


Abstract
The interaction of dopaminergic antagonists with the D1Adopamine receptor was assessed in PC2 cells that transiently express this receptor. The maximal binding and dissociation constants for the D1A dopamine receptor, using the ligand [125I]SCH23982 were 0.38 ± 0.09 nM and 1 to 4 pmol/mg, respectively, when assessed 48 h after transfection with cDNA encoding the rat D1A receptor. Basal adenylyl cyclase activity increased 50 to 60% in membranes of transfected PC2 cells compared with control membranes. The dopaminergic antagonists clozapine, cis-flupenthixol, (+)-butaclamol, haloperidol, chlorpromazine, and fluphenazine inhibited constitutive adenylyl cyclase activity in membranes of cells expressing the D1A receptor. SCH23390, a selective D1 dopamine receptor antagonist, and (−)-butaclamol did not alter basal cyclase activity, whereas dopamine increased enzyme activity in membranes expressing the D1A dopamine receptor. The coupling of D1A receptors with Gs proteins was examined by immunoprecipitation of membrane Gsα followed by immunoblotting with a D1A dopamine receptor monoclonal antibody. Clozapine, cis-flupenthixol, (+)-butaclamol, haloperidol, and fluphenazine but not SCH23390 or (−)-butaclamol decreased D1A receptor-Gsα coupling by 70 to 80%, and SCH23390 was able to prevent the receptor-Gsαuncoupling induced by haloperidol or clozapine. These results indicate that some dopaminergic antagonists suppress basal signal transduction and behave as inverse agonists at the D1A dopamine receptor. This action of the dopamine receptor antagonists may contribute to their antidopaminergic properties that seem to underlie their clinical actions as antipsychotic drugs.
Dopaminergic neuronal systems play important roles in brain function (Kalivas and Stewart, 1991; Williams and Goldman-Rakic, 1995). Dysfunctional dopaminergic neurotransmission has been suggested to be involved in such neurological and psychiatric disorders as Parkinson disease, Tourette syndrome, and schizophrenia (Albin et al., 1989;Knable and Weinberger, 1997). Pharmacological, biochemical, and largely molecular biological studies have identified five different dopamine receptors (Sokoloff and Schwartz, 1995). These receptors have seven transmembrane domains and couple to G proteins. Although the D1A and D1B dopamine receptors stimulate adenylyl cyclase, the D2-like dopamine receptors (D2, D3, D4) inhibit this pathway (Civelli et al., 1993;Seeman and Van Tol, 1994). Several studies have also shown that in brain, activation of phospholipase C is mediated by an as-yet unidentified D1-like dopamine receptor (Undie and Friedman, 1990; Wang et al., 1995; Pacheco and Jope, 1997).
Inhibition of D2 dopamine receptors has frequently been considered as the mechanism by which dopaminergic antagonists exert their therapeutic actions in schizophrenia and Tourette syndrome (Creese et al., 1976; Seeman et al., 1976; Richelson and Nelson, 1984). However, several studies have also shown that the antipsychotic drugs bind to D1A dopamine receptors (Kanba et al., 1994). This effect has been suggested to contribute to their ability to ameliorate the negative symptoms of schizophrenia (Lynch, 1992; Reynolds and Czudek, 1995). Dl receptor antagonists may also be expected to produce fewer neurological adverse effects (Chipkin et al., 1988;Waddington, 1988; Lynch, 1992). Furthermore, an interaction between the Dl and D2 dopamine receptors has been suggested to be of functional importance in determining the output of dopaminergic neurotransmission; thus, inhibition of either receptor by the antipsychotic drugs may contribute to their therapeutic action in schizophrenia (Seeman et al., 1989).
Ligands that bind to G protein-coupled receptors may be classified as agonist, neutral antagonist, or inverse agonist, according to the outcome of their interaction with their receptor (Kenakin et al., 1995). Receptor agonists enhance receptor activity, neutral antagonists block the action of agonists without exerting receptor-mediated effects, and inverse agonists negatively regulate constitutive receptor activity and signaling of G protein-coupled receptors (Schutz and Freissmuth, 1992; Lefkowitz et al., 1993; Kenakin et al., 1995;Milligan et al., 1995; Tiberi and Caron, 1995). Inverse agonists may be particularly useful as therapeutic agents in treating disturbances that are characterized by elevated tonic, agonist-independent, G protein-coupled receptor activity (Bond et al., 1995). In the present study, we attempted to test whether dopaminergic antagonists behave as inverse agonists at the D1A dopamine receptor. For that purpose, we determined the nature of the interaction of dopaminergic antagonists with D1A dopamine receptors by monitoring their effect on adenylyl cyclase activity in membranes obtained from PC2 cells that transiently express D1A dopamine receptors. We also directly assessed the effect of dopaminergic antagonists on D1Adopamine receptor-Gs protein coupling, because this association may determine the functional outcome of the interaction between a ligand and its receptor.
Experimental Procedures
Materials.
[125I]SCH23982 (2200 Ci/mmol), [α-32P]ATP (800 Ci/mmol), and Anti-Gsα protein antiserum (RM/1) were obtained from DuPont NEN (Boston, MA). Anti-D1A receptor monoclonal antibody was obtained from RBI (Natick, MA). [3H]adenosine 3′,5′-cyclic monophosphate (cAMP) was purchased from American Radiolabeled Chemicals, Inc. (St. Louis, MO). RPMI 1640, Lipofectin, and OPTI-MEM I were obtained from GIBCO BRL (Gaithersburg, MD). Pansorbin was purchased from Calbiochem (San Diego, CA). Enhanced chemiluminescence reagents were purchased from Pierce (Rockford, IL). All other chemicals were obtained from Sigma (St. Louis, MO).
PCR Amplification of Rat D1A Dopamine Receptor.
A full-length D1A dopamine receptor cDNA was amplified from rat genomic DNA using oligonucleotide primers corresponding to base pair (bp) −6 to +18 (5′-AGGAAGATGGCTCCTAACACTTCT-3′) and the reverse complement of bp +1324 to +1347 (5′-GCGAGTTCAAGTGGAATGCTGTCC-3′) of the rat D1A dopamine receptor sequence (Monsma et al., 1990; Zhou et al., 1990). The conditions for PCR was 48 s at 94°C, 2 min at 63°C, and 48 s at 72°C, 35 cycles followed by a 7-min extension at 72°C. The reaction products were purified and size-fractionated on a 1% agarose gel. The band of interest was excised, electroeluted, concentrated, and cloned into the pTargeT Mammalian Expression System (Promega, Madison, WI). High-efficiency JM109 competent cells (Promega) were transformed and minipreparations of plasmid DNA were prepared for insert sequencing.
