


Abstract
We have suggested previously that both the negatively and positively charged residues of the highly conserved Glu/Asp-Arg-Tyr (E/DRY) motif play an important role in the activation process of the α1b-adreneric receptor (AR). In this study, R143 of the E/DRY sequence in the α1b-AR was mutated into several amino acids (Lys, His, Glu, Asp, Ala, Asn, and Ile). The charge-conserving mutation of R143 into lysine not only preserved the maximal agonist-induced response of the α1b-AR, but it also conferred high degree of constitutive activity to the receptor. Both basal and agonist-induced phosphorylation levels were significantly increased for the R143K mutant compared with those of the wild-type receptor. Other substitutions of R143 resulted in receptor mutants with either a small increase in constitutive activity (R143H and R143D), impairment (R143H, R143D), or complete loss of receptor-mediated response (R143E, R143A, R143N, R143I). The R413E mutant displayed a small, but significant increase in basal phosphorylation despite being severely impaired in receptor-mediated response. Interestingly, all the arginine mutants displayed increased affinity for agonist binding compared with the wild-type α1b-AR. A correlation was found between the extent of the affinity shift and the intrinsic activity of the agonists. The analysis of the receptor mutants using the allosteric ternary complex model in conjunction with the results of molecular dynamics simulations on the receptor models support the hypothesis that mutations of R143 can drive the isomerization of the α1b-AR into different states, highlighting the crucial role of this residue in the activation process of the receptor.
The α1b-adrenergic receptor (AR) belongs to the superfamily of G protein-coupled receptors (GPCR), which transduce signals across the cell membrane. Stimulation of the α1b-AR by catecholamines can activate proteins of the Gq/11 family, resulting in phospholipase C-mediated production of inositol phosphates (IP).
All GPCR sequences share the presence of seven transmembrane (TM) α-helices connected by alternating intracellular and extracellular hydrophilic loops (Wess, 1997). The seven TM helices contribute to the formation of the ligand binding pocket, whereas amino acid sequences of the intracellular loops mediate the interaction of the receptor with a number of signaling and regulatory proteins, including G proteins, arrestins, and GPCR kinases.
Within the large superfamily of GPCRs, a small number of amino acids are highly conserved throughout evolution. The Glu/Asp-Arg-Tyr motif (E/DRY motif) located at the cytosolic end of helix 3 occurs in the majority of GPCRs belonging to the “rhodopsin-like” subfamily. The high degree of conservation of this motif suggests that it must play an important role in receptor function. Indeed, several experimental and modeling studies highlighted that both negatively and positively charged residues of the E/DRY are involved in the activation process of GPCRs (Oliveira et al., 1994; Wess, 1997).
Recent studies have demonstrated that mutations of the aspartate of the E/DRY motif can induce variable levels of constitutive (agonist-independent) activity for rhodopsin (Cohen et al., 1993;Acharya and Karnik, 1996), the α1b-AR (Scheer et al., 1996, Scheer et al., 1997), and the V2 receptor for vasopressin (Morin et al., 1998). Interestingly, mutations of the homologous aspartate in the gonadotropin-releasing hormone receptor into asparagine enhanced the agonist-induced receptor response (Arora et al., 1997). In the muscarinic M1 receptor, some mutations of the homologous aspartate dramatically decreased receptor expression (Lu et al., 1997). However, for those receptor mutants that were expressed, the agonist-induced receptor response was similar to if not greater than that of the wild-type receptor.
Site-directed mutagenesis studies that have targeted the conserved arginine of E/DRY in different GPCRs have so far mainly identified receptor mutants modestly or severely impaired in their ability to mediate a signaling response. Indeed, it has been demonstrated that mutations of this conserved arginine impaired the response mediated by GPCRs linked to different signaling pathways, including rhodopsin (Franke et al., 1992; Acharya and Karnik, 1996), the M1 and M2 cholinergic receptors (Zhu et al., 1994; Jones et al., 1995), the V2 vasopressin receptor (Rosenthal et al., 1993), the α1b-AR (Scheer et al., 1996), and the gonadotropin-releasing hormone receptor (GnRH-R) (Arora et al., 1997;Ballestreros et al., 1998). These findings suggest that the invariant arginine plays a pivotal role in receptor-mediated activation of downstream signaling proteins. However, the mechanistic role played by the arginine of the E/DRY sequence in receptor activation remains unknown.
Recently, we combined experimental and computer-simulated mutagenesis of the α1b-AR to build a theoretical model of receptor activation (Scheer et al., 1996; Scheer et al., 1997). The comparative molecular dynamics (MD) analysis of the wild-type α1b-AR and several constitutively active receptor mutants highlighted a network of hydrogen-bonding interactions among conserved polar residues forming a “polar pocket” near the cytosol (N63 in helix 1, D91 in helix 2, N344 and Y348 in helix 7) and R143 of the E/DRY motif. We suggested that 1) this set of interactions constrains the receptor in its ground state by controlling the degree of cytosolic exposure attainable by R143 and 2) the main role of R143 is to mediate receptor activation, allowing amino acids of the intracellular loops to attain the right configuration for the formation of a site with docking complementarity with the G protein.
In this study, we performed a series of conservative and nonconservative mutations of R143 to further elucidate its role in the activation process of the α1b-AR. Various substitutions of R143 resulted in receptor mutants with either increased constitutive activity, impairment, or complete loss of receptor-mediated response. Interestingly, all the mutants displayed increased affinity for agonist binding compared with the wild-type α1b-AR. A correlation was found between the extent of the affinity shift and the intrinsic activity of the agonists. The analysis of the receptor mutants conducted using the allosteric ternary complex model suggests that mutations of R143 can drive the α1b-AR into different states, supporting the crucial role of this residue in the activation process of the receptor.
Experimental Procedures
COS-7 Cell Culture and Transfections.
COS-7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and gentamicin (100 μg/ml) and transfected using the DEAE-dextran method. The cDNA encoding the hamster α1b-AR (Cotecchia et al., 1992) and its mutants were subcloned in pRK5. For inositol phosphate determination, COS-7 cells (0.15 × 106) were plated in 12-well plates. For phosphorylation assays, cells were plated in 100-mm dishes (3 × 106 cells). The transfected DNA encoding the receptors was 0.5 to 3 μg/106cells.
Ligand Binding.
Membrane preparations derived from cells expressing the α1b-AR or its mutants and ligand binding assays using [125I]iodo-2-[β-(4-hydroxyphenyl)-ethylaminomethyl]tetralone (HEAT) were performed as described previously (Cotecchia et al., 1992). Prazosin (10−6 M) was used to determine nonspecific binding. [125I]HEAT concentration was 250 pM for measuring receptor expression at a single concentration and 80 pM for competition binding analysis. Saturation analysis and competition curves were analyzed using Prism 2.0 (GraphPAD Software, San Diego, CA).
IP Measurement.
Transfected cells were labeled for 12 h with myo-[3H]inositol at 4 μCi/ml in inositol-free DMEM supplemented with 1% fetal bovine serum. Cells were then preincubated for 10 min in PBS containing 20 mM LiCl, and then stimulated for 45 min with epinephrine. Total inositol phosphates were extracted and separated as described previously (Cotecchia et al., 1992).
32P-Labeling and Immunoprecipitation of the Receptors.
Transfected COS-7 cells were equilibrated in phosphate-free DMEM for 2 h and then incubated in the same buffer containing 32Pi (0.2 mCi/ml) for 2 h at 37°C. The incubation was then continued in the absence or presence of epinephrine as indicated and immunoprecipitation of the phosphorylated receptors was performed as described previously (Lattion et al., 1994). After autoradiography, the32P content of the gel slices containing the immunoprecipitated receptor was quantified (cpm) by liquid scintillation counting. A separate set of dishes were incubated under similar conditions, but in the absence of32Pi to measure receptor binding. The receptors were expressed at similar levels and the32P content (cpm) of different gel slices was directly compared for statistical analysis.
