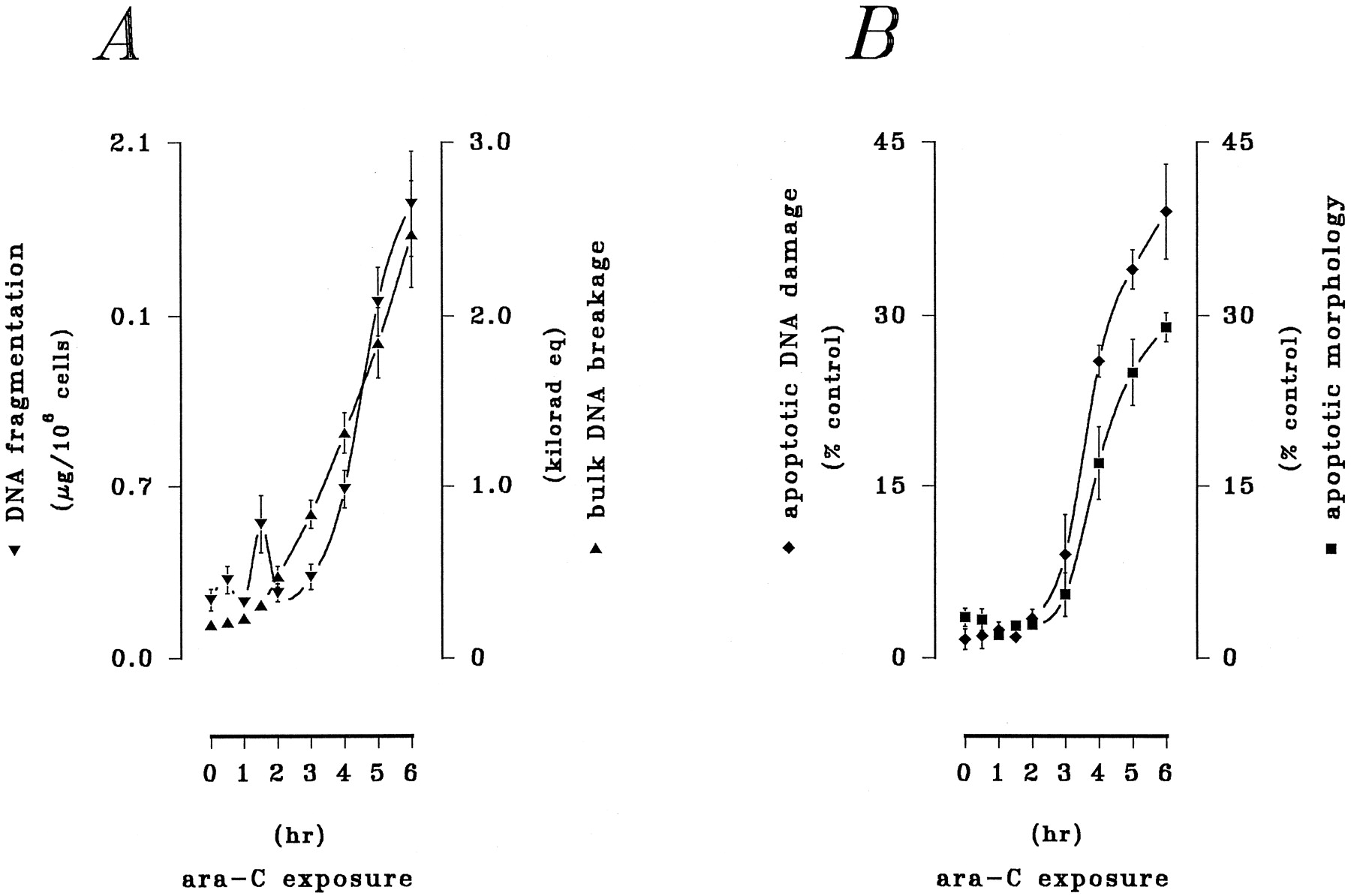
Although exposure to ara-C elicits activation of PKC (Kharbanda et al., 1991), as well as the SAPK (Saleemet al., 1995) and MAPK (Kharbanda et al., 1994) cascades, a simultaneous assessment of these signaling elements in chemomodulation of ara-C action is currently lacking. The current results therefore characterized the contribution of these signaling processes to ara-C-mediated lethality in HL-60 cells. In initial trials, activation of cPKC/nPKC and coordinates changes in SAPK and MAPK activities were examined in response to ara-C in detailed time course studies. Exposure of HL-60 cells to ara-C (10 μm) for 1–6 hr elicited time-dependent increases in the activities of SAPK, MAPK, and cPKC/nPKC that were coincident with the induction of apoptosis (Figs. 1 and2). Bisbenzimide spectrofluorophotometry demonstrated time-dependent degradation of DNA reflected by accumulation of small double-stranded DNA fragments and corresponding double-stranded breakage of bulk DNA (Fig. 1A); significant DNA degradation was detected within 2–3 hr and continued throughout the exposure interval. Examination of apoptotic cells by fluorescent and light microscopy revealed the progressive occurrence of DNA damage and cell death within the population of treated cells (Fig. 1B). In vitro kinase assays demonstrated sequential, but closely coupled, stimulations of cPKC/nPKC and of SAPK and MAPK (Fig. 2). ara-C potently stimulated cPKC/nPKC activity, which increased within 1 hr and remained at a stable, maximum level after 3 hr (Fig. 2A). In addition, ara-C promoted (1) rapid, but transient, activation of SAPK (JNK1/JNK2), which peaked within 2 hr and then subsided to basal levels within the subsequent 1 hr, and (2) slower, but sustained, activation of MAPK (ERK1/ERK2), which peaked within 4 hr and remained elevated throughout the exposure interval (Fig. 2A). The profile of MAPK activation followed that of PKC, with a latency of ≤1 hr.