Sequencing was performed by the Sanger dideoxynucleotide chain termination method using TaqTrack Sequencing System (Promega) on denatured double-stranded DNA. Both strands of the full-length cDNA were entirely sequenced using vector-specific primers or those derived from prior sequencing.
Cell Culture and Expression of Rat D1A Dopamine Receptor.
PC2 cells were plated at a density of approximately 100 to 200/mm2 on collagen coated dishes in RPMI 1640 supplemented with 10% horse serum, 5% fetal calf serum, 50 pg/ml streptomycin, 50 U/ml penicillin. The cells were cultured at 37°C in a water saturated atmosphere of 95% air and 5% CO2 and used for transfection when they reached 40 to 60% confluence.
The rat D1A dopamine receptor cDNA was inserted into the polylinker region of the pTargeT vector (Promega). For transient expression of the D1A dopamine receptor, PC2 cells were transfected using positively charged liposomes and Lipofectin reagent. Lipofectin was mixed with an equal volume of plasmid DNA in buffer containing 10 mM Tris, 1 mM EDTA, pH 8.0, and allowed to stand for 20 min at room temperature. Cells were washed twice and then incubated with 3 ml of OPTI-MEM I, a reduced serum medium. The DNA-Lipofectin mixture was added to cultured cells and incubated for 24 h. The DNA-containing medium was removed and replaced with 3 ml of RPMI 1640 supplemented with 20% fetal bovine serum and incubated for an additional 24 h.
Adenylyl Cyclase Assay.
PC2 cells were harvested after washing with a phosphate-buffered solution. The cells were homogenized in 10 volumes (w/v) of chilled buffer containing 50 mM Tris · HCl, pH 7.4, 2 mM EGTA, and 10% sucrose. The homogenate was centrifuged at 800g for 5 min and the supernatant centrifuged at 49,000g for 20 min. The pellet was washed twice and suspended in 50 mM Tris · HCl, pH 7.4. Protein content was determined by the method of Bradford (1976) using bovine serum albumin as standard. Adenylyl cyclase activity was measured by a modified method of Salomon (1979). Each assay was performed in 250 μl of solution containing 100 mM Tris · HCl, pH 7.4, 2 mM MgCl2, 0.1 mM ATP, 10 mM creatine phosphate, 0.2 mM EGTA, 100 μM 3-isobutyl-1-methylxanthine, 1 μM GTP, 1 mM dithiothreitol, 5 units of creatine phosphokinase, and 1 μCi [α-32P]ATP. Reaction mixture was preincubated at 30°C for 5 min. The assay was initiated by adding 50 μg of membrane protein and carried out at 30°C for 20 min. The reaction was terminated by adding 300 μl of stopping solution containing 2% SDS, 25 mM ATP, and 1.3 mM cAMP. [32P]cAMP was separated from [32P]ATP by Dowex and alumina chromatography. [3H]cAMP was added to each reaction mixture to allow calculation and correction for column recovery. Radioactivity in each sample was determined by liquid scintillation spectroscopy.
Radioligand Binding Assays.
PC2 cells were harvested and homogenized by sonication in buffer containing 5 mM Tris · HCl, pH 7.4, 1 mM EDTA, 0.2 mM PMSF, 14 μg/ml aprotinin. The homogenate was centrifuged at 49,000g for 20 min and the pellets resuspended in binding buffer containing 50 mM Tris · HCI, pH 7.4, 10 mM MgCl2, 1 mM EDTA. For saturation binding experiments, 0.01 to 8 nM [125I]SCH23982 were used. In competition binding experiments, 0.6 nM [125I]SCH23982 was employed. Nonspecific binding was defined as the binding of the radioligand in the presence of 1 μM cis-flupenthixol. The reaction was carried out at 25°C for 120 min and terminated by rapid filtration using a Brandel cell harvester with Whatman GF/C filter followed by washing with ice-cold binding buffer. The radioactivity on the filter was determined in a Beckman gamma counter. Saturation and competition binding curves were analyzed by nonlinear least-square regression using the LIGAND program (Munson and Rodbard, 1980).
Coimmunoprecipitation of D1A Dopamine Receptor with Gsα Protein.
Determination of the linkage between receptor and G protein was carried out as described previously by Wang et al. (1995). Transfected PC2 cells were homogenized in 10 volumes of buffer containing 25 mM HEPES, pH 7.5, 2 mM MgCl2, 1 mM EDTA, 0.2% 2-mercaptomethanol, 50 μg/ml leupeptin, 25 μg/ml pepstatin A, 5 μg/ml aprotinin, 0.01 U/ml soybean trypsin inhibitor, and 0.04 mM PMSF. The homogenate was centrifuged at 800g for 5 min and the supernatant was centrifuged for 10 min at 49,000g. The resulting pellet was washed and resuspended in immunoprecipitation buffer containing 100 mM Tris · HCl, pH 7.5, 200 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 0.2% 2-mercaptomethanol, 50 μg/ml leupeptin, 25 μg/ml pepstatin A, 0.01 U/ml soybean trypsin inhibitor, and 0.04 mM PMSF. Membrane proteins (50 μg) were solubilized in 1 ml of the immunoprecipitation buffer supplemented with 0.2% cholate and 0.5% digitonin. Solubilized tissues were precleaned by the addition of normal rabbit serum (1:100 dilution) at 4°C for 60 min followed by a 30-min incubation with 100 μl of a 10% suspension of protein A-bearing Staphylococcus aureus cells (Pansorbin cells). The suspension was centrifuged and the supernatant was combined with antisera (1:1000 dilution) raised against a specific Gsα peptide and incubated for 3 h followed by an additional 30 min with 100 μl of Pansorbin. After centrifugation, the pellet was suspended in 100 μl of sample preparation buffer and boiled for 5 min. The D1Adopamine receptors in the immunoprecipitates were assessed by immunoblotting using a monoclonal antibody that recognizes D1A dopamine receptor [originally produced and characterized by Hersch et al. (1995)].
Immunoblot Analysis.
