


Abstract
Previously, we demonstrated the involvement of Asn293 in helix VI of the human β2-adrenergic receptor in stereoselective agonist recognition and activation. In the present study, we have further explored the role of this residue by synthesizing derivatives of isoproterenol and clenbuterol, two β-adrenergic receptor agonists. We analyzed their efficacy and affinity on the wild-type and a mutant receptor (Asn293Leu). Each compound had similar efficacy (τ values) on both the wild-type and mutant receptor, although τ values varied considerably among the eight compounds studied. It appeared that one derivative of isoproterenol, but not of clenbuterol, showed a gain in affinity from the wild type to the mutant receptor. This derivative had a methyl substituent instead of the usual β-OH group in the aliphatic side chain of isoproterenol, compatible with the more lipophilic nature of the leucine side chain. Such a “gain of function” approach through a combination of synthetic chemistry with molecular biology, may be useful to enhance our insight into the precise atomic events that govern ligand-receptor interactions.
β2-Adrenergic receptors serve as a prototypic member of the large superfamily of G protein-coupled receptors. Apart from the visual pigment rhodopsin, they were the first to be cloned (Dixon et al., 1986) and subjected to site-directed mutagenesis (Strader et al., 1988, 1989). The latter two studies identified Asp113 in helix III and two serines in helix V (Ser204 and 207) of the human β2-adrenergic receptor as anchor points for the protonated amine function and the catechol group of the endogenous agonist epinephrine, respectively. In the previous two decades, thorough medicinal chemistry had already led to the introduction of an important class of antiasthmatic drugs, all β2-receptor agonists, exemplified by salbutamol and clenbuterol. The nonselective catecholamine isoproterenol has been and still is the reference compound when the pharmacology and molecular biology of β-adrenergic receptors are the subject of investigation. In a recent study, we identified Asn293 in helix VI as a residue that determines the stereoselectivity of catecholamines by virtue of its interaction with the β-OH group in the aliphatic side chain of this class of compounds (Wieland et al., 1996). The hypothesis for this interaction was based on the molecular modeling of the β2-adrenergic receptor for which we used the atomic coordinates of bacteriorhodopsin as a template. Figure1 is a visualization of this receptor model, defining the positions of the four above-mentioned amino acid residues and the levorotamer of isoproterenol.

Model visualizing the binding of (−)-isoproterenol to the human β2-adrenergic receptor. The receptor α-helical backbone (helices I-VII) is viewed from the extracellular side. (−)-Isoproterenol and the receptor residues thought to be involved in ligand binding are shown in red and yellow (Asp113, Ser204, Ser207, and Asn293, respectively).
We thought it worthwhile to further characterize the atomic details of this stereoselective interaction. Therefore, we synthesized “mutated” derivatives of β-adrenergic receptor agonists in the present study and analyzed their effects on the wild-type and aforementioned mutant receptor in which Asn293 was changed for a leucine residue. We reasoned that such an approach (mutant ligands for mutant receptors) might lead to a “gain of function” in terms of affinity. This would provide further physical evidence for a direct interaction between a specific receptor residue and a functional group in the ligand, indeed allowing an interpretation at almost the atomic rather than the molecular level of the ligand-receptor interaction.
Experimental Procedures
Synthesis of Isoproterenol and Clenbuterol Analogs
General Methods and Materials.
Schleicher and Schüll DC Fertigfolien F 1500 LS 254 were used for thin layer chromatography analysis. Compounds were detected under UV light. For structures of compounds, see Fig. 2. Column chromatography was performed on silica gel 60, 230 to 400 mesh (Merck, Darmstadt, Germany). 1H NMR spectra (300 MHz) were recorded at 25°C with a Bruker WM 300 spectrometer.13C NMR spectra (50 MHz) were recorded with a Jeol JNM-FX 200 spectrometer. 1H chemical shifts (δ) are given in parts per million relative to that of Me4Si [CDCl3, MeOD, or dimethyl sulfoxide (DMSO)]. 13C chemical shifts are not included but are available on request from the corresponding author. All mass experiments were performed on a Finnigan MAT900, equipped with a programmable direct insertion probe. Temperature was raised from 30 to 300°C in 5 min. Mass spectra were recorded in electron impact mode with 70 eV. Melting points were uncorrected. All compounds appeared pure on thin layer chromatography, and in1H NMR, 13C NMR, and mass spectrometry.