To compare the phosphorylation among different receptors, we firstly assessed that the efficiency of immunoprecipitation was similar for the wild-type α1b-AR and its mutants. Membranes from COS-7 cells expressing the receptors were photoaffinity-labeled with [125I]iodoazidoprazosin as described previously (Lattion et al., 1994) and immunoprecipitated with antibodies raised against the last 24 amino acids (residues 492–515) or the first 22 amino acids (residues 1–22) of the α1b-AR. After autoradiography, the125I content (cpm) of the gel slices containing the photoaffinity-labeled receptors before and after immunoprecipation were counted to calculate the efficiency of immunoprecipation. The percentage of immunoprecipitated receptor was 89% for the antibody against the carboxyl-terminal sequence used at 1:100 dilution and 70% for that against the amino-terminal sequence at 1:50 dilution. The efficiency of immunoprecipitation was similar for the wild-type and mutated receptors. The experiments described in this study were performed using the antiserum against the carboxyl-terminal sequence of the receptor.
Building of the Receptor Models and MD Simulations.
Modeling of the α1b-AR was achieved following an ab initio procedure as described recently (Fanelli et al., 1998). The building of the receptor model consisted of an iterative procedure starting with a comparative MD study on the transmembrane domains of seven GPCRs (Fanelli et al., 1995) The starting arrangement of the seven helix bundle was successively modified and the model was complicated progressively by adding the intracellular and extracellular domains. A many-step iterative procedure, characterized by the experimental validation of the model in each of the upgrading steps, was employed. The most stringent validation of the model was achieved by challenging its capability to interpret and predict the functional properties of an ever-increasing number of α1b-AR mutants. The latest version of the model used in this study includes new experimental information derived from the electron micrographs of 3-D frog rhodopsin crystals (Unger et al., 1997). The tilt of the helices in the α1b-AR input arrangement previously obtained was slightly modified according to the tilt angles of the seven helices estimated from the map of frog rhodopsin (Baldwin et al., 1997; Unger et al., 1997). In addition, the third intracellular loop (i3) of the α1b-AR was completed by the addition of residues 236 to 284 (Fanelli et al., 1999b). Different input arrangements were built performing translations and rotations of the helices and loops, as well as modifications of side-chain torsion angles. Among the large number of arrangements tested, we selected the input structure of the α1b-AR that, upon MD simulations, produced an average arrangement showing the best agreement with the experimental data available on GPCRs together with high-quality check scores. The selected input structure of the wild-type α1b-AR was used to produce the input structures for the mutants using the molecular graphics package QUANTA 96 (Molecular Simulations, Waltham, MA).
Minimizations and MD simulations of the receptor models were performed using the program CHARMm (Molecular Simulations). Minimizations were carried out using 1500 steps of steepest descent followed by a conjugate gradient minimization, until the root mean square (r.m.s.) gradient was less than 0.001 kcal/mol Å. A distance dependent dielectric term (ε = 4r) and a 12-Å nonbonded cutoff distance were chosen. The “united atom approximation” was used for computational efficiency.
The minimized coordinates of the wild-type α1b-AR and of the A293E constitutively active mutant were then used as starting points for 1050 ps of MD runs. The systems were heated to 300 K with 5°C rise per 6000 steps by randomly assigning velocities from the Gaussian distribution. After heating, the system was allowed to equilibrate for 34 ps and velocities were scaled by a single factor. The system was then subjected to 1ns of MD simulation and the reported results were collected every 0.5 ps. The bond lengths involving hydrogen atoms were constrained according to the SHAKE algorithm, allowing an integration time step of 0.001 ps. Integration of Newton's equation of motion was done using the Verlet algorithm. The secondary structure of the seven-helix bundle was preserved using NOE constraints with a scaling factor of 10. Different combinations of distance constraints were tested. These constraints were applied between the backbone oxygen atom of residue i and the backbone nitrogen atom of residue i + 4, excluding prolines. Because the first 100 ps were representative of the 1-ns period of simulation for both the wild-type α1b-AR and the A293E constitutively active mutant, for all the other mutants considered in this work, MD runs were reduced to 150 ps, using the same heating and equilibration setup as that employed for the longer simulations.
The structures, averaged over the first 100 ps of the equilibrated MD trajectory, were then minimized and used for comparative analysis. Solvent-accessible surfaces were computed with the QUANTA package.
Statistics.
Results are expressed as mean ± S.E. Statistical significance was assessed by paired Student's ttest.
Materials.
COS-7 cells were from American Type Culture Collection (Rockville, MD); DMEM, gentamicin, fetal bovine serum and restriction enzymes from Life Technologies, Inc. (Grand Island, NY);Taq polymerase from Boehringer Mannheim (Mannheim, Germany);125I]HEAT and [3H]inositol from DuPont-New England Nuclear (Boston, MA); epinephrine, norepinephrine, phenylephrine, methoxamine, oxymetazoline, and dopamine were from Sigma (St. Louis, MO); and cirazoline and prazosin from Research Biochemical International (Natick, MA).
Results
Mutation of R143 into Alanine Counteracts the Effect of Constitutively Activating Mutations.
We previously reported that the mutation of R143 to alanine profoundly impaired the α1b-AR-mediated IP response (Scheer et al., 1996). To further challenge the role of R143 in receptor activation, we introduced the mutation of R143 to alanine into either the D142A or the A293E constitutively active receptor. The constitutive activity of the receptor double mutants D142A/R143A and A293E/R143A was dramatically reduced compared with that of the D142A and A293E receptors. In addition, the epinephrine-induced IP response mediated by the D142A/R143A and A293E/R143A mutants was profoundly reduced compared with that of the wild-type α1b-AR (Table1). Thus, the effect induced by the inactivating mutation of R143 into alanine was dominant over that induced by activating mutations of either D142 or A293, resulting in significant impairment of both the constitutive and agonist-induced activity of the receptor.
IP accumulation in COS-7 cells was measured after incubation in the absence (Basal) or presence of 100 μM epinephrine (Epi) for 45 min. The basal IP is expressed as the percentage increase of IP levels over those of mock-transfected cells (control) in the absence of Epi. Rmax indicates the Epi-induced accumulation of IP expressed as percentage increase above the basal levels of cells expressing the individual receptors. Results of receptor expression and IP accumulation are the mean ± S.E. of several independent experiments, whose number is indicated in parentheses. TheK i of epinephrine (Epi), norepinephrine (Ne), phenylephrine (Phe), and cirazoline (Cir) were assessed in competition experiments using 80 pm [125I] HEAT . The K ivalues are from two to five independent experiments that did not differ by more than 40%.
Effects of Mutations of R143 on the Receptor-Mediated IP Response.
To further investigate its mechanistic role in receptor function, R143 was mutated into several amino acids differing in their charge, size, or hydropathy index (Table 1). All the mutated receptors were expressed in COS-7 cells at levels between 0.2 and 0.6 pmol/mg of protein. The receptor mutants were tested for their ability to mediate epinephrine-stimulated IP accumulation in transfected COS-7 cells.
The charge-conserving mutation of R143 into lysine not only preserved the maximal agonist-induced response of the α1b-AR, but also conferred high degree of constitutive activity to the receptor (Table 1 and Fig.1). In cells expressing the R143K mutant, the basal IP accumulation was over 200% above that of cells expressing the wild-type receptor. This constitutive activity of the R143K mutant was inhibited by the α1-antagonist prazosin (results not shown). The R143H mutant, carrying also a charge-conserving mutation of R143, displayed a small degree of agonist-independent activity and was modestly impaired in its ability to mediate the epinephrine-induced IP response (Table 1 and Fig. 1).

IP response of the α1b-AR and its mutants carrying substitutions of R143. COS-7 cells were transfected with the cDNA encoding the wild-type α1b-AR (WT) or its mutants R143K, R143H, R143D, R143A, R143N, R143I, and R143E. Cells were incubated in the absence (Bas) or presence of 10−4 M epinephrine (Epi) for 45 min. Total IP were measured as described underExperimental Procedures. Receptor expression measured in membrane preparations was in the range of 0.2 to 0.3 pmol/mg of protein for all receptors. The results are the mean ± S.E. of three independent experiments.