Time course of ara-C-mediated cytotoxicity. HL-60 cells were exposed to ara-C (10 μm) for 0–6 hr. A, Apoptotic DNA damage. Spectrofluorophotometric analyses for the total accumulation of double-stranded DNA fragments (▾) or corresponding double-stranded breakage of bulk DNA (▴) were performed as described in Experimental Procedures. Values reflect the mean ± standard error of triplicate determinations and are expressed as a percentage of untreated controls. Data shown are from a representative study performed six times with comparable results. B, Apoptotic cell death. Cells in fixed preparations were either (1) stained with fluorescein isothiocyanate-dUTP in the presence of terminal deoxynucleotidyl transferase and examined by fluorescent microscopy to identify apoptotic cells by DNA damage (♦) or (2) stained with modified Wright-Giemsa and examined by conventional light microscopy to identify apoptotic cells by cytoarchitectural traits (▪), as explained in Experimental Procedures. For each assay, values reflect the mean ± standard error of apoptotic cells noted on three fields of 100 or more cells and are expressed as a percentage of untreated controls. Results are from a representative study performed three times with comparable outcomes.

Activation of PKC and the SAPK and MAPK cascades by ara-C. HL-60 cells were exposed to ara-C (10 μm) for 0–6 hr as before. Activities of cPKC/nPKC and of both SAPK and MAPK were determined by in vitro assays as described in Experimental Procedures. A, Particulate (i.e., membrane-associated) cPKC/nPKC (♦) activity. B, SAPK (p46-JNK1/p54-JNK2; ▾) and MAPK (p42-ERK1/p44-ERK2; ▴) activities. In each case, values reflect the mean ± standard error of duplicate determinations and are expressed as a percentage of basal activity detected in untreated controls. Data shown are from a representative study performed three times with comparable results.
Effects of cPKC/nPKC modulation on ara-C action and modulation of MAPK/SAPK.
To assess the susceptibility of ara-C action to the influences of lipid messengers and potential consequences for MAPK and SAPK activation, synthetic diradylglycerols and sphingoid bases were evaluated with respect to their capacity to alter ara-C-related apoptosis and modulate MAPK/SAPK activity (Fig.3). Brief (30-min) exposure to diglyceride (2.5 μm) alone sharply increased activities of cPKC/nPKC (by 370%) and ERK1/ERK2 (by 220%), whereas the activity of JNK1/JNK2 was not significantly modified. Parallel exposure to sphingosine (750 nm) alone decreased cPKC/nPKC (by ≥90%) and ERK1/ERK2 (by 57%) but moderately increased JNK1/JNK2 (by 44%). Modulation of these kinase activities by synthetic lipids was closely associated with reciprocal alterations in the genotoxic potential of ara-C, as shown in Figs. 4 and5. First, exposure to diglyceride (10 μm) alone for 6 hr failed to promote DNA damage but attenuated ara-C-related accumulation of DNA fragments by 64% and breakage of bulk DNA by 61% (Fig. 4A). Second, exposure to sphingosine (750 nm) alone for 6 hr failed to promote DNA damage and cell death but markedly augmented ara-C-related accumulation of DNA fragments by 190% and bulk DNA breakage by 215% (Fig. 5A). DNA damage was closely associated with both induction of apoptosis and loss of clonogenic potential. ara-C (10 μm) triggered apoptosis in 33% of the treated cell population and decreased clonogenic survival commensurately (i.e., by 48%). Diglyceride moderately limited drug-induced apoptosis (to only 19%) and partially restored the potential for clonogenic growth (to 68%) (Fig. 4B), whereas sphingosine potently augmented drug-induced apoptosis (to 94%) and diminished clonogenicity (to ≤5%) in parallel exposures (Fig. 5B). Cell survival was unaffected by a 6-hr exposure to either lipid alone (not shown).

Acute PKC and SAPK/MAPK responses to diglyceride and sphingosine. HL-60 cells were exposed to synthetic preparations of diglyceride (2.5 μm) and sphingosine (750 nm) for 45 min. Relative activities of cPKC/nPKC the SAPK and MAPK cascades were then determined by in vitro assays as described in Experimental Procedures. A, Particulate cPKC/nPKC (■) activity. B, SAPK (p46-JNK1/p54-JNK2; ▨) and MAPK (p42-ERK1/p44-ERK2; ▩) activities. In each case, values reflect the mean ± standard error of duplicate determinations and are expressed as a percentage of basal activity detected in untreated controls. Data shown are from a representative study performed four times with comparable results.

Attenuation of ara-C-related apoptosis by diglyceride. HL-60 cells were exposed to ara-C (10 μm) in the absence or presence of synthetic 1,2-sn-dioctanoylglycerol (10 μm) for 6 hr. Apoptotic DNA damage and cell death were assessed as before. A, Accumulation of double-stranded DNA fragments (▨) and occurrence of double-stranded breakage of bulk DNA (▩) as described; for both measurements, values represent the mean ± standard error of quadruplicate determinations. B, Induction of apoptosis (▪) and suppression of clonogenicity (▥). All values represent the mean ± standard error of triplicate determinations. Data shown are from a representative study performed four times with comparable results.