Membrane proteins (25 μg) were solubilized in sample preparation buffer and were separated by SDS-polyacrylamide gel electrophoresis (12%). Proteins were transferred electrophoretically to nitrocellulose membrane. The completeness of the transfer was checked by Coomassie blue staining of the gel. The membranes were incubated at 4°C overnight with 10% nonfat dry milk in phosphate-buffered saline containing 0.1% Tween 20 (0.1% PBST) to block nonspecific sites. In some cases, 1 μg/ml of goat antirabbit IgG was added to the blocking solution to reduce immunostaining of the IgG band. The membrane was washed with 0.1% PBST and incubated for 2 h either with Gsα antiserum at 1:2000 dilution or with specific D1A dopamine receptor antibody at 1:1000 dilution. The unbound antibody was washed out with 0.1% PBST. The blot was incubated for 60 min with 1:10,000 dilution of horseradish peroxidase-conjugated antirabbit IgG (for Gsαprotein blot) or anti-mouse IgG (for D1A dopamine receptor blot) followed by washing in 0.3% PBST and in 0.1% PBST, respectively. The immunoreactive proteins were detected by the enhanced chemiluminescence Western blot detection system and visualized by exposure to X-ray film.
Data Analysis.
Data are presented as mean ± S.E. Two-tailed analysis of variance (ANOVA) was employed to compare the data among the groups followed by Newman-Keuls test. Significance was considered at P < .05.
Results
Functional Expression of D1A Dopamine Receptors in PC2 Cells.
The expression of D1A dopamine receptors was determined in membranes obtained from transfected PC2 cells by saturation binding using the specific D1dopamine receptor antagonist, [125I]SCH23982, as ligand. Specific [125I]SCH23982 binding was not detected in membranes of untransfected PC2 cells. After 48 h of transfection with rat D1A dopamine receptor cDNA, [125I]SCH23982 binding was saturable and the dissociation constant (K d) was calculated to be 0.38 ± 0.09 nM, with a maximal binding constant (B max) of 1 to 4 pmol/mg protein (Fig.1A).

Specific D1 dopamine receptor binding in PC2 cell membranes expressing D1A dopamine receptor. A, saturation binding was assessed by incubating 50 μg of PC2 cell membrane protein with 0.01 to 8 nM [125I]SCH23982 at 25°C for 120 min. Nonspecific binding was defined as binding in the presence of 10 μM cis-flupenthixol. B, displacement of specific [125I]SCH23982 binding was tested by incubating 50 μg of PC2 cell membrane protein with 0.6 nM [125I]SCH23982 in the presence of increasing concentrations of the indicated dopaminergic ligands. The assay was carried out 25°C for 120 min. Reaction was terminated by rapid filtration. The results shown are representative of three independent experiments.