All the other receptors carrying mutations of R143 were dramatically impaired in their ability to mediate agonist-induced IP response. Whereas the R143A, R143D, or R143N receptors could still mediate a very small agonist-induced response, the R143I and R143E mutants displayed an almost complete loss of activity (Table 1 and Fig. 1). In contrast to R143K and R143H receptors, neither R143A, R143N, R143I, nor R143E displayed any constitutive activity. Only the R143D mutant showed a small but significant 40% increase of basal IP accumulation compared with the wild-type receptor, even though its ability to mediate agonist-induced response was considerably impaired.
Phosphorylation Properties of Receptors Mutated at Position 143.
A recent study reported that moderately uncoupled rhodopsin mutants carrying mutations of the arginine of the E/DRY motif displayed a higher level of light-induced phosphorylation than that of the wild-type receptor (Shi et al., 1998). Two of these mutants also displayed enhanced interactions with rhodopsin kinase and arrestin in the absence of 11-cis-retinal. These findings suggested the intriguing hypothesis that the lower level of transducin activation might result from enhanced phosphorylation and desensitization of the rhodopsin mutants. A small increase in basal phosphorylation was also reported for a V2 vasopressin receptor mutant, in which the mutation of the homologous arginine to histidine resulted in receptor uncoupling (Innamorati et al., 1997). Thus, to further characterize the properties of the α1b-AR mutants carrying mutations of R143, we measured the phosphorylation of the R143A, R143E, and R143K receptors in intact cells.
As shown in Fig. 2, for the constitutively active R143K receptor, both the basal and agonist-induced phosphorylation levels were significantly increased by 50% above those of the wild-type α1b-AR. The enhanced basal phosphorylation observed for the R143K mutant is reminiscent of the properties of other constitutively active adrenergic receptor mutants (Ren et al., 1993; Pei et al., 1994), which might represent substrates for GPCR kinases even in the absence of the agonist.

Phosphorylation of the α1b-AR and its mutants carrying substitutions of R143. COS-7 cells were transfected with the cDNA encoding the wild-type α1b-AR (WT) or its mutants R143A, R143E, and R143K. After labeling with32Pi, cells were incubated in the absence (Bas) or presence of 10−4 M epinephrine (Epi) for 15 min. Immunoprecipitation of the phosphorylated receptors and quantification of their 32P content was performed as described underExperimental Procedures. Receptor expression was about 0.3 pmol/mg of proteins for all receptors. Receptor phosphorylation is expressed as percent of the control, which indicates the32P content (cpm) of the WT receptor in the absence of epinephrine. The results are the mean ± S.E. of four independent experiments. a, P < .05 compared with the basal phosphorylation of each respective receptor; b, P< .05 compared with the basal phosphorylation of the WT.
For both the activation-defective mutants R143A and R143E, epinephrine did not significantly increase receptor phosphorylation above the basal level. However, basal phosphorylation was 40% greater than that of the wild-type receptor for the R143E, but not for the R143A mutant.
The fact that both the R143A and R143E mutants were impaired in their ability to undergo agonist-induced phosphorylation seems to rule out the possibility that enhanced phosphorylation and desensitization is the mechanism underlying decreased receptor-mediated response for mutations of R143. Indeed, these findings suggest that the R143E might represent a receptor conformation, which favors its phosphorylation in the absence of the agonist, despite the impairment in its signaling response.
These results also support the view that mutations of GPCRs can have diverging effects on the activation and regulatory properties of the receptor. We have recently shown that two constitutively active mutants of the α1b-AR displayed opposing phosphorylation features (basal phosphorylation was enhanced for the A293E, but not for the D142A mutant) (Mhaouty-Kodja et al., 1999). Here we report that the activation-deficient R143E mutant displays increased basal phosphorylation in a manner similar to that observed for some uncoupled mutants of rhodospin (Shi et al., 1998) and V2 receptor (Innamorati et al., 1997).
Structural-Dynamic Features of the Receptors Carrying Mutations of R143.
To further elucidate the structural and dynamic role of R143, MD simulations were performed on the α1b-AR model carrying mutations at R143. We have used a recently described upgraded three-dimensional model of the α1b-AR that incorporates several new pieces of experimental information available on GPCRs (Fanelli et al., 1998,1999b). To gain further insight into the receptor-G protein interface, the up-graded α1b-AR model included the entire i3 loop of the receptor (residues 236–284).
Following the same computational approach as reported previously (Scheer et al., 1996; Scheer et al., 1997; Fanelli et al., 1999b), the trajectories of the receptor mutants were compared with those of the wild-type α1b-AR on the one hand and with those of the constitutively active mutants D142A and A293E on the other. Consistent with the model of α1b-AR activation previously proposed, the average minimized structure of the wild-type receptor in the upgraded model is stabilized by a network of H-bonding interactions involving highly conserved polar amino acids in the helix bundle. Among these, the charge reinforced H-bonding interaction between D91 (helix 2) and R143 (helix 3) constitutes one of the constraining interactions in the receptor ground state. In the constitutively active mutants D142A and A293E, the motions of the helices, although triggered differently, induce the breakage of the D91-R143 interaction. Furthermore, there is a rearrangement of the cytosolic domains characterized by the opening of a solvent exposed site formed by the second intracellular loop (i2), the cytosolic extension of helices 5 and 6 as well as amino acids 242 to 259 forming an α-helical segment in the middle of the i3 loop (Fig.3). We recently proposed that this crevice, which is characterized by a large, solvent-accessible surface, represents a site with good electrostatic and shape complementarity with the G protein (Fanelli et al., 1999b).

Receptor models. The four panels display the results of MD simulations carried on the wild-type α1b-AR (ground state), the constitutively active mutants A293E and R143K, and the activation-deficient mutant R143E. All views are in a direction perpendicular to the helix main axis. The seven helices 1, 2, 3, 4, 5, 6, and 7 are colored in blue, orange, green, pink, yellow, sky blue, and violet, respectively. The i1, i2, and i3 loops are colored in green, white, and cyclamen, respectively, whereas the e1, e2, and e3 loops are colored in carnation. Each panel shows two views of the average minimized structure of each receptor together with several amino acids (colored according to their location) in the environment of the residue at position 143. In the middle of each panel, the solvent-accessible surface areas computed on i2, i3, and the cytosolic extensions of helices 5 and 6 are displayed.
Another “hallmark” of these mutation-induced active structures is the cytosolic exposure of cationic residues, including the pair R288 and K291 at the cytosolic extension of helix 6 (Fig. 3). These positively charged amino acids play an important role in receptor-mediated signaling as suggested experimentally by the fact that the double mutation of the R288-K291 pair to either alanines or glutamates impairs the α1b-AR-mediated IP response (Table 1).
As shown in Fig. 3, substitution of R143 with lysine produces an average arrangement that shares some structural similarities with the constitutively active mutants A293E and D142A (i.e., the cytosolic exposure of R288 and K291) as well as the configuration of the cytosolic crevice formed by the i2 loop, the cytosolic extension of helices 5 and 6, and the stretch of amino acids 242 to 259 in the middle of the i3 loop (Fig. 3). In fact, the shorter amino acid side chain that results from mutation of the arginine to lysine does not favor the interaction of this residue with D91 (helix 2), thus mimicking the breakage of the D91-R143 interaction observed in the A293E and D142A mutants.
Similar structural changes are also found in the weakly constitutively active mutant R143H, in which the protonated H143 is surrounded by a cluster of aromatic amino acids (F83, Y144, Y348, and F355), whereas D91 is involved in H-bonding interactions with N63 and N344 (results not shown). This interaction pattern results in the opening of a site in the cytosolic domains that is similar to that found in the structures of the constitutively active mutants D142A and A293E. However, the lower constitutive activity of R143H with respect to R143K may be attributable, at least in part, to the fact that a fraction of uncharged H143 might exist at physiological pH because it is close to positively charged amino acids that may contribute to decrease its pK a.