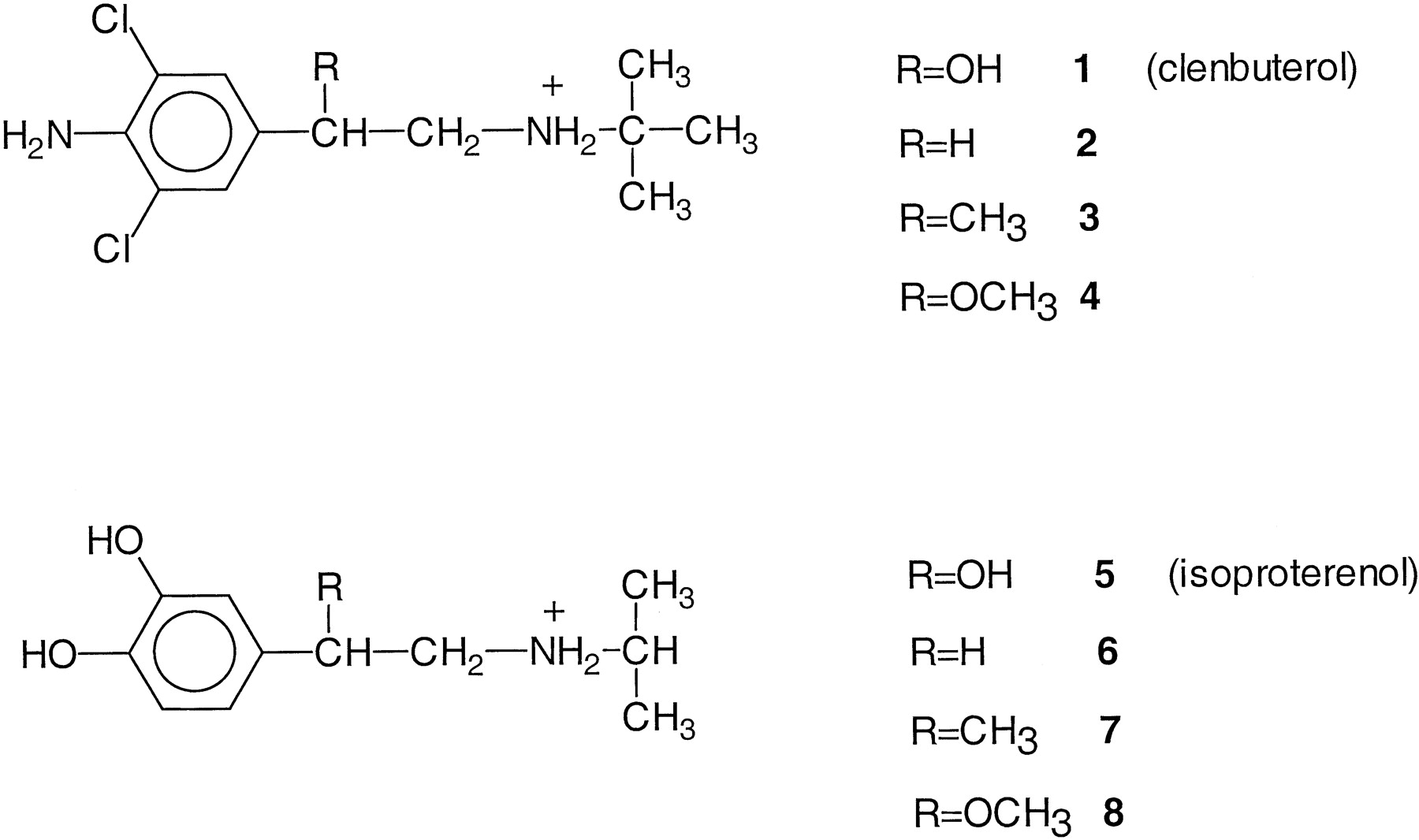
Structural formulae of clenbuterol and isoproterenol and their derivatives used in this study.
4-Amino-3,5-dichlorobenzylcyanide (9).
To a stirred solution of commercially available 4-aminobenzylcyanide (0.5 g, 3.8 mmol) in acetic acid (10 ml) was added dropwise N-chlorosuccinimide (1.1 g, 8.4 mmol) in acetic acid (5 ml). After 17 h at room temperature, the reaction mixture was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 (25 ml) and extracted with water (10 ml), dried (MgSO4), and concentrated. The residue was purified by column chromatography on silica gel (CH2Cl2) to give pure 9 (0.46 g, 60%). 1H NMR (MeOD) δ 3.65 (s, 2H, CH2CN), 7.15 (s, 2H, Harom). MS: m/z 200 (M+).
4-Amino-3,5-dichloro-α-methylbenzylcyanide (10).
Compound9 (0.4 g, 2.0 mmol) was dissolved in tert.-BuOH (10 ml). To this solution was added potassiumtert-butanolate (0.2 g, 2 mmol) dissolved intert-BuOH (5 ml), and subsequently MeI (0.3 g, 2.2 mmol). After 4 h at room temperature, the reaction mixture was diluted with CH2Cl2 (20 ml) and the organic layer extracted with water (10 ml), dried (MgSO4), and concentrated. Purification of the remaining oil on silica gel [1:0 to 0:1 light petroleum (bp 40–60°C)/ether] afforded 10 (0.3 g, 67%).1H NMR (CDCl3) δ 1.54 (d, 3H, CH3, J = 7.2 Hz), 3.60 (q, 1H, CH), 7.18 (s, 2H, Harom).
3,4-Dimethoxy-α-methylbenzylcyanide (11).
A solution of potassium tert-butanolate (8.3 g, 73 mmol) intert-BuOH (100 ml) was added at once to a solution of commercially available 3,4-dimethoxybenzylcyanide (10 g, 57 mmol) and MeI (13.3 g, 57 mmol) in tert-BuOH (100 ml). The temperature was maintained at 35–38°C for 60 min. Then the reaction mixture was concentrated and the residue diluted with CH2Cl2 (250 ml). The organic layer was washed with water (125 ml), dried (MgSO4), and concentrated to give 11(13 g, 85%). 1H NMR (CDCl3) δ 1.06 (t, 3H, CH 3, J = 7.2 Hz), 3.67 (t, 1H, CH, J = 7.2 Hz), 3.86 (s, 6H, 2× OCH3), 6.83 (dd, 1H, Harom,J o 8.1 Hz, J m2.0 Hz), 7.25 (d, 1H, Harom,J m 2.0 Hz), 7.28 (d, 1H,J o 2.0 Hz).
General Procedure for One-Pot Reduction-Transimination-Reduction.
To a cooled (−70°C) solution of cyanide (5 mmol) in dry ether (40 ml) was added 1 M diisobutylaluminum hydride in cyclohexane (10 ml, 10 mmol). After stirring at −70°C for 3 h, dry MeOH (15 ml) was added. The cooling bath was removed and the amine (25 mmol) added. Stirring was continued for 2 h during while the temperature was allowed to rise to room temperature. The mixture was stirred for another 2 h, and 1 N HCl (50 ml) was added. The organic layer was extracted with an additional 1 N HCl (30 ml). The aqueous layer was made alkaline with 5 N NaOH and then extracted with CH2Cl2 (3 × 25 ml). The combined organic layers were dried (K2CO3) and evaporated. The residue was purified on silica gel (1:0 to 9:1 CH2Cl2/MeOH).