Functional expression of D1A dopamine receptors in PC2 cells was also assessed by measuring basal and dopamine-stimulated adenylyl cyclase activity. Basal adenylyl cyclase activity in membranes of transfected cells was significantly increased compared with untransfected PC2 cells (22.2 ± 0.7 versus 14.5 ± 0.6 pmol/min/mg). Dopamine (100 μM) stimulated adenylyl cyclase 2-fold in transfected PC2 cell membranes, whereas the production of cAMP in membranes of untransfected cells was not affected by dopamine. The dopamine-induced stimulation of adenylyl cyclase was inhibited by the specific D1 dopamine receptor antagonist, SCH23390 (Table 1).
Dopamine-stimulated adenylyl cyclase activity in PC2 cells expressing D1A dopamine receptors
Effects of Dopaminergic Antagonists on Constitutive Adenylyl Cyclase Activity.
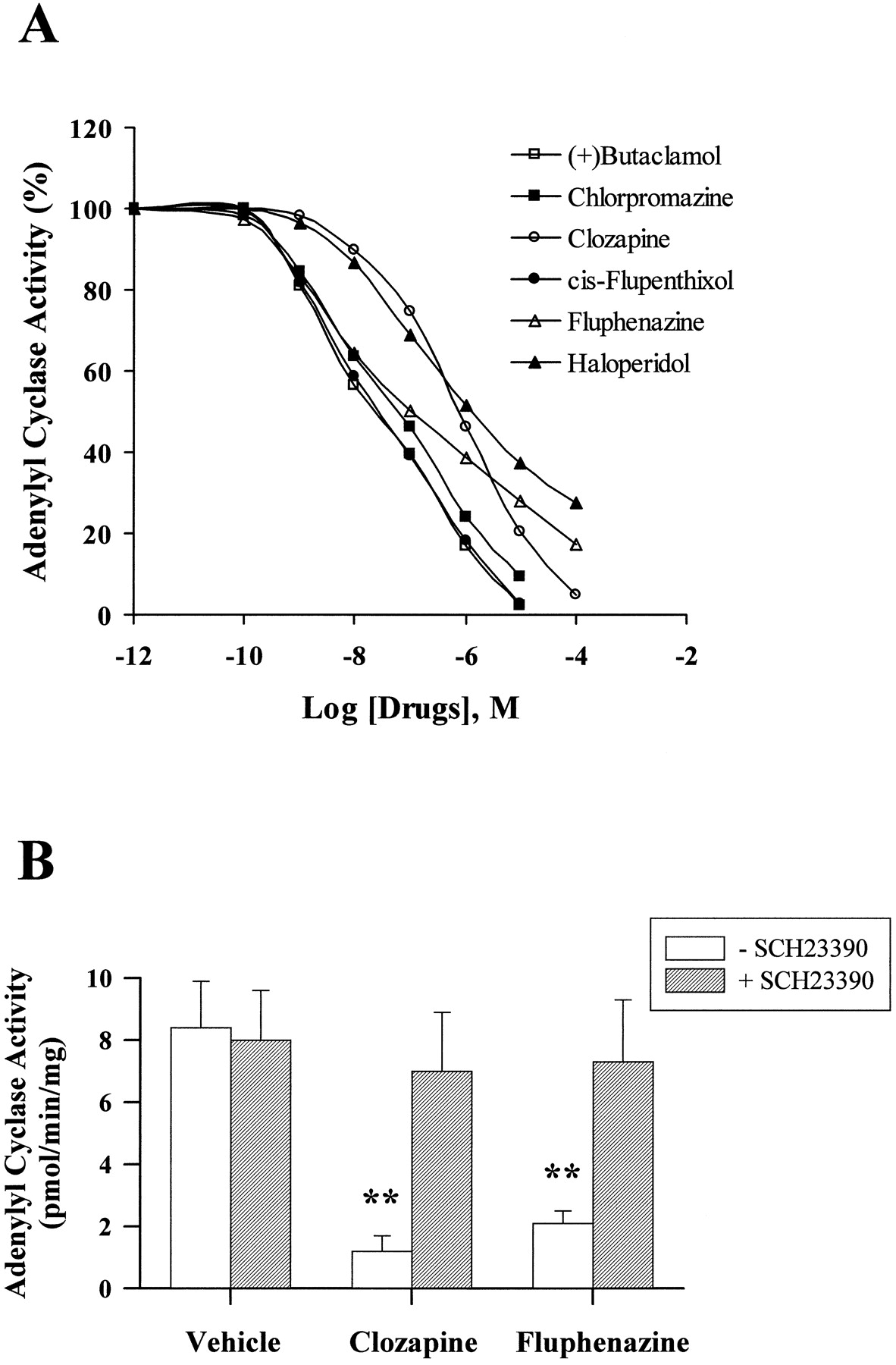
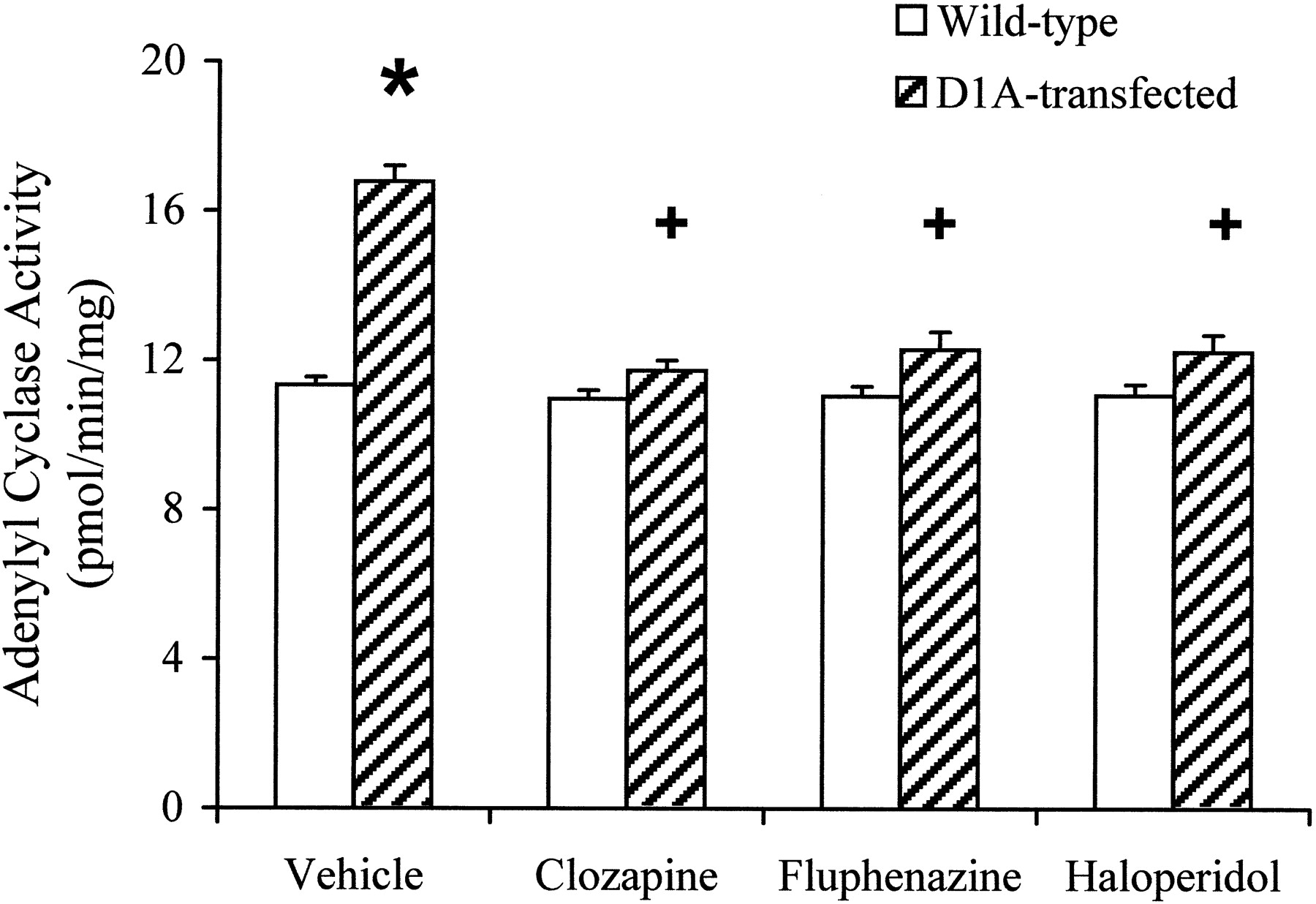
Inverse antagonist activity of dopaminergic antagonists was measured by monitoring the effect of compounds on D1A receptor-dependent constitutive adenylyl cyclase activity, which was defined by subtracting basal adenylyl cyclase activity in wild-type PC2 cells from that in PC2 cells expressing D1A receptors. Although previously classified as dopaminergic antagonists, (+)-butaclamol, chlorpromazine, clozapine, fluphenazine, cis-flupenthixol, and haloperidol inhibited constitutive adenylyl cyclase activity in a dose-dependent fashion (Fig. 2A). (+)-Butaclamol, chlorpromazine, clozapine, and cis-flupenthixol completely inhibited constitutive activity at concentrations between 10 to 100 μM, whereas fluphenazine and haloperidol achieved maximal inhibitions of 78.9% and 76.9%, respectively (Table2). The potencies of clozapine and haloperidol were lower than that of the other antipsychotic drugs in inhibiting constitutive adenylyl cyclase activity (Table 2). Constitutive adenylyl cyclase activity was not affected by the inactive isomer of butaclamol, (−)-butaclamol, by the selective D1 dopamine receptor antagonist SCH23390, or by the selective D2 dopamine receptor antagonistl-sulpiride. The selective D1 dopamine receptor antagonist SCH23390 (Fig. 2B) inhibited the effects of fluphenazine and clozapine on constitutive adenylyl cyclase activity. In contrast, the dopaminergic antagonists clozapine, cis-flupenthixol, and haloperidol did not alter adenylyl cyclase activity in membranes obtained from untransfected wild-type PC2 cells (Fig.3).