Amplification of ara-C-related apoptosis by sphingosine. HL-60 cells were exposed to ara-C (10 μm) in the absence or presence of syntheticd-erythro-sphingosine (850 nm) for 6 hr. Apoptotic DNA damage and cell death were assessed as described in Experimental Procedures. A, Total accumulation of double-stranded DNA fragments (▨) and occurrence of double-stranded breakage of bulk DNA (▩) as before; for both measurements, values represent the mean ± standard error of quadruplicate determinations. B, Induction of apoptosis (▪) and suppression of clonogenicity (▥). All values represent the mean ± standard error of triplicate determinations. Data shown are from a representative study performed four times with comparable results.
Steric aspects of diglyceride and sphingosine action.
In related studies, steric aspects of the chemomodulatory properties of these lipids indicated that the reciprocal actions of diglyceride and sphingosine on ara-C-related cytotoxicity were mediated proximally through cPKC/nPKC. First, attenuation of ara-C-related apoptosis by diglyceride was exclusively associated with the 1,2-sn-substituted isomer, whereas the 2,3-sn-substituted and 1,3-rac-substituted species were ineffective (Table 1), consistent with the established selectivity of this species for activation of cPKC/nPKC (Nomura et al., 1986; Go et al., 1987). Second, the d and l forms oferythro and threo enantiomers of sphingosine comparably potentiated ara-C-induced apoptosis (Table2), consistent with the documented lack of stereoselectivity of sphingoid bases in inhibition of cPKC/nPKC (Merrill et al., 1989). Moreover, sphingosine and dihydrosphingosine were equivalent in their capacity to potentiate ara-C lethality (not shown), further supporting involvement of cPKC/nPKC (Merrill et al., 1989). Acute modulation of ara-C by lipid messengers thus seemed to be directly related to alterations in cPKC/nPKC activity and presumably in the status of a critical downstream cytoprotective system.
Steric aspects of diglyceride action
Steric aspects of sphingosine action
MAPK in ara-C action.
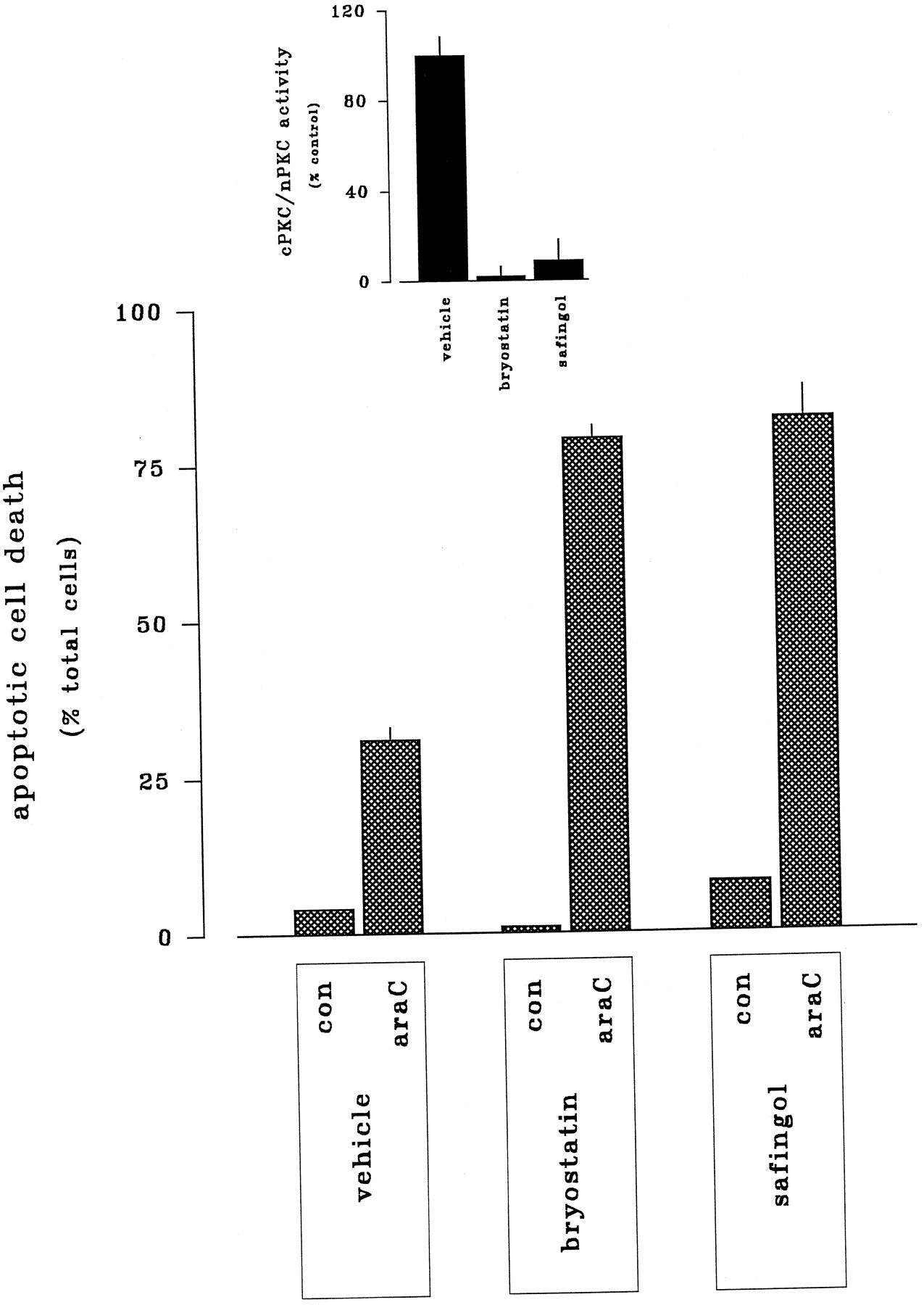
The preceding observations indicated that alteration of ara-C-mediated lethality by diradylglycerols and sphingoid bases involved selective targeting of conventional and novel isoforms of PKC, a phenomenon similar to the transmodulation of ceramide-mediated cell death that we described previously (Jarviset al., 1994a, 1996). This in turn raised the possibility that analogous pharmacological reductions in net PKC activity could enhance the toxicity of ara-C. As shown in Fig.6, the cytotoxic capacity of ara-C, manifested by the induction of apoptotic cell death or the suppression of clonogenic survival, was comparably enhanced (i.e., by ∼65%) through (1) preexposure to bryostatin 1 (10 nm) or (2) coexposure to safingol (825 nm). Both of these responses were associated with reductions in assayable cPKC/nPKC activity. Thus, membrane-associated cPKC/nPKC activity was decreased ≥96% by bryostatin 1 and ≥72% by safingol (Fig. 6, inset), demonstrating that active, lipid-driven cPKC/nPKC activity was substantially reduced by both treatments. Furthermore, Western analyses revealed that expression of cPKCα (the dominant PKC isoform expressed in HL-60 cells) was virtually absent in bryostatin-treated cells but intact in safingol-treated cells (not shown). Taken together, these observations indicated that the effects of bryostatin 1 and safingol derived from down-regulation and inhibition of cPKC/nPKC, respectively.

Potentiation of ara-C-induced apoptosis by bryostatin 1 and safingol. HL-60 cells were incubated for 6 hr in the absence or presence of ara-C (10 μm) in conjunction with either (1) 24-hr pretreatment bryostatin 1 (10 nm) or (2) 6-hr cotreatment safingol (1 μm). Apoptotic cell death was assessed by fluorescent microscopy as before. Inset, HL-60 cells were exposed either to bryostatin 1 (10 nm) for 24 hr or to safingol (1 μm) for 6 hr; changes in basal cPKC/nPKC activity (defined as membrane-associated, phorboid-sensitive activity) were determined by in vitro as described in Experimental Procedures.
Parallel trials sought to establish the downstream importance of the MAPK and SAPK cascades in the actions of bryostatin 1 and safingol (Fig. 7). Basal MAPK and SAPK activities were not discernibly modified by treatment with bryostatin 1 for 24 hr or safingol for 6 hr alone. As noted above, ara-C stimulated both ERK1/ERK2 (by 365%) and JNK1/JNK2 (by 310%). These responses exhibited strikingly dissimilar sensitivities to experimental reductions in cPKC/nPKC, however. MAPK responses to ara-C were completely suppressed by either pretreatment with bryostatin 1 or cotreatment with safingol, consistent with the premise that potentiation of ara-C action arises from blockade of drug-induced MAPK activation. In marked contrast, the SAPK response to ara-C was unaffected by either cPKC/nPKC down-regulation by bryostatin 1 or cPKC/nPKC inhibition by safingol, indicating that potentiation of ara-C-related cell death did not involve modulation of SAPK.
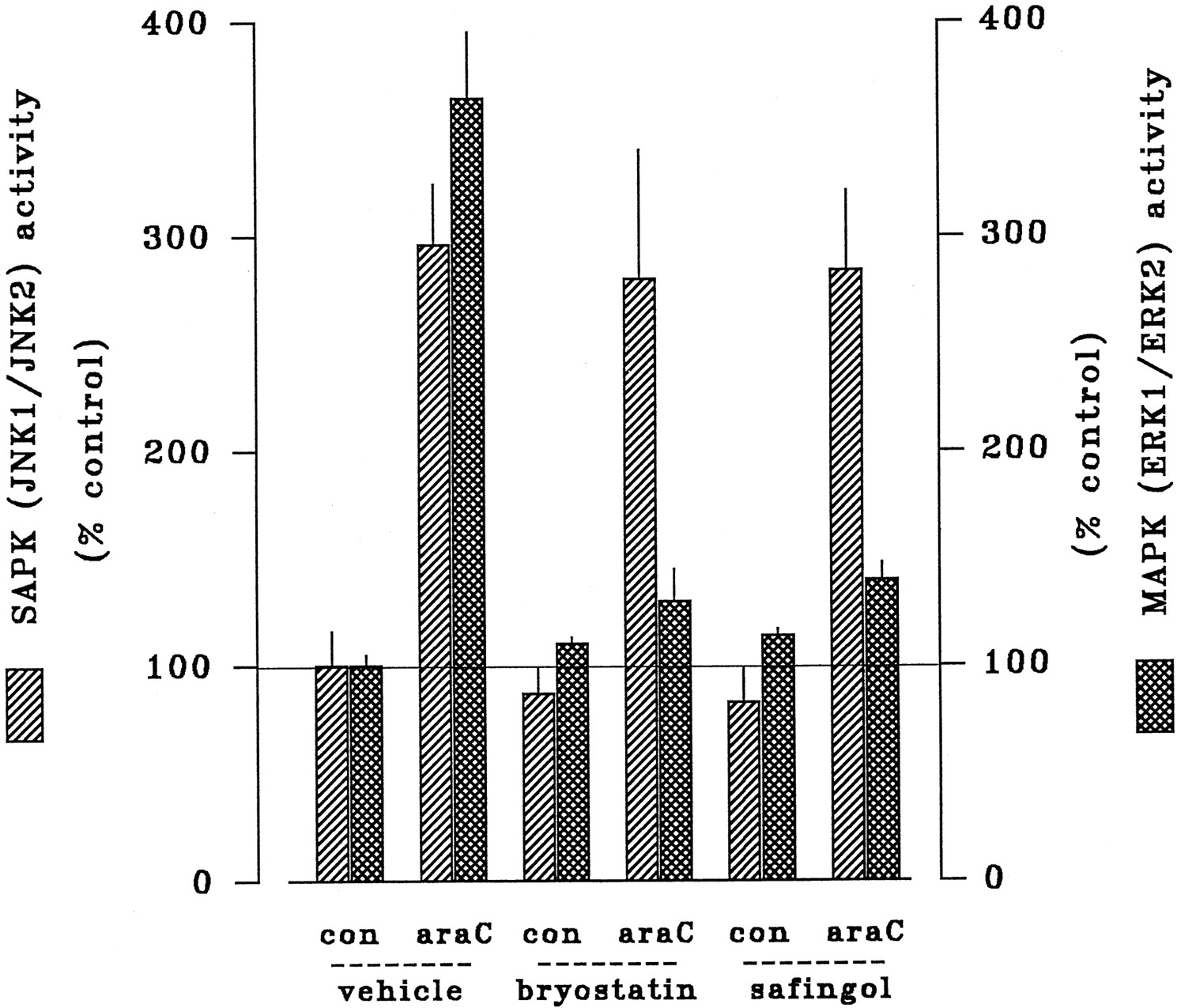
Effects of bryostatin 1 and safingol on ara-C-induced activation of MAPK and SAPK. HL-60 cells were incubated for 2 hr in the absence or presence of ara-C (10 μm) in conjunction with either (1) 24-hr pretreatment bryostatin 1 (10 nm) or (2) 6-hr cotreatment safingol (1 μm). Relative activities of SAPK (p46-JNK1/p54-JNK2; ▨) and MAPK (p42-ERK1/p44-ERK2; ▩) were determined by in vitroassays as before. In each case, values reflect the mean ± standard error of duplicate determinations and are expressed as a percentage of basal activity detected in untreated controls. Data shown are from a representative study performed three times with comparable results.