In the R143D mutant, which also displays a low level of constitutive activity, the interactions of D91 resemble those found in the R143H mutant (results not shown). However, although the cytosolic domains of R143D reveal a site exposed to the solvent, K291 in the i3 loop is less exposed to the cytosol than in the highly constitutively active mutant R143K because it interacts with the aspartate substituted at position 143. This might, at least in part, explain why the R143D mutant is only weakly constitutively active.
A striking feature of all the activation-deficient mutants (R143E, R143A, R143I, and R143N) is their failure to permit the translocation of positively charged amino acids of the i3 loop (i.e., R288, K291) toward the cytosol. In the case of R143E shown in Fig. 3, the i2 and portions of the i3 loop seem to delimit a site accessible to the solvent reminiscent of that observed in the active structures. However, two main features predict that the R143E mutant is inactive: 1) K291 is buried because of its interaction with E143 and 2) the conformation of the i3 loop is such that the stretch of amino acids 242 to 259 is arranged differently than that of the active receptor forms and might therefore not create a good electrostatic and shape complementarity with the G protein.
In accordance with the experimental findings (Table 1), the computer-simulated structures of the mutants D142A/R143A or A293E/R143A displayed none of the structural/dynamic “hallmarks” of either of the constitutively active mutants, D142A or A293E (results not shown). This suggests that the integrity of R143 is crucial in maintaining the structural-dynamic features of the receptor, which correlate with the high levels of constitutive activity induced by the mutations at either position 142 or 293.
The mechanistic role of the arginine belonging to the E/DRY motif has also been the focus of a recent study on the receptor for the GnRH-R (Ballestreros et al., 1998). This study has proposed that the inactive state of the receptor is characterized by a salt bridge between the arginine of the E/DRY motif and the adjacent aspartate, whereas the interaction between the same arginine and the conserved aspartate in helix 2 is a structural peculiarity of the active receptor state. The following arguments might explain why the mechanism of receptor activation proposed by Ballestreros et al. differs from the conclusions of our studies: 1) the model of the “inactive” receptor state proposed by Ballestreros et al. was inferred from Monte Carlo simulations on the isolated helix 3 of the GnHR-R, not on the entire receptor model. Given the absence of any other strongly attractive electrostatic field in the isolated helix 3, these calculations obviously revealed the propensity of the arginine of the E/DRY sequence to perform a charge reinforced H-bonding interaction with the adjacent aspartate. 2) In contrast, the model of the “active” receptor state proposed by Ballestreros et al. was not based on the results of computer simulations, but on those from site-directed mutagenesis studies on the GnRH-R and other GPCRs.
Mutations neutralizing the charge of the conserved aspartate, which is on helix 2 in most GPCRs and on helix 7 in the GnRH-R, can impair the signaling ability of several receptors. Ballestreros et al. interpret these findings as an evidence that in the active state of GPCRs, the aspartate might be engaged in a salt bridge with the arginine of the E/DRY sequence. In contrast, we propose that in the active states of the α1b-AR, the arginine of the E/DRY sequence is not engaged in a strong salt bridge interaction. Our hypothesis is consistent with the experimental results of the present study showing that replacing the fully conserved arginine with lysine, histidine, and aspartate, which are unlikely to interact with the conserved aspartate on helix 2, can result in constitutively active forms of the α1b-AR. Our hypothesis is also in accordance with recent findings showing that the mutations of the arginine of E/DRY with alanine produces a constitutively active form of the oxytocin receptor (Fanelli et al., 1999a).
On the basis of the above findings, one should also consider the hypothesis that the pattern of interactions involving the conserved aspartate in helix 2 (in helix 7 for the GnRH-R) might not be identical in all GPCRs. This is supported by the example of rhodopsin in which the mutation of the homologous aspartate (D83) into asparagine does not impair the transition from the MI to the MII state. In addition, spectroscopic experiments on rhodopsin have demonstrated that D83 is protonated both in the dark and in the MII state and is only weakly H-bonded in MII (Rath et al., 1993; Fahmy et al. 1993). Thus, D83 does not seem to play a major role either as an acceptor or donor group in the active state of rhodopsin.
Effects of Mutations at Position R143 on the Ligand Binding Properties of the Receptor.
To further characterize the receptor mutants at R143, ligand binding studies were performed on membranes from COS-7 cells expressing the wild-type α1b-AR or the mutated receptors (Table 1). Saturation binding analysis of [125I]HEAT indicated that the K d values were similar at the wild-type and mutated receptors, ranging from 80 to 120 pM (results of two independent experiments not shown). In contrast, the affinity of various agonists differed among the receptors.
All the receptor mutants displayed higher affinity for α-adrenergic agonists compared with the wild-type α1b-AR (Table 1). Consistent with its increased binding affinity, the potency of epinephrine at the R143K mutant was greater than at the wild-type receptor (EC50 values from one experiment were 2.9 × 10−8 M and 5 × 10−9 M at the α1b-AR and R143K mutant, respectively). In contrast, no significant differences were observed for the different receptors in the binding affinities of the antagonist prazosin (results not shown).
Surprisingly, the inactive mutants R143E and R143I exhibited the highest affinity for agonists in this series of receptor mutants. In particular, the affinity of the R143E mutant for epinephrine was 250-fold higher than that of the wild-type α1b-AR and even 10-fold higher than that of the constitutively active receptor R143K. Thus, the agonist-binding affinities of the R143 mutants seem to be negatively correlated with their activation properties. As shown in Table 1, the higher the affinity of the receptor mutant for catecholamine, the lower its ability to mediate agonist-induced IP response and vice versa.
Comparison of the Constitutively Active Receptors R143K and A293E with the Inactive Mutant R143E.
To better understand the nature of the increased affinity for agonist binding of the activation-defective R143 mutants, we compared the ligand-binding properties of the inactive receptor R143E with those of the two constitutively active mutants, R143K and A293E. First, we assessed the intrinsic activities of seven structurally different agonists; second, we measured the binding affinity of the various agonists at the wild-type α1b-AR, R143E, R143K, and A293E receptors.
The intrinsic activities for different agonists were computed by dividing the maximal IP response induced by a saturating concentration of each ligand with that of epinephrine in Rat-1 fibroblasts permanently expressing wild-type α1b-AR (Table2). It has been reported previously that both in human embryonic kidney 293 and SK-N-MC cells permanently transfected with the cDNA encoding the α1b-AR, the maximal IP response to norepineprhine was positively correlated with the expression of the receptor (Theroux et al., 1996). Similar findings were obtained in Rat-1 fibroblasts permanently expressing different levels of the α1b-AR (results not shown). These observations seem to rule out the presence of receptor reserve in cells permanently expressing the α1b-AR, suggesting that the measurement of intrinsic activity reflects the efficacy of different ligands.
IP accumulation was measured in Rat-1 cells permanently expressing the wild-type α1b-AR at 1 pmol/mg of protein after stimulation with agonists at a concentration of 10−4 M (epinephrine, norepinephrine, cirazoline, and oxymetazoline) or 10−3 M (phenylephrine, methoxamine, and dopamine) for 45 min. The intrinsic activity (IA) of the agonists was computed by dividing the maximal IP response induced by each ligand with that of epinephrine. The IA values are from three independent experiments. The K i of different agonists was assessed in membranes from COS-7 cells expressing the wild type α1b-AR or its mutants A293E, R143K, and R143E at about 0.2 pmol/mg of protein. The K ivalues are from two to five independent experiments that did not differ by more than 40%.
As shown in Table 2, with the exception of the imidazoline derivative oxymetazoline, all the agonists displayed greater affinity for the mutated receptors than for wild-type α1b-AR. Interestingly, for all the receptor mutants, there was a direct relation between the intrinsic activity of each agonist and the extent of its affinity shift (i.e., the ratio between theK i value at the mutated and wild-type α1b-ARs). The higher the intrinsic activity of the agonist, the greater was the affinity shift induced by the various mutations (Fig. 4). The two constitutively active mutations, A293E and R143K, produced similar affinity shifts for all the agonists tested, whereas the shifts induced by R143E were much greater. The affinity of epinephrine for the wild-type as well as for the three receptor mutants (A293E, R143K, and R143E) was not influenced by 10−4 M guanosine-5′-O-(3-thio)triphosphate (results not shown).