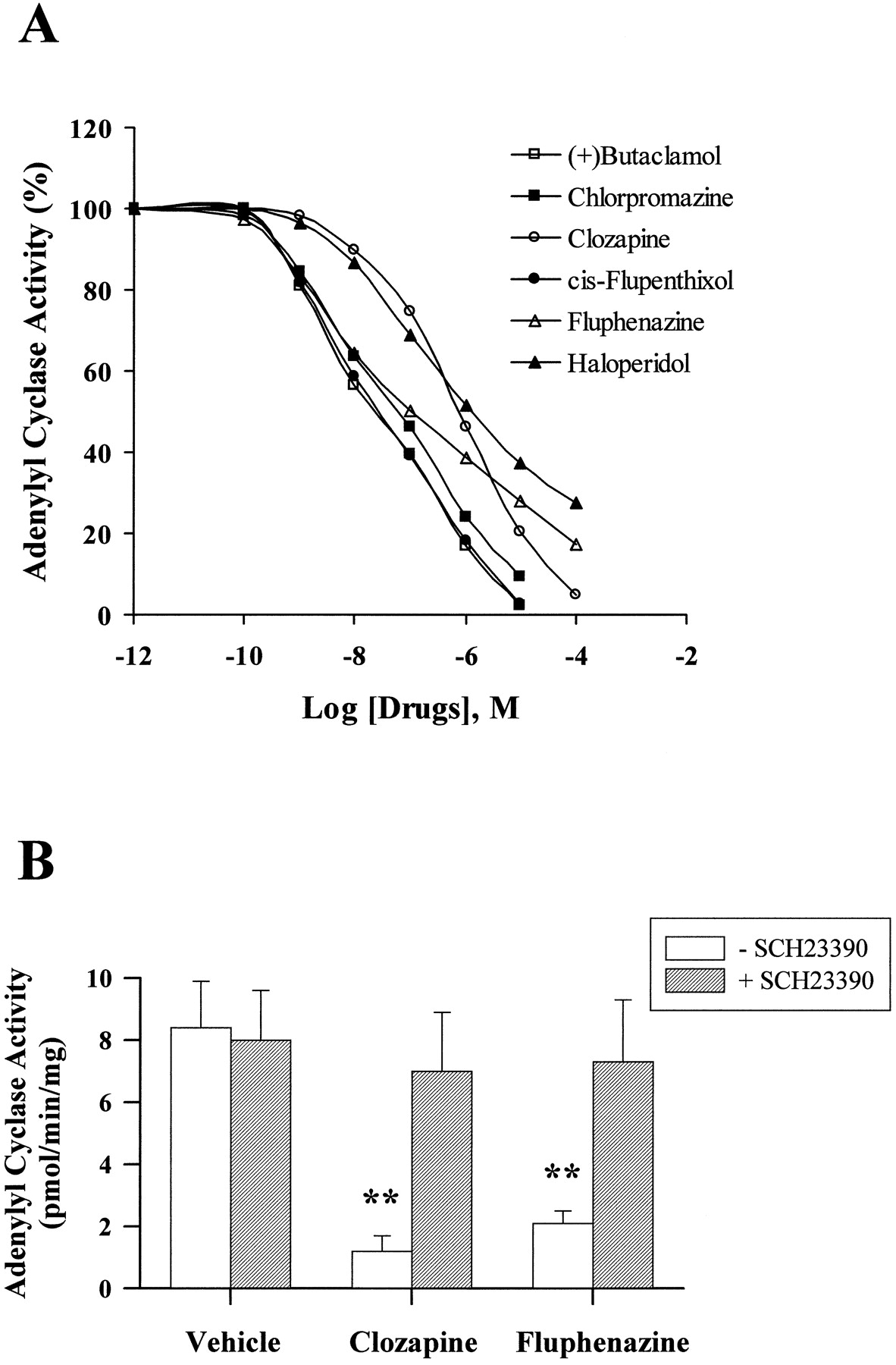
Effect of dopaminergic antagonists on constitutive adenylyl cyclase activity in PC2 cell membranes The constitutive activity was defined by subtracting basal adenylyl cyclase activity in wild-type PC2 cells from that in PC2 cells expressing D1Areceptors. A, membrane protein (50 μg) obtained from PC2 cells expressing D1A dopamine receptors was incubated at 30°C for 10 min with dopaminergic antagonists in the presence of 1 μCi [α-32P]ATP and formed [32P]cAMP was determined. The antagonists (+)-butaclamol, chlorpromazine, clozapine, fluphenazine, cis-flupenthixol, and haloperidol, in doses ranging from 0.1 nM to 100 μM, inhibited constitutive adenylyl cyclase activity. SCH23390, (−)-butaclamol, andl-sulpiride, tested at 10 μM each, were ineffective in inhibiting cyclase activity. B, PC2 cell membranes were preincubated with 10 μM SCH23390 at 30°C for 10 min before addition of buffer or 10 μM clozapine or fluphenazine and adenylyl cyclase activity was assessed. The D1 dopamine receptor antagonist SCH23390 inhibited clozapine- or fluphenazine-mediated inhibition of constitutive adenylyl cyclase activity. The results are the mean ± S.E. obtained from four to six independent experiments. **,p < .01 compared with the vehicle treatment.
The potencies and efficacies of dopaminergic antagonists in inhibiting constitutive adenylyl cyclase activity

Effect of dopaminergic antagonists on basal adenylyl cyclase activity in membranes of wild-type PC2 cells and D1A dopamine receptor-expressing PC2 cells. Membrane proteins (50 μg) were incubated at 30°C for 10 min with dopaminergic antagonists in the presence of 1 μCi of [α-32P]ATP and formed [32P]cAMP was determined. Clozapine, fluphenazine, and haloperidol, in doses of 10 μM decreased basal adenylyl cyclase activity in PC2 cells expressing the D1A dopamine receptor but not in the untransfected wild-type PC2 cells. Each bar represents the mean ± S.E. obtained from four individual experiments. *, p < .05 compared with the basal adenylyl cyclase activity in untransfected wild-type PC2 cells. +, p < .05 compared with the vehicle treatment.
Binding of Dopaminergic Antagonists to D1A Dopamine Receptor.
The binding affinities of the dopaminergic compounds for the D1A dopamine receptor were assessed by their ability to compete for [125I]SCH23982 binding sites in PC2 cell membranes expressing D1Adopamine receptors. (+)-Butaclamol, haloperidol,cis-flupenthixol, fluphenazine, chlorpromazine, and clozapine inhibited [125I]SCH23982 binding in a dose-dependent manner, yielding inhibitory dissociation constants (K i) in the range of 0.8 to 60 nM (Fig. 1B, Table 3). On the other hand, (−)-butaclamol and SCH23390 displayed a large difference in their affinities for the D1A receptor (K i = 9 μM and 0.28 nM, respectively). Binding properties of these dopaminergic compounds at the D1A dopamine receptor were consistent with those reported in previous studies on brain membranes (Dearry et al., 1990;Kanba et al., 1994; Sugamori et al., 1994).
Ki values for ligands in competing for [125I]SCH23982 binding to D1A dopamine receptors in transfected PC2 cell membranes
Effects of Dopaminergic Antagonists on D1A Dopamine Receptor-G Protein Coupling.