PKC activity and ara-C metabolism.
Because PKC has been implicated in the biosynthesis of ara-C-derived nucleotides (i.e., through phosphorylation by dCyd kinase) in leukemia cells (Wang and Kucera, 1994), other trials determined whether lipid-mediated augmentation of ara-C action reflected increased production of ara-CTP. Both diglyceride and sphingosine were without discernible effect on cellular ara-CTP levels (Table 3), indicating that lipid-dependent changes in the response to ara-C could not be attributed to increased conversion of the prodrug to its lethal derivative. Similarly, neither bryostatin 1 nor safingol significantly altered ara-CTP levels (Table 4), demonstrating that augmented responses to ara-C associated with these agents did not stem from increased ara-C nucleotide formation.
Insensitivity of ara-CTP metabolism to diglyceride and sphingosine
Insensitivity of ara-CTP metabolism to bryostatin 1 and safingol
Enhancement of ara-C-induced apoptosis by AMF.
The ability of bryostatin 1 and safingol to augment ara-C-induced apoptosis seemed to be directly linked to loss of MAPK activity downstream of cPKC/nPKC suppression. To test this possibility directly, additional studies evaluated the potentiation of ara-C action by pharmacological interference with MAPK function using AMF, a potent and highly selective inhibitor of MEK1 (Alessi et al., 1995; Dudleyet al., 1995) (Fig. 8A). Exposure to AMF alone over a broad range of concentrations (1–100 μm) for 3 hr resulted in a pronounced concentration-related decline in basal MAPK activity (decreased maximally by ≥65%, with an EC50 value of ≤5 μm). Coexposure to AMF and ara-C (10 μm) completely suppressed drug-related stimulation of MAPK, reducing activity to well below basal levels. Additional studies evaluated the capacity of AMF, at a concentration sufficient to prevent ara-C-related MAPK activation (5 μm), to potentiate the cytotoxicity of ara-C. AMF sharply augmented the ability of ara-C to promote DNA damage (Fig. 8B), augmenting both the accumulation of double-stranded DNA fragments (by 236%) and corresponding double-stranded breakage of bulk DNA (by 194%). Cell survival also was affected by AMF (Fig. 8C). The induction of apoptosis by ara-C was sharply increased in the presence of AMF (from 29% to 98%); drug-related inhibition of clonogenicity was similarly enhanced by AMF (reducing the surviving fraction from 61% to ≤2%). The ability of AMF to potentiate the toxicity of ara-C was comparable to that noted above for bryostatin 1 or safingol.