2-(4-Amino-3,5-dichlorophenyl)-1-(tert-butylamino)ethane (2).
Prepared as described above, starting from cyanide9 and with tert-BuNH2 as the amine in a yield of 45%. 1H NMR (DMSO) δ 1.27 (s, 9H, 3× CH3-tert-Bu), 2.78 (m, 2H, CH 2NH), 3.02 (m, 2H, CH 2CH2NH), 7.21 (s, 2H, Harom).
The hydrochloride salt of compound 2 was prepared from the free base. The solid was recrystallized (MeOH-CH2Cl2): mp 224–226°C. MS: m/z 260 (M+), and used as such in the test systems.
2-(4-Amino-3,5-dichlorophenyl)-1-(tert-butylamino)propane (3).
Prepared as described above, starting from cyanide10 and with tert-BuNH2 as the amine in a yield of 51%. 1H NMR (CDCl3) δ 1.07 (s, 9H, 3 × CH3-tBu), 1.21 (d, 3H, CH3, J = 6.4 Hz), 2.66 (m, 2H, CH 2NH), 2.69 (m, 1H, CHCH2NH), 7.05 (s, 2H, Harom).
The hydrochloride salt of 3 was prepared from the free base. The solid was recrystallized (MeOH-diisopropylether): mp 190–193°C. MS: m/z 274 (M+), and used as such in the test systems.
2-(3,4-Dimethoxyphenyl)-1-(iso-propylamino)ethane (12).
Prepared as described above, starting from dimethoxybenzylcyanide (Brussee et al., 1978) and withiPrNH2 as the amine in a yield of 89%. 1H NMR (CDCl3) δ 1.05 (d, 6H, 2× CH3-iPr,J = 6.2 Hz), 2.71–2.89 (m, 5H, CH 2CH2NH, CH 2NH, CH-iPr), 3.86, 3.87 (2× s, 6H, 2× OCH3), 6.75 (dd, 1H, Harom, J o 8.2 Hz,J m 1.9 Hz), 6.80–6.85 (m, 2H, Harom).
2-(3,4-Dimethoxyphenyl)-1-(iso-propylamino)propane (13).
Prepared as described above, starting from cyanide 11 and with iPrNH2 as the amine in a yield of 91%. 1H NMR (CDCl3) δ 1.02 (d, 3H, CH3-iPr,J = 6.2 Hz), 1.05 (d, 3H, CH3-iPr, J = 6.3 Hz), 1.25 (d, 3H, CH3, J = 7.2 Hz), 2.42–2.95 (m, 4H, CHCH2NH, CH 2NH, CH-iPr), 3.85, 3.87 (2 × s, 6H, 2 × OCH3), 6.78 (dd, 1H, Harom, J o 8.1 Hz,J m 2.1 Hz), 6.70–6.75 (m, 2H, Harom).
General Procedure for Demethylation.
The dimethoxy derivative (2 mmol) was dissolved in 48% HBr (10 ml) and the solution was refluxed for 17 h. Then the solvent was evaporated and the last traces of the solvent were removed with the aid of repeated addition and evaporation of toluene (3 × 25 ml). The residue was purified by column chromatography on silica gel (EtAc/MeOH/NH4OH, 85/15/1, v/v).
2-(3,4-Dihydroxyphenyl)-1-(iso-propylamino)ethane (6).
Prepared as described above, starting from dimethoxy derivative 12 in a yield of 52%. 1H NMR (DMSO) δ 1.21 (d, 6H, 2 × CH3-iPr, J = 6.6 Hz), 2.72 (m, 2H, CH 2NH), 3.02 (m, 2H, CH 2CH2NH), 3.30 (q, 1H, CH3-iPr, J = 6.2 Hz), 6.49 (dd, 1H, Harom,J o 8.0 Hz, J m2.1 Hz), 6.63 (d, 1H, Harom,J m 2.1 Hz), 6.67 (d, 1H, Harom, J o 8.0 Hz). MS: m/z 195 (M+).
The fumaric acid salt of 6 was prepared from the free base. The solid thus obtained was >97% pure and was used as such in the test systems.
2-(3,4-Dihydroxyphenyl)-1-(iso-propylamino)propane (7).
Prepared as described above, starting from dimethoxy derivative (compound 13) in a yield of 63%. 1H NMR (MeOD) δ 1.28 (d, 3H, CH3,J = 6.1 Hz), 1.30 (d, 6H, 2× CH3-iPr, J 6.2 Hz), 2.95 (m, 1H, CHCH2NH), 3.12 (m, 2H, CH 2NH), 3.32 (q, 1H, CH-iPr,J = 6.2 Hz), 6.62 (dd, 1H, Harom,J o 8.1 Hz, J m2.2 Hz), 6.72 (d, 1H, J m 2.2 Hz), 6.76 (d, 1H, J o 8.1 Hz). MS: m/z 209 (M+).
The fumaric acid salt of 7 was prepared from the free base. The solid thus obtained was >97% pure and was used as such in the test systems.
2-(4-Amino-3,5-dichlorophenyl)-2-[(methyl)oxy]-1-(tert-butylamino)ethane (4).