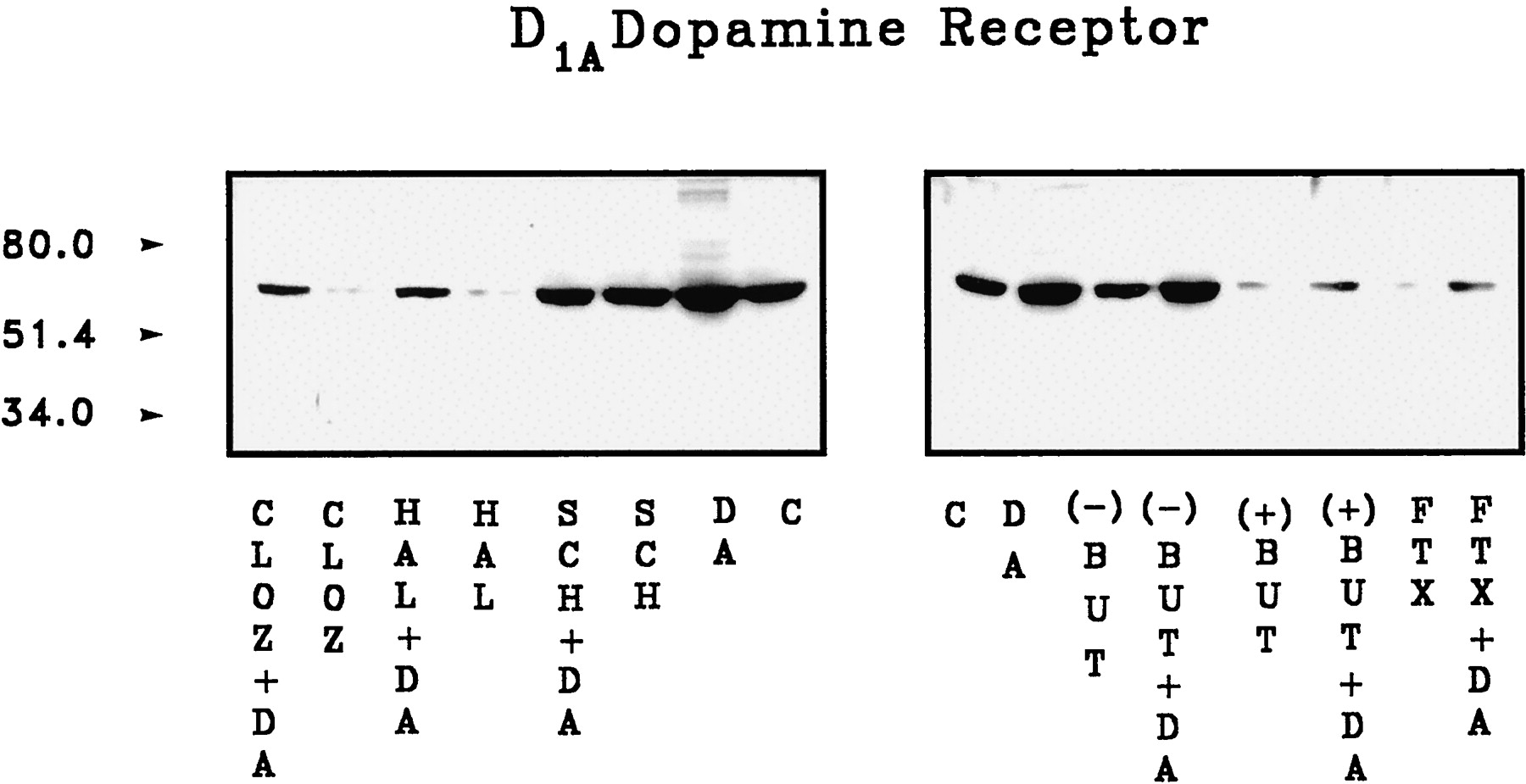
The coupling of D1A dopamine receptors to Gs protein was assessed by monitoring D1A dopamine receptor protein that coimmunoprecipitated with Gsα protein. The D1A dopamine receptor protein was detected in immunoblots using a monoclonal D1A dopamine receptor antibody. The specificity of this antibody was supported by the fact that a single 60-kDa band was observed in membranes of D1A dopamine receptor-transfected PC2 cells but not in membranes of untransfected wild-type PC2 cells (Fig.4). Significant basal coupling of receptor with Gsα protein was found in membranes of PC2 cells expressing D1A dopamine receptors. Receptor-Gsα coupling was enhanced by exposing the membranes to 1 μM dopamine. On the other hand, treatment with 100 μM 5′-guanylylimidodiphosphate [Gpp(NH)p] reduced D1A dopamine receptor-Gsα coupling (Fig. 4). (+)-Butaclamol,cis-flupenthixol, clozapine, haloperidol, and fluphenazine at concentrations of 1 μM reduced D1A dopamine receptor-Gsα protein coupling by 70 to 80%. In contrast, no apparent effect on coupling was found when 1 μM (−)-butaclamol or 1 to 10 μM SCH23390 were tested (Fig.5A). Furthermore, as shown in Fig. 5B, clozapine and haloperidol reduced D1A dopamine receptor-Gsα protein coupling in a dose-dependent manner. The effects of these compounds on coupling were not attributable to immunoprecipitation efficiency because comparable Gsα protein levels were detected when the immunoblots were probed with Gsα antiserum (Fig. 5, A and B, bottom).