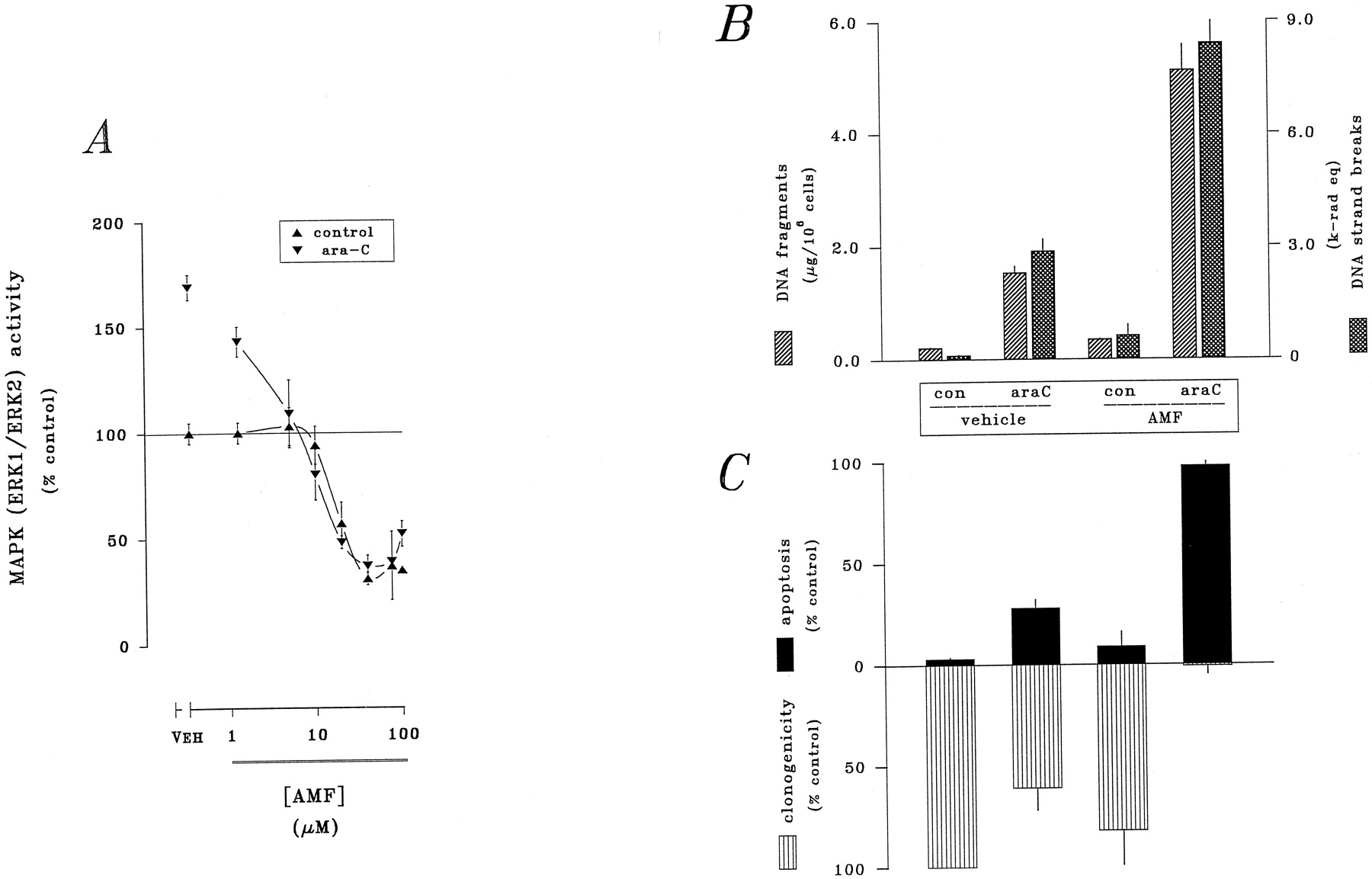
Suppression of MAPK and potentiation of ara-C action by AMF. HL-60 cells were exposed to ara-C (10 μm) in the absence or presence of AMF (2.5 μm). A, MAPK (p42-ERK1/p44-ERK2 was determined by in vitro assay after 3 hr; all values reflect the mean ± standard error of duplicate determinations and are expressed as a percentage of basal activity present in untreated controls. Apoptotic DNA damage and cell death were assessed after 6-hr exposure. B, Accumulation of double-stranded DNA fragments (▨) and occurence of double-stranded breakage of bulk DNA (▩); for both measurements, values represent the mean ± standard error of quadruplicate determinations. C, Induction of apoptosis (▪) and suppression of clonogenicity (▥); values represent the mean ± standard error of triplicate determinations. Data shown are from a representative study performed four times with comparable results.
SAPK in ara-C action.