Clenbuterol (0.5 g, 1.8 mmol) was suspended in MeOH (10 ml) and hydrogen chloride was passed in with stirring at 10°C until a clear solution resulted. The reaction mixture was concentrated and the hydrochloride salt thus obtained (87% yield) was recrystallized (MeOH-diisopropylether): mp 212–214°C. 1H NMR (MeOD) δ 1.34 (s, 9H, 3 × CH3-tBu), 3.07 (m, 2H, CH 2NH), 3.28 (s, 3H, OCH3), 4.28 (t, 1H, CHCH2NH, J = 5.8 Hz), 7.23 (s, 2H, Harom). MS: m/z 290 (M+).
2-(3,4-Dihydroxyphenyl)-2-[(methyl)oxy]-1-(isopropylamino)ethane (8).
Prepared as described for compound 4 starting from (±)-isoproterenol in a yield of 85%. The hydrochloride salt thus obtained was >97% pure and was used as such in the test systems.1H NMR (MeOD) δ 1.33 (d, 3H, CH3-iPr), J = 6.5 Hz), 1.35 (d, 3H, CH3-iPr,J = 6.1 Hz), 3.05 (dd, 1H, CHCHHNH,J = 12.9 Hz, J = 3.5 Hz), 3.13 (dd, 1H, CHCHHNH, J = 12.9 Hz, J = 10.3 Hz), 3.25 (s, 3H, OCH3), 3.31 (q, 1H, CH-iPr), 4.35 (dd, 1H, CHCH2NH, J = 3.5 Hz,J = 10.3 Hz), 6.70 (dd, 1H, Harom, J o 8.1 Hz,J m 1.9 Hz), 6.80 (d, 1H, Harom, J m 1.9 Hz), 6.81 (d, 1H, Harom,J o 8.1 Hz). MS: m/z 225 (M+).
Expression of Wild-Type and Mutant β2-Adrenoceptors in Stable CHO Cells
CHO cell lines stably expressing either wild-type or mutated β2-adrenoceptor cDNA (encoding a single point mutation at amino acid position 293 from asparagine to leucine) (Wieland et al., 1996) were grown in monolayers on Dulbeccos’s modified Eagle’s medium (DMEM F-12) supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all purchased at Pan Systems, Berlin, Germany) and 0.5 mg/ml of G418 (Gibco Laboratories, Eggenstein, Germany) in a 7.5% CO2 incubator at 37°C. Clones with comparable densities (∼0.2 pmol/mg membrane protein) were selected for the experiments.
Preparation of Crude Cell Membranes
Cells were washed three times with ice-cold phosphate-buffered saline, scraped in 50 ml of ice-cold lysis buffer (5 mM Tris-HCl, 2 mM EDTA, pH 7.4), homogenized with an Ultra Turrax for 30 s at full speed and centrifuged at 1000g for 10 min (4°C). The supernatants were then centrifuged at 50,000g for 15 min (4°C), and the resulting pellets were resuspended in an appropriate volume of incubation buffer (50 mM Tris-HCl, pH 7.4) to give a final concentration of 2 mg/ml. Protein content was determined by the method of Bradford (1976) with bovine serum albumin (Sigma Chemical Co., Deisenhofen, Germany) as the standard.
Radioligand Binding Assay
For inhibition assays, 0.1 ml of fresh membranes was incubated in triplicates with 50 pM 125I-cyanopindolol (125I-CYP) and various concentrations of isoproterenol/clenbuterol (Sigma Chemical Co.) and their derivatives (100 nM–1 mM) in the presence of 100 μM Gpp(NH)p (Sigma Chemical Co.) for 1 h at 37°C. The latter addition was used to uncouple the β2-adrenergic receptors from Gs and thereby generate monophasic competition curves for agonists. Nonspecific binding was determined in the presence of 10 μM (−)-propranolol (Sigma Chemical Co.). The incubations were terminated by filtration through Whatman GF/C filters and washing with ice-cold 50 mM Tris-HCl, pH 7.4.
Adenylyl Cyclase Assays
Adenylyl cyclase activity was determined as described in Wieland et al. (1996). Crude cell membranes were prepared freshly immediately before the assay as described above. Incubations contained 40 to 50 μg of protein; 50 mM Tris-HCl, pH 7.4; 500 μM RO 20–1724; 100 μM cAMP; 2 mg/ml BSA; 1 mM MgCl2; 5 mM creatine phosphate; 0.4 mg/ml creatine kinase, 1 μM GTP; 100 μM [α-32P]ATP (0.2 μCi/tube; Amersham Corp., Braunschweig, Germany); and the desired concentrations of the test compounds in a final volume of 100 μl. Incubations were done for 30 min at 30°C. Adenylyl cyclase activities measured in the presence of 100 μM (−)-isoproterenol were set to 100%. Basal activities were not subtracted.
UV-Spectrometry
Isoproterenol and clenbuterol (100 μM in 0.1 M KCl) were analyzed at 25°C, under N2 atmosphere and in the dark, essentially to prevent oxidation of isoproterenol during measurements. Solutions were adjusted to either pH 5 or pH 11 by addition of minute amounts of HCl or NaOH, respectively. UV spectra were recorded on an Aminco DW-2A UV/VIS spectrophotometer.
Molecular Modeling
The receptor-ligand model presented in Fig. 1 was developed exactly as described in Wieland et al. (1996). The figure was generated with programs MOLSCRIPT and RASTER3D.
Data Analysis
Determination of ligand-binding parameters was performed by nonlinear curve fitting with SCTFIT and plotted with KALEIDAGRAPH, respectively. K D values of125I-CYP were 7.1 and 7.3 pM for the wild-type and mutant receptor, respectively (Wieland et al., 1996).
Concentration-response curves of adenylyl cyclase experiments were fitted to the operational model developed by Black and Leff (1983) as described in Lohse (1990) to obtain an estimate of the transducer ratio τ:
Results
Chemistry.