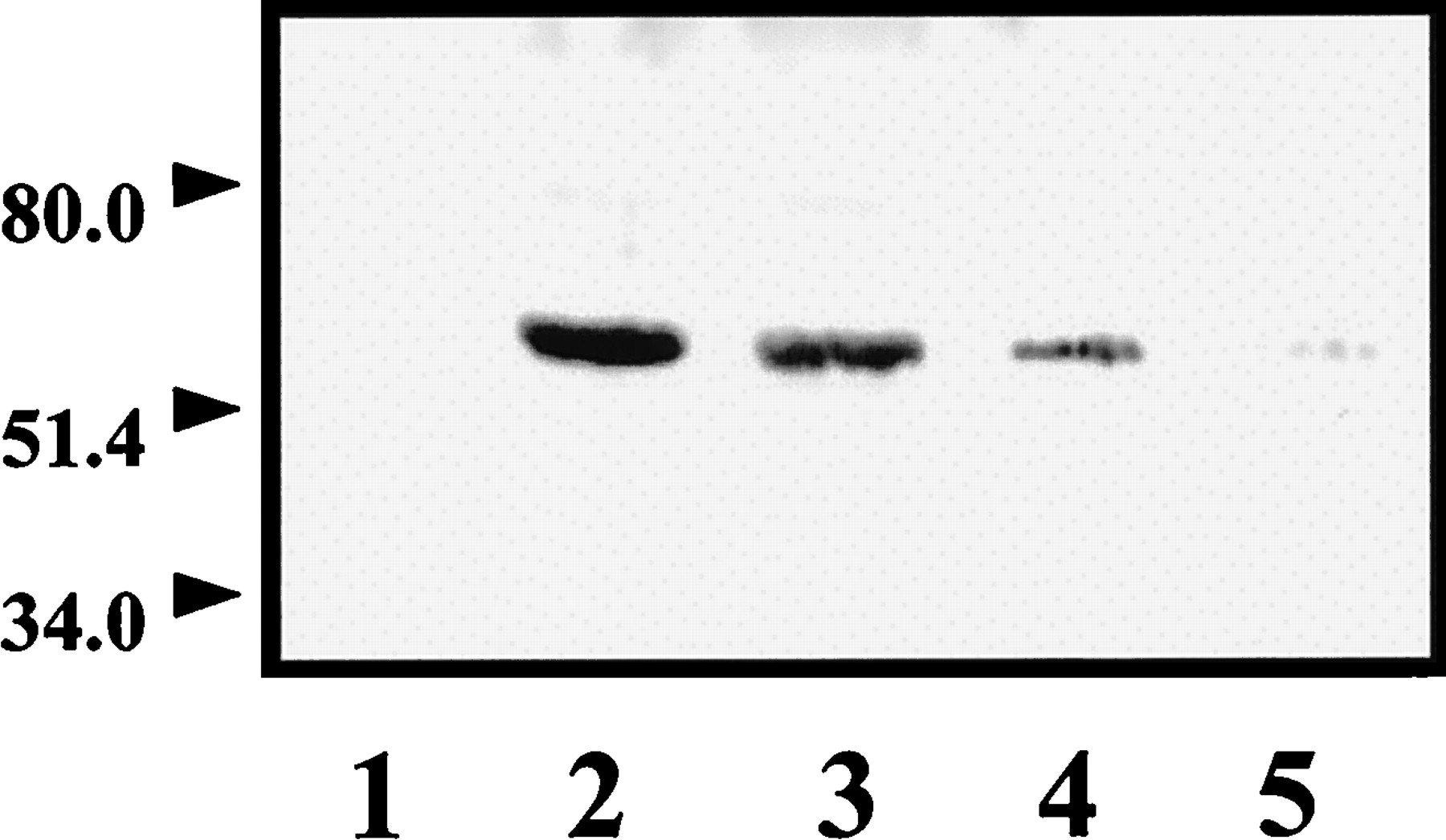
D1A dopamine receptor immunoblots of wild-type PC2 cells and D1A dopamine receptor-expressing PC2 cells. Membrane proteins (50 μg) obtained from wild-type PC2 cells (lane 1) and D1A dopamine receptor-expressing PC2 cells (lane 2) were immunoblotted with an anti-D1A dopamine receptor antibody. In the immunoprecipitation experiment, 200 μg of membrane protein obtained from PC2 cells expressing D1Adopamine receptors was incubated at 30°C for 5 min in the absence (lane 3) or presence (lane 4) of 1 μM dopamine. Membrane protein (200 μg) incubated at 30°C for 15 min with 100 μM Gpp(NH)p was shown in lane 5. Membranes were solubilized, immunoprecipitated with Gαs antiserum, and 50 μg of original membrane proteins were immunoblotted with an anti-D1A dopamine receptor monoclonal antibody. The data indicate the absence of detectable D1A dopamine receptor protein in wild-type PC2 cell membrane, whereas D1A dopamine receptor cDNA-transfected cells show a clear 60-kDa band. D1A dopamine receptor protein was also detected in Gsα immunoprecipitates of transfected cell membranes; the amount of receptor protein coprecipitated with Gsα protein was increased by stimulation with dopamine and reduced by exposure to Gpp(NH)p.

Effect of dopaminergic antagonists on D1A dopamine receptor-Gαs protein coupling. A, membrane protein (200 μg) obtained from PC2 cells expressing D1A receptors was incubated at 30°C for 5 min without (C) or with 1 μM (−)-butaclamol [(−)BUT], (+)-butaclamol [(+)BUT], cis-flupenthixol (FTX), clozapine (CLOZ), haloperidol (HAL), fluphenazine (FPZ), or with 1 or 10 μM SCH23390 (SCH). Membranes were solubilized, immunoprecipitated with Gαs antiserum and the immunoprecipitates subjected to immunoblotting with an anti-D1A dopamine receptor monoclonal antibody (top) or with Gsα antiserum (bottom). The effects of increasing concentration (1–1000 nM) of haloperidol or clozapine are shown in B. The results indicate that (+)-butaclamol,cis-flupenthixol, clozapine, haloperidol, and fluphenazine reduce D1A dopamine receptor-Gsαcoupling by 70–80%, whereas (−)-butaclamol and SCH23390 do not affect the coupling. The results are representative of four independent experiments.
The coupling of D1A dopamine receptor with Gsα protein was enhanced by incubating membranes with dopamine, reflecting an increase in receptor-stimulated transmembrane signaling. Clozapine, haloperidol, (+)-butaclamol,cis-flupenthixol, and SCH23390 blocked dopamine-enhanced D1A dopamine receptor-Gsαprotein coupling. Furthermore, the combination of dopamine and the inverse agonists resulted in coupling at a level lower than basal. However, the neutral antagonist SCH23390 prevented the dopamine-induced effect on D1A dopamine receptor-Gsα protein coupling without reducing coupling to a level below control (Fig.6).

Effects of dopaminergic antagonists on dopamine-induced increase in D1A dopamine receptor-Gαs protein coupling. Membrane protein (200 μg) obtained from PC2 cells expressing D1A dopamine receptors was incubated at 30°C for 5 min with 1 μM (−)-butaclamol [(−)BUT], (+)-butaclamol [(+)BUT], cis-flupenthixol (FTX), clozapine (CLOZ), haloperidol (HAL), and SCH23390 (SCH) in the absence (C) or presence of 1 μM dopamine (DA). Membranes were solubilized, immunoprecipitated with Gαs antiserum and the immunoprecipitates subjected to immunoblotting with an anti-D1A dopamine receptor monoclonal antibody. The results indicate that (+)-butaclamol, cis-flupenthixol, clozapine, haloperidol, and SCH23390 inhibit DA-induced increase in D1A dopamine receptor-Gsα coupling, whereas (−)-butaclamol does not show this inhibitory action. The results are representative of four independent experiments.