Ceramide generation and activation of SAPK have been implicated in the apoptotic response to ara-C in HL-60 cells (Strum et al., 1994; Saleem et al., 1995). A mechanism coupling ara-C action to enzymatic processes that mediate ceramide formation (e.g., hydrolysis of sphingomyelin by sphingomyelinase or de novo synthesis from free dihydrosphingosine by ceramide synthase) has not been identified, however. Similarly, a mechanism underlying recruitment of the SAPK cascade by ara-C has not been established, nor is there evidence that SAPK activity contributes directly to the drug’s antileukemic actions. Increased production of ceramide (via sphingomyelinase or ceramide synthase) and downstream activation of the SAPK cascade have been implicated in the cytotoxicity of several antineoplastic agents (Strumet al., 1994; Bose et al., 1995; Saleem et al., 1995). The functional importance of ceramide in the lethal response to ara-C has not been definitively established; therefore, alternate pathways for ceramide generation and their possible relationship to ara-C-related SAPK activation were examined (Table 5). Exposure to ara-C produced a 253% increase in cellular ceramide levels (from 94 to 335 pmol/106 cells) within 30 min and a corresponding 345% stimulation of JNK1/JNK2 activity at 2 hr; after 6 hr, apoptotic cell death was noted in ∼30% of the cell population. Cotreatment with 3-O-methoxy-N-octanoyl-sphingosine-1-phosphorylcholine (methoxysphingomyelin), a synthetic lipid inhibitor of neutral/acidic sphingomyelinase (Lister et al., 1995), abolished ara-C-dependent production of ceramide and reduced JNK1/JNK2 activity by 65%. In contrast, cotreatment with fumonisin B1, a mycotoxin inhibitor of ceramide synthase (Merrill et al., 1993), produced a modest increase in SAPK activity but failed to failed to modify the effects of ara-C on ceramide availability or JNK1/JNK2 activity. However, neither methoxysphingomyelin nor fumonisin B1 modified drug-induced apoptosis. Because the antiproliferative properties of some agents are mediated through autocrine elaboration of TNFα (Lillyet al., 1991) and consequent activation of neutral and acidic sphingomyelinases through the type-1 TNF receptor (TNFR1; CD120a) (Wiegmann et al., 1994), SAPK activation by ara-C was tested in conjunction with experimental blockade of endogenous TNFα (Table 6). Ara-C (10 μm) increased cellular ceramide levels by 327% (from 71 to 393 pmol/106 cells) within 30 min; this response was associated with a 646% increase in JNK1/JNK2 activity at 2 hr and induction of apoptotic cell death in ∼30% of treated cells. Neutralization of endogenous TNFα by cotreatment with either (1) anti-TNF mAb or (2) soluble ligand-binding domain of CD120a (type I TNF receptor) eliminated the ara-C-stimulated increase in ceramide generation and significantly reduced drug-related JNK1/JNK2 activity, but neither of these manipulations modified the drug’s lethal capacity. Parallel control studies showed that the apoptotic responses to exogenous rhTNFα were completely eliminated by these treatments (not shown). These findings indicated that recruitment of SAPK activity by ara-C derived at least in part from the actions of endogenous TNFα but suggested that these events did not underlie the drug’s lethal effects. Collectively, these findings indicated that the SAPK response to ara-C resulted from production of ceramide by sphingomyelinase (but not ceramide synthase), presumably via the action of autocrine TNFα but suggested that ceramide-driven SAPK activity was not required for the lethal effects of ara-C.
Sensitivity of ara-C action to inhibitors of ceramide generation
Effects of TNFα neutralization on the response to ara-C
Discussion
The specific effector system coupling ara-C-induced DNA damage to the initiation of cell death has not yet been established. Treatment of myeloid leukemic cells with ara-C is associated with (1) generation of diradylglycerols (Kucera and Capizzi, 1992; Strum et al., 1994), (2) stimulation of cPKC/nPKC activity (Kharbanda et al., 1991), and (3) recruitment of the MAPK cascade (Kharbandaet al., 1994). Other observations demonstrate that pharmacological suppression of ara-C-related cPKC/nPKC activity enhances drug-induced apoptosis (Jarvis et al., 1994c; Grantet al., 1994, 1996), suggesting that the diglyceride-driven cPKC/nPKC does not underlie the apoptotic influence of ara-C. On the contrary, we have proposed that activation of this pathway instead mediates an antiapoptotic influence that reduces drug-induced cell death and therefore represents an intrinsically self-limiting component of ara-C action (reviewed in Grant, 1997). Other investigators have reached similar conclusions (Whitman et al., 1997). Thus, inhibition or down-regulation of cPKC/nPKC may eliminate a critical subcellular target for diglyceride, thereby preventing recruitment of an antiapoptotic downstream effector. Although the signaling system involved in these processes remains to be identified, the MAPK cascade represents a plausible candidate target.
The present data provide direct support for these hypotheses. Preliminary studies demonstrated that (1) exogenous diglycerides activated the MAPK cascade and attenuated ara-C action, whereas (2) exogenous sphingoid bases potently suppressed MAPK activity and amplified the drug’s toxicity. Thus, modulation of cPKC/nPKC and (MAPK further downstream) by physiological lipid messengers reciprocally altered ara-C-related cell death. In similar fashion, pharmacological reductions in cPKC/nPKC produced substantial increases in ara-C lethality. Specifically, induction of apoptotic DNA damage and cell death by ara-C was enhanced through either (1) inhibition of cPKC/nPKC by acute coexposure to safingol or (2) down-regulation of cPKC/nPKC by chronic preexposure to bryostatin. Moreover, reduction of assayable cPKC/nPKC activity and potentiation of ara-C-mediated lethality seemed to be closely coupled. Potentiation of ara-C action was not associated with altered ara-C metabolism, again consistent with a lowered threshold for initiation cell death.
Although the precise mechanism underlying the general antiapoptotic influence of PKC has not been identified, it is significant that reductions of cPKC/nPKC by either bryostatin 1 or safingol were accompanied by downstream suppression of ERK activity. In marked contrast, there was no evidence of corresponding alterations in JNK activity. These findings strongly suggest that collapse of MAPK signaling downstream from suppression of cPKC/nPKC is directly involved in pharmacological enhancement of ara-C action. Consistent with this conclusion, the cytotoxicity of ara-C was sharply enhanced after disruption of the MEK-ERK module by AMF. Thus, pharmacological agents that decrease MAPK activity indirectly through suppression of PKC (i.e., bryostatin, safingol), as well as those that bypass cPKC/nPKC and suppress MAPK more proximally (i.e., AMF), equivalently abrogated the MAPK response to ara-C. The role of MAPK activity in cell survival and various other cellular processes (e.g., proliferation) is well documented (Xia et al., 1995). The present observations provide strong, if indirect, evidence of a role for the MAPK cascade in determining the sensitivity of myeloid leukemia cells to antineoplastic agents. Significantly, these findings raise the possibility that pharmacological interference with the MEK-ERK module may prove useful in increasing the susceptibility of leukemic cells to ara-C and potentially other antineoplastic agents. Studies examining this approach are under way.