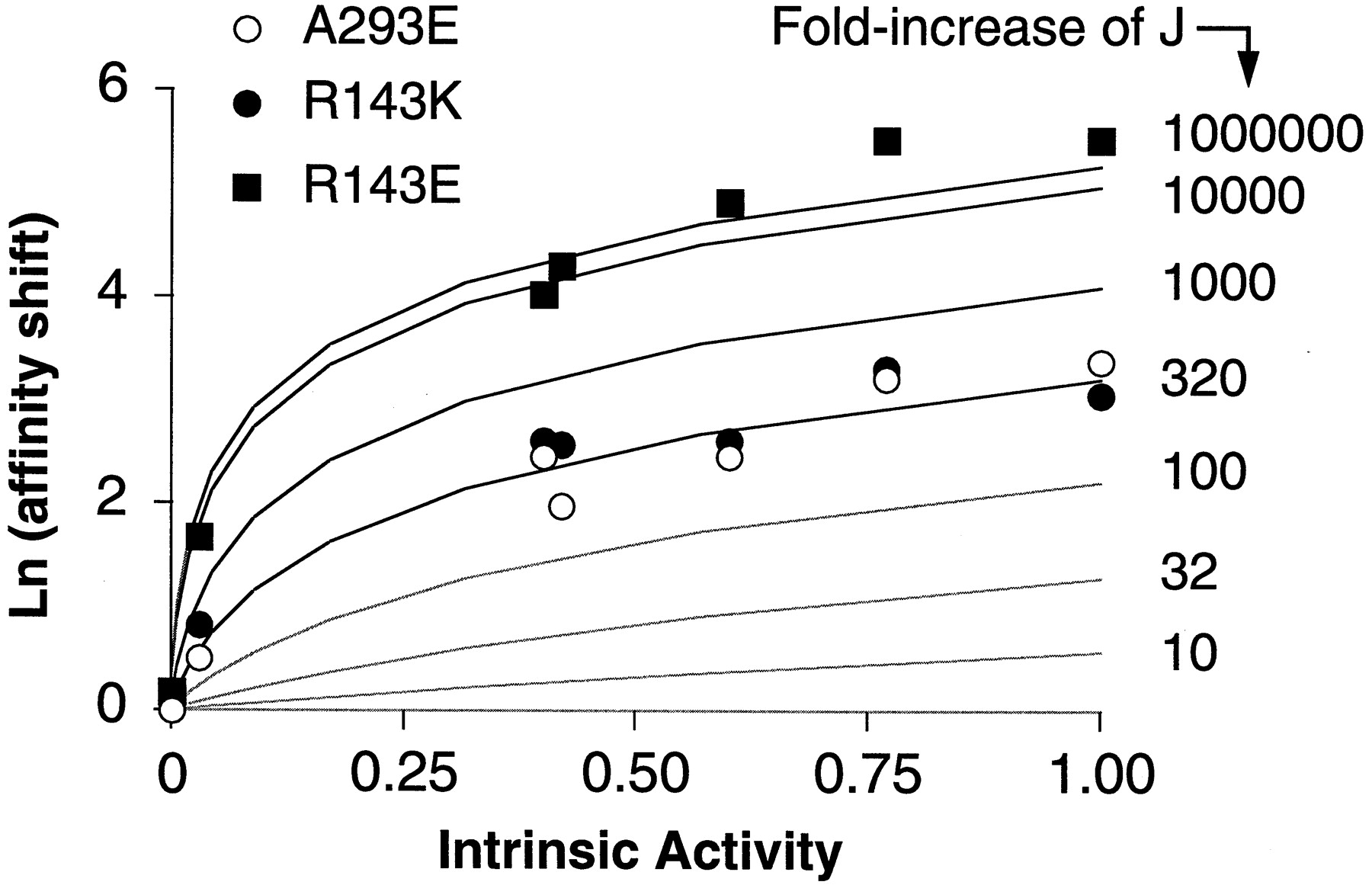
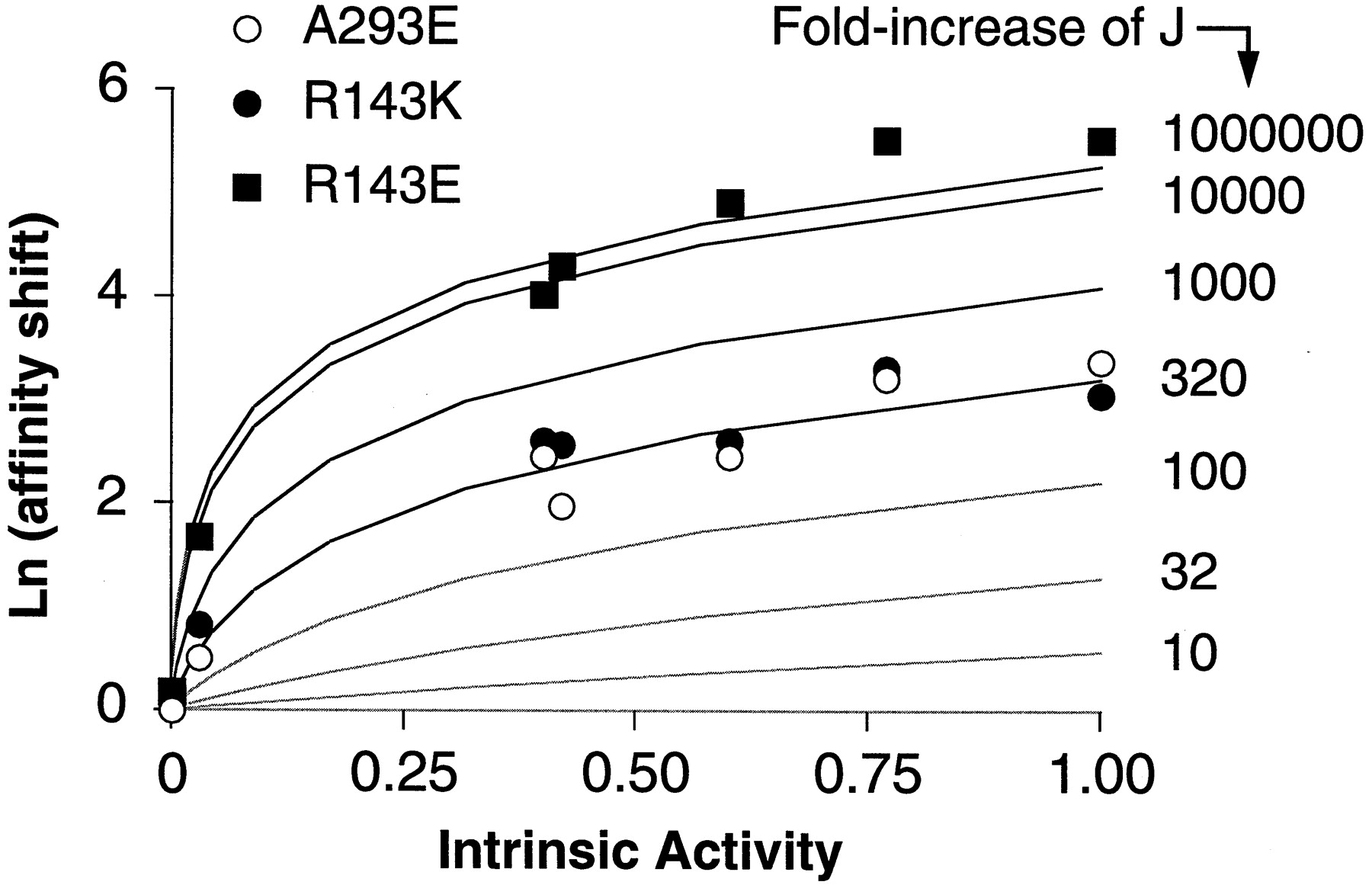
Relationship between the intrinsic activity and the affinity shift for various ligands at the receptor mutants A293E, R143K, and R143E. The values of K i and intrinsic activities are from Table 2. The affinity shift, expressed as Ln units, is the ratio between the K i value at the mutated and wild-type α1b-ARs. The experimental results were overlaid on the theoretical curves shown in Fig. 5 in the .