The clenbuterol derivatives 2–4 were prepared as follows. First, commercially available 4-aminobenzylcyanide was chlorinated with N-chlorosuccinimide to give its 3,5-dichloro derivative 9, which was converted into the desired end product 2 by a one-pot reduction/transimination/hydride reduction sequence in a yield of 45% (Zandbergen et al., 1992). Methylation of derivative 9 with methyl iodide in the presence of potassium tert-butoxide gave methyl derivative 10 in a yield of 67%. Conversion of the latter into clenbuterol analog 3 was achieved by the one-pot reduction/transimination/hydride reduction sequence. The protected isoproterenol analogs 12 and 13were likewise obtained. Demethylation of analogs 12 and 13 in aqueous HBr gave the unprotected isoproterenol analogs 6and 7. The methoxy derivatives 4 and 8were synthesized by HCl/MeOH treatment of clenbuterol and isoproterenol, respectively.
Biology.
The parent compounds clenbuterol and isoproterenol and their derivatives modified at the β-OH position (Fig. 2) were tested in radioligand binding studies and adenylate cyclase assays. For that purpose the wild-type β2-adrenergic receptor was expressed in CHO cells and membranes were prepared, as was done for a mutant β2-adrenergic receptor in which an asparagine residue in the sixth transmembrane domain was changed into a leucine (N293L) (Wieland et al., 1996). All clenbuterol and isoproterenol derivatives were capable of displacing125I-CYP, a radiolabelled antagonist, from both wild-type and mutant receptors (Fig. 3and Table 1). TheK i values listed in Table 1 show that the methoxy analogs 4 and 8 had highest affinity on the wild-type receptor among the derivatives. All derivatives had lower affinities for the wild-type receptor than their parent compounds isoproterenol and clenbuterol. This latter observation held also on the mutant receptor. Here, the methyl derivative 7 was most potent among the isoproterenol derivatives, whereas both the unsubstituted (2) and methoxy derivative 4 were the most potent clenbuterol analogs.

Stereospecific binding of isoproterenol derivatives to CHO cell membranes containing wild-type and mutant β2-adrenergic receptors. Data are means ± S.E. (n = 3). Top, competition for 125I-CYP binding to the wild-type β2-adrenergic receptor by various isoproterenol derivatives (6, ○; 7, ●; 8, ▪). Bottom, competition for 125I-CYP binding to the mutant N293L β2-adrenergic receptor by various isoproterenol derivatives (6, ○; 7, ●; 8, ▪).
Affinities of clenbuterol/isoproterenol and their derivatives (all racemates, except (−)-isoproterenol) for wild-type and N293L mutant β2-adrenergic receptors
More interesting, however, were the differential affinities at the two receptors, as quantified by the ratios given in Fig.4. Apparently, clenbuterol and each of its analogs had essentially equal affinity for wild-type as well as mutant receptor because all ratios were equal or close to unity. However, results were different for isoproterenol and its analogs. In our previous study (Wieland et al., 1996; data also in Table 1) it had been observed that (−)-isoproterenol, but not clenbuterol, lost its stereoselective recognition of the receptor upon the N293L mutation, due to the decrease in affinity on the mutant receptor of this stereoisomer. A 6-fold gain in affinity, however, was observed for the methyl derivative 7, suggesting an improved fit of this ligand on the mutant over the wild-type receptor.

Affinity ratios (K i, wild-type/K i, N293L) for all eight compounds, calculated from the data in Table 1. Data are means ± S.E. (n = 3).
Next, activation of adenylyl cyclase in the cell membranes was studied. Concentration-response curves of the various ligands were obtained on membranes containing either the wild-type or the N293L mutant receptors. In both types of membranes, (−)-isoproterenol stimulated adenylyl cyclase activity by 10- to 15-fold, and in agreement with earlier data (Wieland et al., 1996), there was no difference in the extent of stimulation between wild-type and mutant receptors. However, the position of the isoproterenol stimulation curve was shifted to the right by more than one order of magnitude in the mutant receptors (Fig.5). Smaller rightward shifts (2–3-fold) were seen with isoproterenol derivatives 6 and 8. Again, the exception was the isoproterenol methyl derivative7, displaying an ∼3-fold leftward shift. Both clenbuterol (1) and its methoxy analog 4 showed a small leftward shift as well; it proved impossible to estimate EC50 values for the other low-efficacy clenbuterol derivatives 2 and 3.

Concentration-response curves (adenylyl cyclase activation) for all compounds on both wild-type and N293L mutant β-adrenergic receptors. Maximal stimulation by (−)-isoproterenol was set to 100%. A typical experiment is shown, which was repeated at least three more times in duplicate with similar results.