Although the MAPK cascade seems to oppose the cytotoxicity of ara-C, a role for the SAPK cascade in ara-C action is uncertain. ara-C promotes generation of ceramide, which mediates activation of the SAPK cascade in various settings (Westwick et al., 1995; Verheijet al., 1996; Jarvis et al., 1997); furthermore, both activation of SAPK (Saleem et al., 1995) and induction of c-Jun/AP1 have been associated with the apoptotic response to ara-C (Kharbanda et al., 1990; Brach et al., 1992). Several lines of evidence argue against the involvement of this stress pathway in ara-C action. First, ara-C-related increases in ceramide accumulation and JNK activity were limited by blockade of autocrine TNFα (by anti-TNFα mAb or rhsTNFR), whereas cytotoxicity was preserved, indicating that recruitment of the SAPK cascade was not essential to ara-C action. Second, the sphingomyelinase inhibitor methoxysphingomyelin abolished ara-C-related ceramide generation and partially limited downstream activation of JNK yet failed to modify drug-induced apoptosis; in contrast, the ceramide synthase inhibitor fumonisin B1 was entirely without effect. This suggested that drug-related SAPK activity involved generation of ceramide from hydrolysis of sphingomyelin (rather than de novo synthesis from dihydrosphingosine) and suggested that neither pathway for ceramide generation contributed to ara-C lethality. Third, cPKC/nPKC-directed treatments that enhanced ara-C action (e.g., down-regulation by bryostatin, inhibition by safingol) failed to augment drug-dependent SAPK activity. Collectively, these observations indicate that the ceramide-driven SAPK responses associated with ara-C action do not underlie the drug’s lethal capacity but may instead represent a consequence of apoptotic cell death. This is consistent with other findings from our laboratory demonstrating that dominant-negative ablation of the primary SAPK substrate c-Jun (which disables normal AP1-dependent trans-activation) abrogates ceramide-mediated apoptosis (Verheij et al., 1996; Jarviset al., 1997) but does not compromise ara-C lethality (Grantet al., 1996). In conclusion, the present data demonstrate that the lethal actions of ara-C are partially limited by the intrinsic capacity of this agent to activate the MAPK cascade downstream of cPKC/nPKC. The cytoprotective effector engaged by the MAPK cascade that is ultimately responsible for antagonizing ara-C-induced cell death remains to be identified; among several plausible candidates is the transcription factor nuclear factor-κB, the activation of which reportedly opposes drug-mediated apoptosis (Mayo et al., 1997). Further support for involvement of the cPKC/nPKC → Raf-1 → MEK1 → ERK1/ERK2 sequence in cytoprotective responses to ara-C is provided by the findings that cPKC/nPKC down-regulation (by bryostatin) or inhibition (by safingol) was associated with corresponding reductions in ERK activity and increases in drug-associated lethality. Alternatively, disruption of the MEK-ERK module by AMF exerted a similar influence. Finally, the discordances between stimulation of JNK activity and induction of apoptosis by ara-C suggest that recruitment of the SAPK cascade (presumably via ceramide) represents a secondary event in ara-C-induced cell death. These findings underscore the potential utility of interrupting MAPK cascade signaling in attempts to augment drug-induced apoptosis and raise the possibility that specific elements within this signaling system represent suitable targets for chemomodulatory interventions in future trials.
Acknowledgments
This work was supported primarily by Research Grants CA63753 and CA77141 from National Cancer Institute (S.G.) and 6405–97 (S.G.) from the Leukemia Society of America and HL16660 from National Heart, Lung, and Blood Institute (R.B.). W.D.J. is recipient of National Research Service Award CA09380 from National Cancer Institute. F.A.F. is recipient of National Research Service Award HL09241 from National Heart, Lung, and Blood Institute. P.D. is supported by Research Grant IN-105V from American Cancer Society and by the V-Foundation. Additional funding was provided by Cancer Center Support Core Grant CA16059 to the Massey Cancer Center.
Footnotes
- Received March 23, 1998.
- Accepted July 28, 1998.
-
Send reprint requests to: Dr. W. David Jarvis, Medical College of Virginia, MED-HEM/ONC, Box 980230 MCV Station, Richmond, VA 23298-0230.
Abbreviations
- AMF
- 2′-amino-3′-methoxy-flavone (PD-98059)
- ara-C
- 1-β-d-arabinofuranosylcytosine
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal receptor-activated kinase
- SAPK
- stress-activated protein kinase
- JNK
- c-Jun NH2-terminal kinase
- cPKC
- conventional protein kinase C subfamily
- nPKC
- novel protein kinase C subfamily
- rhTNFα
- recombinant human tumor necrosis factor-α
- rhsTNFR-I
- soluble type-I (p55) tumor necrosis factor receptor
- antiTNFα mAb
- tumor necrosis factor-α-directed monoclonal antibody
- PBS
- phosphate-buffered saline
- EGTA
- ethylene glycol bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid
- HEPES
- 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}