Analysis of the Relation Between Ligand Intrinsic Activity and Shift of Affinity.
As explained in the , the two-state allosteric model of receptor activation predicts that if a mutation alters the stability constant for the conversion of the inactive (R) to the active state (R*) (and thus basal activity), it also changes ligand binding affinity. The extent and direction of such an “allosteric” shift in affinity depends on ligand efficacy; therefore, it must be related to the intrinsic activity of the ligand (Samama et al., 1993).
To examine the relation between ligand intrinsic activity and increase in affinity induced by the three mutations, the affinity shifts caused by the mutations R143E, R143K, and A293E for the seven different agonists were plotted as a function of the agonist intrinsic activities measured at wild-type receptor (Fig. 4). The pattern of such a relationship closely resembles that predicted theoretically according to the allosteric receptor model as detailed in the (Fig. 5B). By overlaying theoretical curves on the experimental data (Fig. 4), we can estimate that all three mutants behave as if their J (i.e., the isomerization constant) value was enhanced by the mutation starting from a very small preexisting value of the wild-type receptor. The increase in isomerization constant is similar for the constitutively active mutants A293E and R143K, but it seems to be much greater for the activation-deficient mutant R143E. Therefore, compared with the constitutively active mutants R143K and A293E, the inactive mutant R143E exhibits a similar pattern but with a much greater efficacy-related shift in binding affinity. This means that R143E, although unable to activate the G protein, seems to have an isomerization equilibrium that is displaced toward the active form R* to an even greater extent than constitutively active receptors themselves. How can such paradox be explained?

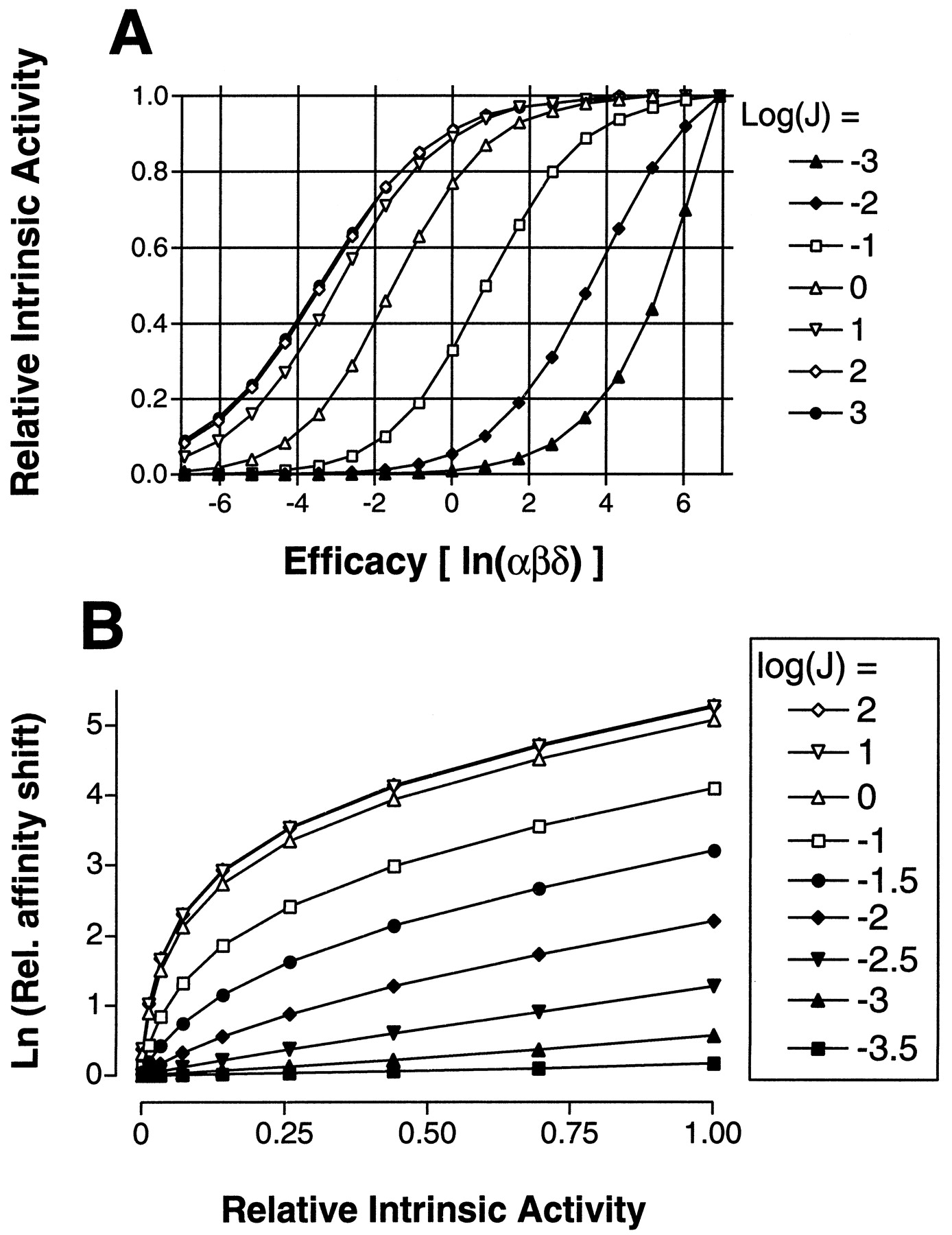
A, relationship between apparent intrinsic activity and “true” thermodynamic efficacy according to the two-state allosteric model. Data were generated according to eqs. 2b. Maximal effects for ligands of different efficacy (αβδ, as given on the x-axis) were computed for different values of J (shown as log values in the legend) and converted into “relative” intrinsic activity by dividing all of them for theE max of the ligand with the highest efficacy(αβδ =1000). B, effect of changingJ on the relation between shift in affinity and relative intrinsic activity. Shifts were calculated (eq. 4b) for different values of J (legend) and are relative to that of “wild-type” receptor (J=0.0001) for ligands of different efficacy as in A. Relative intrinsic activity (calculated as in A) refers to the “wild-type” receptor.
One simple explanation may be that the mutation of R143 to glutamic acid disrupts the interactions occurring at the receptor-G protein interface to such an extent that the association between the two proteins is severely reduced. In terms of the parameters of the allosteric model (see ), this implies that the R143E mutation enhances J but concurrently reduces the binding affinity M to the point that no receptor-mediated signaling can occur. However, because a reduction of M that could abolish signaling would also strongly reduce the affinity enhancement induced by J (see eqs. 2 and 4 in the ), this hypothesis could not explain the very large shift in agonist binding affinity observed for the R143E mutant.
In the full version of the allosteric model (“cubic ternary complex model”) (Weiss et al., 1996a), receptor association to the G protein and formation of a signaling competent complex can be partly dissociated if we assume that only the R*-bound G protein species can trigger the transduction pathway. In this case, efficacy has ligand-dependent and ligand-independent components (see ). The ligand-independent part (γ) controls the proportion of GPCR available for signaling, whereas the affinity constant M determines the extent of overall receptor association to G. Mechanistically, a reduction of γ at constant M can be interpreted as an impairment of the process that couples R-G association to G protein activation. As illustrated in the , if Jand γ are changed into opposite directions, being M constant, the result is a divergent effect on ligand affinity and ligand-mediated activation (Fig. 6). Thus, an alternative explanation is that the R143E mutation enhances J to a much greater extent than R143K and A293E mutations, but unlike those constitutive activating mutations, it also produces a dramatic reduction of the constant γ. This can produce very high agonist affinity and total loss of receptor-mediated signaling, as shown by simulations according to the model (Fig. 6). From a structural point of view, it is equivalent to state that R143E may be converted into a state that fully matches the high-affinity agonist binding conformation and also binds to the G protein, but in which the network of interactions that transmit conformational perturbation through the G protein interface is disrupted.