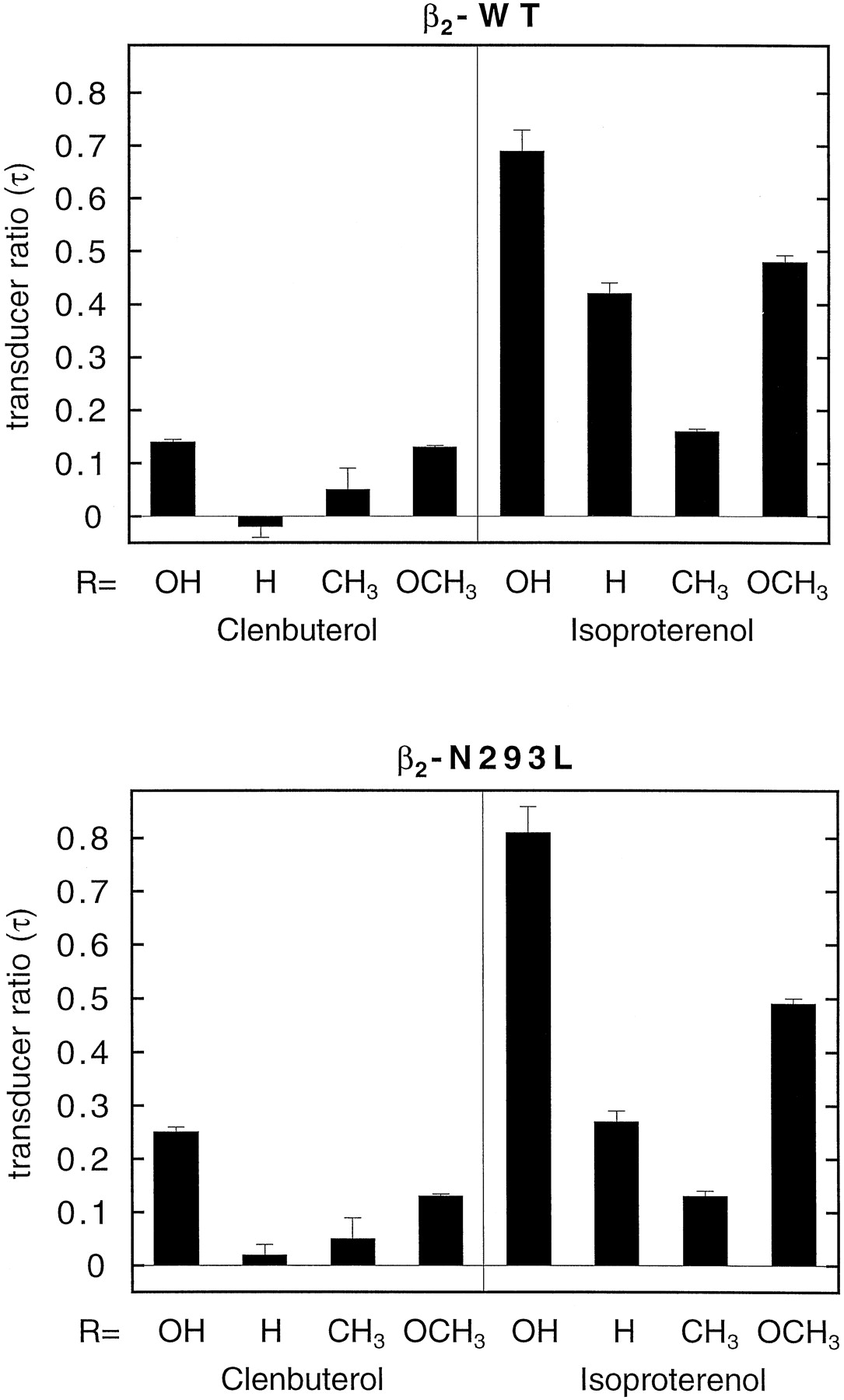
Intrinsic activities were estimated from these data by fitting them to the operational model as described in Experimental Procedures (the fitted curves shown in Fig. 5 were obtained with this model) to obtain the transducer ratios τ. These values are depicted in Fig. 6. There were no obvious differences in results obtained for wild-type and mutant receptors. Compared with (−)-isoproterenol, all compounds were partial agonists, and the extent of agonism was very similar for wild-type and mutant receptors. Clenbuterol (1) had a slightly higher τ value in the mutant receptors, whereas those of 6 and 7were slightly lower. These data indicate that there was no correlation between the alterations in affinities as seen in the binding experiments and the intrinsic activities.

Transducer ratios τ (see Experimental Procedures) for all eight compounds on both wild-type and N293L mutant receptors. Data are means ± S.E. (n = 4–5).
UV-Spectroscopy.
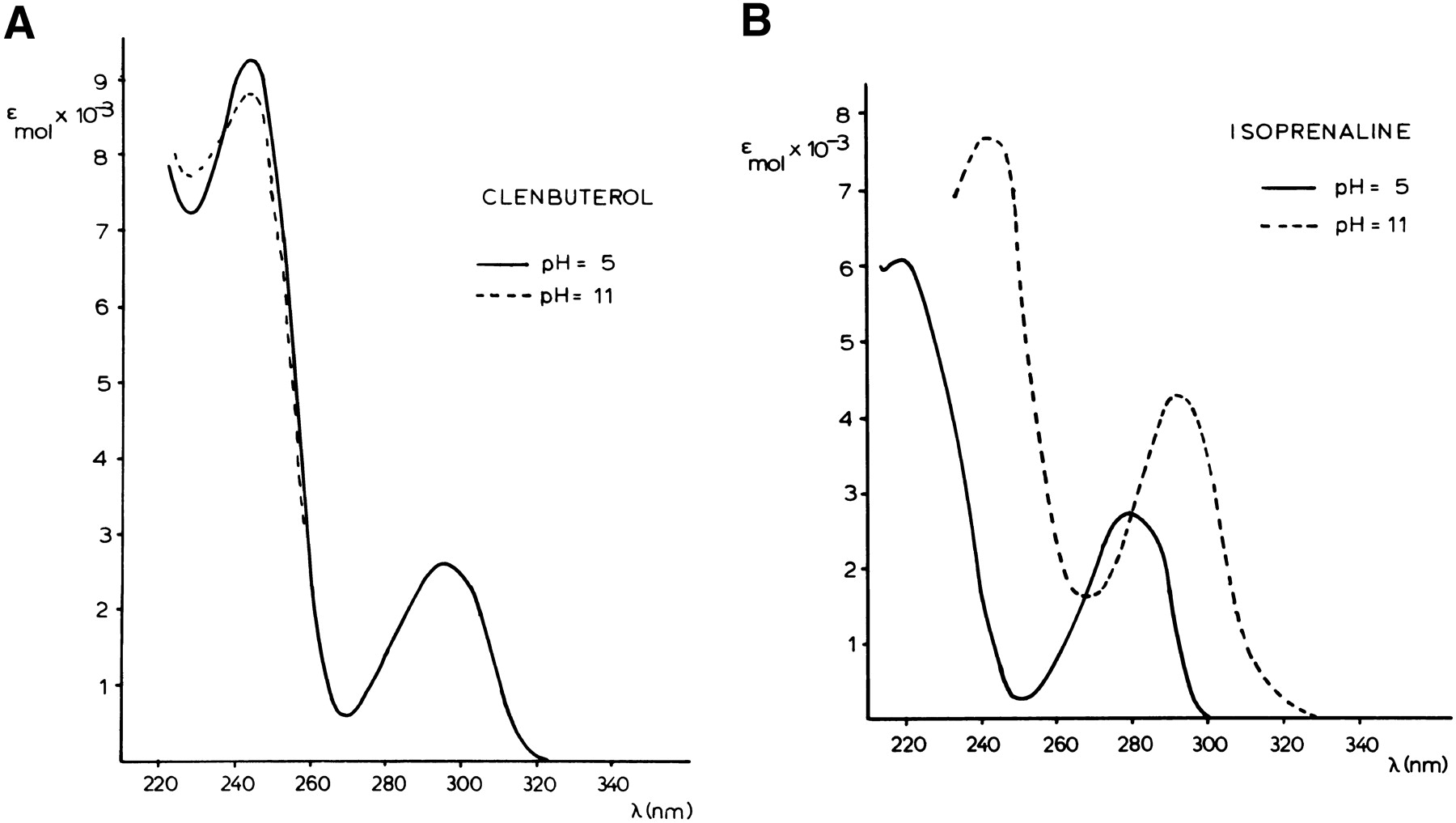
The UV spectra of isoproterenol and clenbuterol were recorded at two pH values. At pH 5, the aromatic ring systems of both compounds are neutral, whereas at pH 11 the catechol ring of isoproterenol is negatively charged, and no changes occur in clenbuterol’s ring system (IJzerman et al., 1984). As a result, the UV spectra of clenbuterol at both pH values are almost coinciding (Fig.7A), whereas isoproterenol’s spectra are different (Fig. 7B). Interestingly, at pH 11 the spectra of isoproterenol and clenbuterol are virtually identical, whereas at pH 5 the UV spectra of both do not match.

UV spectra of clenbuterol (A) and isoproterenol (B) at pH 5 and pH 11 (εmol: molar extinction; λ: wavelength).
Discussion
In Fig. 1, a possible binding mode of (−)-isoproterenol, the prototypic β-adrenergic receptor agonist, to the human β2-adrenergic receptor is visualized. This representation is entirely based on the receptor model developed byWieland et al. (1996), but viewed from a different angle. In that previous study, we demonstrated the involvement of Asn293 in the stereospecific agonist recognition and activation of the human β2-adrenergic receptor, through its interaction with the β-OH group in the ligand side chain (see also Fig. 2). In particular, catecholamines such as isoproterenol proved highly sensitive to a mutation from asparagine to leucine on this position. The mutant receptor displayed strongly diminished affinity for the active (−)-stereoisomer of isoproterenol, whereas the (+)-isomer was less affected. However, clenbuterol, a structurally dissimilar agonist (Fig. 2), did not appear to suffer from the mutation.