Discussion
Overexpression of G protein-coupled receptors enhances basal receptor activity, thus facilitating the assessment of the nature of the interaction of a ligand with its receptor-linked signal transduction pathway (Adie and Milligan, 1994; Barker et al., 1994;Chidiac et al., 1994; Bond et al., 1995; Barr and Manning, 1997). In the present experiments, the introduction of D1Adopamine receptors into PC2 cells, which ordinarily do not express this receptor, resulted in increased agonist-independent constitutive adenylyl cyclase activity. In this system, adenylyl cyclase activity was enhanced by receptor stimulation with dopamine and was inhibited, in a receptor-specific manner, by several dopaminergic antagonists, including (+)-butaclamol, chlorpromazine, clozapine, fluphenazine,cis-flupenthixol, and haloperidol. The D1A dopamine receptor-dependent constitutive adenylyl cyclase activity was, however, not affected by other dopaminergic antagonists such as the selective D1dopamine receptor antagonist SCH23390, the inactive isomer of butaclamol, (−)-butaclamol, or the D2 dopamine receptor antagonist l-sulpiride. The selective action of these dopaminergic antagonists on constitutive adenylyl cyclase activity was further supported by the fact that these antagonists did not affect adenylyl cyclase activity in untransfected wild-type PC2 cells. This profile of action of the tested compounds at the D1A dopamine receptor supports their classification as agonist (dopamine), inverse agonist [chlorpromazine, (+)-butaclamol, clozapine, fluphenazine, cis-flupenthixol, and haloperidol], or neutral antagonist (SCH23390). These compounds demonstrated a range of affinities for the D1Adopamine receptor that did not parallel their action on cyclase activity. Nonetheless, the inverse agonist activity of the dopaminergic agents is related to their affinity for the D1Adopamine receptor, because the specific and neutral D1dopamine receptor antagonist SCH23390 prevented their actions. Thus, the antipsychotic dopaminergic antagonists do not behave as simple competitive antagonists but also exert inverse agonist activity at the D1A dopamine receptor. These drugs, therefore, may be expected to be highly effective in inhibiting signals transduced by D1A dopamine receptors even at low levels of receptor occupancy compared with the simple competitive receptor blockers. A recent study has demonstrated that some dopaminergic antagonists also act as inverse agonists at D2dopamine receptors (Hall and Strange, 1997). It is possible, therefore, that the therapeutic actions of antipsychotic drugs may be mediated by inverse agonist activity at either the D1A or D2 dopamine receptor or both. Thus, inverse agonist properties of dopaminergic antagonists should be considered together with their affinities for the dopamine receptors to more fully understand the mechanism of action of these drugs in the pharmacotherapy of schizophrenia and other disorders that may be related to a hyperdopaminergic state.
The two-state model of receptor activation suggests that G protein-coupled receptors exist in active (R*) and inactive (R) forms that are in dynamic equilibrium (Leff, 1995). The active form may exist in the absence of receptor occupancy, reflecting spontaneous or constitutive receptor activity (Lefkowitz et al., 1993). The active form of G protein-coupled receptors has a high binding affinity for receptor agonists and is believed to be precoupled to G proteins (Lefkowitz et al., 1993). Agonists elicit increased receptor-G protein coupling, whereas inverse agonists might be expected to affect a reduction in receptor-G protein coupling. In the present study, a coimmunoprecipitation technique was used to directly assess the effect of dopamine, Gpp(NH)p, and potential inverse agonists on D1A dopamine receptor-G protein coupling. Although the agonist, dopamine, increased the coupling of D1A dopamine receptors to Gsα protein, Gpp(NH)p and the dopaminergic inverse agonists were found to reduce the basal association of D1A dopamine receptors with Gsα protein, and the neutral D1A dopamine receptor antagonist SCH 23390 failed to directly influence this coupling, although it prevented the interaction of inverse agonists with the D1Areceptors. Furthermore, the inverse agonists blocked the dopamine-induced increase in D1A receptor-G protein coupling, resulting in a coupling level lower than that found under basal conditions, suggesting that the uncoupling induced by the inverse agonists can not be easily surmounted by receptor stimulation. When we consider these results in the framework of the two-state receptor model, in which active and inactive conformations of a receptor exist in equilibrium, the compounds that bind to the D1A dopamine receptors are classified as agonists if they increase the receptors that are in the active state. Inverse agonists shift the equilibrium in favor of the inactive form of the receptor, and neutral antagonists do not change the ratio of active to inactive receptors. Thus, an increase in receptors that are in the active state by dopamine enhances the coupling of D1A dopamine receptors to their associated G protein; inverse agonists, on the other hand, are more likely bound to the inactive form of the D1A receptor and shift the equilibrium toward the inactive form of the receptor, dissociating precoupled receptors from Gs protein. Neutral antagonists do not alter the ratio of active and inactive receptors or the coupling state of the D1A receptor to Gs protein. Westphal and Sanders-Bush (1994) have previously demonstrated that inverse agonists bind to the uncoupled form of the 5-HT2C receptor with a higher affinity than to the G protein-coupled form of the receptor, and agonists have a high affinity for the coupled rather than the uncoupled form of the receptor. The present results directly demonstrate that D1A dopamine receptor inverse agonists negatively regulate the coupling of the D1A dopamine receptor with Gs proteins. This is probably mediated by their binding to the inactive form of the receptor and shifting the equilibrium of receptors from the active to the inactive form.
In conclusion, the results of this study indicate that some classical dopaminergic antagonists inhibit constitutive adenylyl cyclase activity and behave as inverse agonists of D1A dopamine receptors. The mechanism for the apparent inverse agonist activity of these dopaminergic antagonists is related to their ability to reduce D1A dopamine receptor-Gsprotein coupling. This newly described action of the antipsychotic drugs should be considered as an alternative mechanism or an additional action of these agents that contributes to the therapeutic action of these drugs in the treatment of neuropsychiatric disorders.
Footnotes
- Received February 15, 1999.
- Accepted August 9, 1999.
-
Send reprint requests to: Eitan Friedman, Ph.D., Department of Pharmacology, MCP Hahnemann School of Medicine, 3200 Henry Avenue, Philadelphia, PA 19129-1137. E-mail:friedmane{at}mcphu.edu
-
This study was supported by United States Public Health Service Grants NS29514, from the National Institute of Neurological Disorders and Stroke, and T32-AG00131, from the National Institute on Aging.
Abbreviations
- bp
- base pair(s)
- PMSF
- phenylmethylsulfonyl fluoride
- PBST
- phosphate-buffered saline/Tween 20
- Gpp(NH)p
- 5′-guanylylimidodiphosphate
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}