Simulation of the effect of “activating mutations” on biological responses (top) and binding isotherms (bottom) according to the two-state allosteric model. The simulated system consists of one receptor (10 pM), one G protein (10 pM), and two ligands, one of which is an antagonist (αβδ=1) and is present at a fixed “radiotracer” concentration (8 pM, K = 1 × 1011/M), whereas the other (agonist, αβδ=2790, K = 2 × 106/M) varies as indicated on thex-axis. The affinity constant for the interaction between receptor and G protein (M) was set as 3 × 1010/M. The concentrations of bound and free species were computed by a numerical procedure as explained in Samama et al. (1993). Biological response is given as fraction of G protein-boundR* form of the receptor; binding is expressed as fraction of bound “tracer” over its total concentration. Wild-type receptor is assumed to have a low initial J value (0.001). Two kinds of mutations are compared. Left, the mutations produce only enhancements of J without altering γ. Right, the mutations increase J to the same extent, but also decrease γ.
Discussion
In this study, we combined site-directed mutagenesis of the α1b-AR, computational simulations of receptor dynamics, and thermodynamic analysis of the receptor's pharmacological properties to provide hypotheses about the role played by R143 belonging to the conserved E/DRY motif in the activation process of the receptor.
Role of R143 in Receptor Activation.
Several lines of evidence support the conclusion that R143 of the α1b-AR is an important residue in the process of receptor activation. First, the inactivating mutation of R143 to alanine severely impaired both the constitutive and agonist-induced activity of the A293E and D142A mutants (Table 1). Secondly, a number of mutations of R143 resulted in a profound impairment of the agonist-induced receptor response (Fig.1). Finally, the fact that the R143A and R143E mutants were unable to undergo agonist-induced phosphorylation seems to exclude enhanced phosphorylation and desensitization as the main mechanism underlying the decrease in the receptor-mediated response for the mutants carrying neutral or negative amino acids at position 143.
Our findings suggest that a positive charge at position 143 of the α1b-AR is required for the agonist-induced receptor-mediated response. In fact, mutations of R143 with either neutral or negatively charged amino acids (Ala, Asn, Ile, Asp, and Glu) profoundly impaired the agonist-induced receptor response, which was preserved only in the R143K mutant (Fig. 1).
R143 Plays a Role in the Isomerization of the Receptor.
Our findings provide evidence that mutations of R143 enhance the stability constant (J), which governs the transition of the α1b-AR between the inactive (R) and active (R*) states (Samama et al., 1993).
First, evidence is provided by the fact that mutations of R143 can increase the constitutive activity of the receptor. In fact, the R143K mutant displayed several features characteristic of constitutively active receptors, including increased constitutive activity (Fig. 1), increased affinity for agonists (Table 1), and increased basal as well as agonist-induced phosphorylation (Fig. 2). A small increase in the constitutive activity could also be observed for the R143H and R143D mutants.
Second, evidence is provided by the fact that all the receptors mutated at R143 displayed high affinity for agonists. In particular, for both the constitutively active R143K and the activation-deficient R143E mutants, the affinity shift was positively correlated with the intrinsic activity of each agonist. Similar findings were also obtained for the previously described constitutively active mutant A293E (Fig.4). The analysis of the pharmacological properties of the mutated receptors using the full version of the allosteric model of receptor activation (“cubic” ternary complex model) (Weiss et al., 1996a) could provide some hypothesis about the effects of the mutations ( ).
The relationship between the affinity shift and the intrinsic activity of the ligands observed for the A293E, R143K, and R143E mutants suggests that the isomerization equilibrium constant of the receptor (J), starting from a very small pre-existing value of the wild-type receptor, is progressively increased for the different mutants [i.e., wild-type α1b-AR ≪ A293E = R143K ≪ R143E (Fig. 4)].
For A293E and R143K, the increase of the J value is consistent with the fact that these mutants are constitutively active [i.e., the isomerization equilibrium is displaced toward the active (R*) states]. However, the apparent large increase of J for the R143E mutant is in patent conflict with the lack of activity of this receptor (Fig. 1). As pointed out under Results, a concurrent reduction of the receptor affinity for the G protein (M) caused by the mutation could explain the loss of signaling, but it was not consistent with the large enhancement of agonist binding affinity observed for the R143E mutant. In the context of the “cubic ternary complex model” (Weiss et al., 1996a), the behavior of the R143E mutant can be explained by the fact that mutation of R143 to glutamic acid enhances J but also dramatically reduces the constant γ, which represents the effect of the G protein on the J constant. Such constant has a double connotation in the context of the model. First, it represents the contribution of the G protein in stabilizing the R* form of the receptor. However, because we assume that R*G and HR*G, but not RG and HRG, are biologically active species, γ also represents the interconversion into active state of receptor-G protein complexes and implicitly alludes to the process of receptor-mediated G protein activation. Thus, from a mechanistic point of view, a reduction of γ can either mean diminished affinity of R* for G or impaired ability of R* to activate G. We may speculate that the R143E isomerizes to a state that favors high affinity for agonists but is unable to either bind the G protein or induce its activation. Direct measurements of the G protein-binding properties of this mutant will be necessary to elucidate this point.
The results of MD simulations of the α1b-AR and its mutants support the hypothesis that R143 plays a role in receptor isomerization to different states. Based on the structural similarities among different constitutively active α1b-AR mutants carrying mutations of D142 or A293, we consider the opening of a cytosolic crevice as well as the exposure of a number of cationic residues, including R288 and K291 toward the cytosol, as some of the “hallmarks” that contribute to define the “active” states of the receptor (Fig. 3) (Fanelli et al., 1999b). The important role of the cationic R288 and K291 in receptor activation is supported by our experimental findings showing that mutations of these residues into either alanines or glutamates can impair the α1b-AR-mediated IP response (Table 1).
In all the structures carrying mutations of R143, the amino acids substituted at this position (Lys, His, Asn, Ile, Ala, Asp, and Glu) are no longer able to perform the constraining interaction with D91 in helix 2. Consequently, in most of these structures, the i2 and portions of the i3 seem to delimit a site exposed to the cytosol. However, only in the R143K, R143H, and R143D mutants, which display various levels of constitutive activity, does the arrangement and the orientation of the cytosolic loops form structures that are similar to those described previously for the “active” forms of the receptor (compare R143K and A293E in Fig. 3) (Fanelli et al., 1999b). On the other hand, in the activation-deficient mutants, the arrangement of the cytosolic loops is different than that of the active receptors, A293E and R143K (Fig. 3). Based on the structural features of R143E, we speculate that this mutant might represent an “active-like” state of the receptor (i.e., a conformational state that might interact with the G protein but is unable to mediate the activation of the G protein).
The fact that the agonist affinity shift for the R143E mutant was greater than for all the other mutated receptors suggests that mutation of R143 to glutamic acid results in the highest degree of isomerization of the α1b-AR. Interestingly, the R143E displayed a small but significant increase in basal phosphorylation, that was not observed for the other activation-deficient mutant, R143A (Fig. 2). Thus, R143E might represent a substrate for GPCR kinases and/or other regulatory proteins in the absence of agonist, despite being impaired in the receptor-mediated response. We speculate, therefore, that the small increase in basal phosphorylation might be another feature related to the isomerization of the receptor to an “active-like” state upon mutation of R143 to glutamic acid.
Conclusions.
Our findings demonstrate that different mutations of R143 in the α1b-AR result in apparently divergent functional effects. However, the results of thermodynamic analysis and of MD simulations of the receptor mutants could provide some hypothesis suggesting that R143 in the α1b-AR plays a role in receptor isomerization toward different states, which are best represented by the mutants R143K and R143E. The mutation of R143 to lysine seems to trigger receptor isomerization to an “active” state characterized by high affinity for agonists, increased basal and agonist-induced activity, enhanced basal and agonist-stimulated phosphorylation, and receptor conformers that predict productive receptor-G protein coupling. On the contrary, the mutation of R143 to glutamic acid seems to induce the isomerization of the receptor to an “active-like” state that shares some properties with the “active” forms (including high affinity for agonists, enhanced basal phosphorylation, and the opening of a cytosolic crevice between the i2 and i3 loops) but is severely impaired in its ability to mediate a response. The other mutants of R143 share features of both the R143K and R143E receptors.