In the present study, we focused on this apparent discrepancy at the agonist ligand binding site in more detail. By “mutating” isoproterenol and clenbuterol, we sought to complement our analysis of the receptor-agonist interaction. Hence, we synthesized a number of isoproterenol and clenbuterol analogs, all varied at the β-OH position in the side chain (Fig. 2). Radioligand binding studies and adenylate cyclase assays on both wild-type and mutant receptors were performed with the two parent compounds and their derivatives. We reasoned that the mutation of an asparagine residue into a leucine would cause a change in the characteristics of part of the ligand binding site from essentially hydrophilic with hydrogen-bonding capacity to far more lipophilic. As a consequence, certain analogs of isoproterenol should be able to recognize the mutant receptor better than the wild-type receptor, and we hypothesized that replacement of the β-OH group in the aliphatic side chain of the ligand by a more lipophilic group might do in this respect. Such a gain of function also would complement and corroborate the findings in our previous study (Wieland et al., 1996), and, moreover, help us in appreciating atomic rather than molecular mechanisms in receptor recognition.
Indeed, we succeeded in making an isoproterenol derivative (7) that displayed higher affinity for the mutant receptor (Table 1; Fig. 4). Apparently, a methyl substituent (7) instead of a hydrogen atom (6) or a methoxy group (8) is an appropriate point of interaction for the leucine side chain. The affinity of 7 for the N293L mutant receptor was comparable to isoproterenol’s affinity, given the fact that7 is a racemate and isoproterenol was tested as the active enantiomer only. Thus, the ∼100-fold affinity difference on the wild-type receptor between isoproterenol and its methyl derivative7 was fully canceled on the mutant receptor. Interestingly,7 did not prove superior to isoproterenol on the mutant receptor, suggesting that the receptor environment around the β-position in the aliphatic side chain of the ligands remains hydrophilic in nature, despite the presence of the leucine side chain.
It might be argued that such a gain in affinity as observed for7 could be due to more indirect effects, e.g., changes in overall receptor conformation. However, in this case a direct effect is more probable due to the other observations we made. First, the two ligands clenbuterol and isoproterenol behaved differently. The ratios shown in Fig. 4 demonstrate that hardly any gain or loss of function was found for clenbuterol and its derivatives, suggesting that Asn293 is not relevant for the recognition of this β-adrenergic receptor agonist. If the effect observed for isoproterenol were due to general changes in receptor conformation, a similar change would have been expected for clenbuterol and analogs. Second, the data from the adenylate cyclase experiments show that the intrinsic activities on the two receptors are almost similar for each derivative (Fig. 5). Also, the transducer ratios τ (Fig. 6) hardly varied between the wild-type and the mutant receptors. Apparently, the transduction mechanism for the wild-type and the mutant receptor had not drastically changed. Such a change would have been more probable if the mutant receptor had acquired a different overall conformation.
The mutated ligands were all less effective than their parent compounds because maximal cyclase activation was not observed (Fig. 5) and their τ values were lower (Fig. 6). Apparently, the β-OH group is preferred over the other substituents for full receptor activation. This finding corroborates the observations in our previous study (Wieland et al., 1996) that this position is not only important for stereoselective receptor recognition but also for receptor activation. The adenylyl cyclase experiments confirmed the preference of7 for the mutant receptor. Its EC50value on the mutant receptor was 20 ± 9 μM compared with 58 ± 5 μM on the wild-type receptor.
The present study also provides evidence that there is not one single agonist binding site on the β2-adrenergic receptor, based upon the above-mentioned observations that the recognition of clenbuterol and analogs is different from isoproterenol and derivatives. Hence, the critical involvement of Ser204 and Ser207 on helix V in agonist binding (Fig. 1), as elegantly shown by Strader et al. (1989), may be limited to catecholamines. These two amino acids are supposed to interact with the two catechol-OH groups via hydrogen bonding and, as a consequence, the ligand is directed to position its β-OH group in the vicinity of Asn293. In binding clenbuterol, Asn293 does not seem to play a significant role, and, thus, Ser204 and Ser207 are hypothesized to be of less importance, too. Preliminary experiments in which we analyzed the binding of a series of agonists to a “double mutant” β2-adrenergic receptor (Ser204Ala, Ser207Ala) strongly corroborated this notion (data not shown). Isoproterenol was >100-fold more potent on the wild-type receptor, whereas clenbuterol’s affinity was only slightly diminished on the double mutant receptor (∼3-fold).
Another line of evidence for this hypothesis came from the UV experiments (Fig. 7). In hydrogen bonding to the two serine residues, the two catechol-OH groups in isoproterenol may undergo a weakening of their O—H bond. This gradual transition from a fully intact OH group to a situation that resembles a deprotonated form is mimicked in the UV recordings at pH 5 and pH 11, respectively. Under slightly acidic conditions (pH 5) the catechol structure is intact, whereas basic conditions (pH 11) promote the dissociation of a proton from the catechol OH groups (IJzerman et al., 1984). Clenbuterol’s UV spectrum, unlike that of isoproterenol, is not pH dependent. The electronic characteristics of the aromatic ring systems in clenbuterol and isoproterenol (as exemplified by their UV spectra) are almost coinciding at pH 11. It has been shown that the electronic features of the aromatic ring system in β-adrenergic receptor agonists are strongly correlated to their intrinsic activity (IJzerman et al., 1986). Apparently, clenbuterol’s dichloro-anilino ring system may have the appropriate characteristics for productive receptor interaction without the need to undergo some sort of proton abstraction. This would make a direct interaction with the two serine residues unnecessary, and allow a different orientation of clenbuterol in the receptor.
Interestingly, β-receptor antagonists having an oxypropanolamine side chain as in propranolol and in the radioligand used in the present study (125I-CYP), also show a stereoselective interaction through their side chain OH group with an asparagine residue, but now in helix VII. This has been convincingly demonstrated, in particular on several subtypes of the 5-hydroxytryptamine receptor, because compounds such as propranolol and pindolol display nanomolar affinity for these receptors, too (Guan et al., 1992). In a recent study, we applied a similar strategy as described above to a series of propranolol analogs (Kuipers et al., 1997). Asn386 (helix VII) of the human 5-hydroxytryptamine1A receptor was mutated into a valine rather than a leucine residue. It was found that a propranolol derivative with methoxy instead of hydroxy in the oxypropanolamine side chain showed a 25-fold affinity gain on the mutant receptor. Valine has a branched side chain with one -CH2 group less than leucine, which might explain this receptor’s preference for the bigger methoxy group compared with the methyl substituent in isoproterenol.
In summary, our study demonstrates that by simultaneous mutations in both ligands and receptors, gain of function can be established. To our knowledge, this is the first study of its kind focusing on agonist-receptor interactions. This approach is helpful to rule out mutations that have indirect effects on this interaction; it is a first step in the unraveling of ligand-receptor interactions at atomic detail. It is expected that future elucidation of the three-dimensional architecture of G protein-coupled receptors combined with the approach outlined in the present study, i.e., a combination of synthetic chemistry with molecular biology, will enhance our insight in the atomic events that govern the ligand-receptor interaction.
Acknowledgments
We thank Dr. T. Costa (ISS, Rome) for helpful discussions.
Footnotes
- Received February 23, 1999.
- Accepted July 22, 1999.
-
Send reprint requests to: Adriann P. IJzerman, Ph.D., Receptor Medical Chemistry, Leiden/Amsterdam Center for Drug Research, P.O. Box 9502, 2300RA Leiden, the Netherlands. E-mail:ijzerman{at}lacdr.leidenuniv.nl
-
This study was supported by grants from the European Community (programs EUROCEPTOR and InverseA).
Abbreviations
- CDCl3
- deuterated chloroform
- MeOD
- deuterated methanol
- DMSO
- dimethylsulfoxide
- CYP
- cyanopindolol
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}