Several of the features displayed by the α1b-AR mutated at R143 are similar to those reported for other GPCRs carrying mutations of the arginine of the E/DRY motif. Mutations of this conserved arginine impaired the response mediated by rhodopsin (Franke et al., 1992; Acharya and Karnik, 1996), the V2 vasopressin receptor (Rosenthal et al., 1993), and the GnRH-R (Arora et al., 1997;Ballestreros et al., 1998). In rhodopsin, however, the ability of the arginine mutants to activate transducin seemed to be influenced by the experimental conditions (membrane versus detergent-extracted proteins) used in different laboratories (Cohen et al., 1993). In the M1-muscarinic receptor (Jones et al., 1995) and in the 5-HT7 receptor (Obosi et al., 1997), mutations of the arginine caused both increased agonist affinity and reduced receptor-mediated response. On the other hand, the replacement of the arginine with histidine in the β2-AR increased the affinity of the mutated receptor for agonists without impairing receptor signaling (Seibold et al., 1998).
In conclusion, we consider that the apparent polymorphism of the phenotype, which can be associated with mutations of the arginine of E/DRY motif in different GPCRs, does not invalidate the hypothesis that this residue might play an important role in the activation process of various receptors. However, the true mechanistic role of the arginine of the E/DRY motif in the activation process of GPCRs remains to be determined.
A recent study on rhodopsin suggested that the homologous arginine (R135) is directly involved in promoting GDP release from transducin (Acharya and Karnik, 1996). This was mainly demonstrated by the fact that the rhodopsin mutant R135G could still bind a peptide derived from the carboxyl tail of transducin but failed to mediate light-induced GDP release. These findings seem to support the hypothesis of our MD simulations on the α1b-AR mutants showing that the conformational state of R143E might interact with the G protein but is unable to mediate the activation of the G protein.
Our experiments on the α1b-AR cannot directly assess whether mutations of R143 affect receptor-G protein binding and/or receptor-mediated G protein activation. However, our findings support the hypothesis that main role of the arginine of the E/DRY motif is to mediate the conversion of the receptor into the active state and dictate the configuration of the flexible cytosolic loops involved in the receptor-G-protein recognition process.
G Protein Interaction of a Two-State Allosteric Receptor.
Let's consider the interaction of a ligand (H) and a transducer protein (G) with a receptor that can exist in at least two interconverting conformations: the constrained (or inactive) formR and the relaxed (or active) form R*. At equilibrium, the system is described by the following set of reversible transitions and association reactions, each of which marks a numbered path in the reaction scheme drawn below (Scheme FS1).

The system is completely described by three “unconditional” constants and four allosteric “coupling” coefficients. The two second-order equilibrium constants K (path 1) andM (path 2) describe the associations of ligand and G protein to the R form of the receptor, respectively, whereas the first-order constant J (path 9) specifies the equilibrium between the two receptor states. Greek letters indicate the free-energy coupling factors. They state the conservation of free energy for concurrent chemical perturbation on the same protein. Pair-wise coupling between hormones or G protein association and receptor isomerization are given by β and γ, respectively. The coupling between the two association processes is given by α,whereas δ defines the linkage among the three concurrent processes (Weber, 1972).
As indicated in the scheme, there are four receptor species bound to the G protein; two involve the “constrained” form (RG and HRG) and two, the “relaxed” form (R*G and HR*G). Assuming that what is measured as response is proportional to the G protein-bound receptor species, the predictions of the model depend on whether we consider signaling competent all receptor G protein complexes or only those involving theR* form. For γM ≫ M and γJ ≫ J, G protein is always prevalently bound to R*; thus, the model can be simplified on the assumption that only the R* form of the receptor can bind G protein (Samama et al., 1993). The equilibrium scheme then reduces to only two thermodynamic cycles (indicated by thick arrow lines in Scheme FS1).
However, if we imagine that the R*-bound forms of the G protein are the only signaling competent species, trapping of receptor into “inactive” RG complexes becomes an important variable, and the predictions of the six-cycle model (cubic model; Weiss et al., 1996b) can differ significantly from those of the two-cycle simplification. The underlying assumption in this case is that the receptor can bind the G protein without triggering its activation, thus neither RG nor HRG contribute to biological activity. The parameter γ (i.e., the G protein effect on the transition R → R*) becomes a primary factor determining the emergence of biological output from the system, because it describes the conversion into active form of receptor-G protein complexes. Thus, by attributing activity only to R*-coupled G protein forms, γ acquires an additional connotation and hints to the process of receptor-mediated G protein activation.
Efficacy and Apparent Intrinsic Activity.
Efficacy is a pharmacological concept that describes the intrinsic property of a ligand to trigger biological output upon binding to the receptor. In chemical thermodynamics, efficacy is given by the sign and magnitude of the free energies that drive the system toward the active receptor species at equilibrium. Thus, the free-energy coupling constants are definitions of ligand's efficacy.
In the simplified form of the model, the two ligand-dependent coupling factors α and β are all that is needed to define efficacy (Samama et al., 1993). But in the full model, efficacy has both ligand-dependent and -independent elements (Weiss et al., 1996b). The ligand-dependent components include—in addition to α and β—the “triple” interaction δ. The ligand-independent contribution is the constant γ, which provides a measure of receptor/G protein “efficacy” (i.e., to which extent and direction G protein binding “pushes” receptor isomerization and vice versa).
In classical receptor theory, intrinsic activity can be equated to intrinsic efficacy when the relationship between stimulus and response is linear (Kenakin, 1987). If so, efficacy can be experimentally measured as the ratio between responses induced by any two agonists at their maximally effective concentrations. But if the receptor undergoes a ligand-driven conformational shift, is this assumption still correct? To answer such a question, we shall now examine the relation between apparent intrinsic activity and ligand efficacy according to the two-state model.
Relation between Apparent Intrinsic Activity and Efficacy.
Let's first define fractional effect (Ef
) as the ratio between G protein-bound receptor species and the total concentration of receptor:
Relation between Maximal Effect and Affinity.
We now define fractional ligand occupancy to the receptor (Of
) as the ratio of ligand-bound receptor forms over the total concentration of receptor. For both versions of the model, this can be written with a general hyperbolic relation as follows: Equation 5bAll these relations clearly indicate that both biological effect and shift of affinity are efficacy-dependent parameters, and it makes no sense to assume that they provide independent information in the interpretation of experimental results. Also, the assumption that the ratios of maximal effects measured for two ligands only reflect the ratio of their efficacies is obviously wrong. In fact, if we callA and B any two ligands of different efficacy (subscripting their constants accordingly), their relative maximal effects (intrinsic activity) can be derived by computing the ratiosE
B/E
A according to equation 2b and substituting terms from equation 4b:
Equation 5bAll these relations clearly indicate that both biological effect and shift of affinity are efficacy-dependent parameters, and it makes no sense to assume that they provide independent information in the interpretation of experimental results. Also, the assumption that the ratios of maximal effects measured for two ligands only reflect the ratio of their efficacies is obviously wrong. In fact, if we callA and B any two ligands of different efficacy (subscripting their constants accordingly), their relative maximal effects (intrinsic activity) can be derived by computing the ratiosE
B/E
A according to equation 2b and substituting terms from equation 4b:
Mutations That Induce Constitutive Activity.
For both models, mutations that enhance constitutive activity can result from a shift in the intrinsic stability constant J. As indicated from equations 2 and 4, the increase of J produces linked enhancements of both apparent affinity and apparent intrinsic activity of the system. In Fig. 5A, we show how “relative” intrinsic activity (i.e., the ligand maximal effect relative to that of a “full agonist”) is related to the “thermodynamic” efficacy of the system (the product αβδ). As expected, the relation is not linear and depends on the value of J. At low J, only the range of positive intrinsic activity is detectable, and ligands with negative efficacy (inverse agonists) are indistinguishable from neutral antagonists. The situation is inverted as J becomes larger.
Let us now imagine a “wild-type” receptor that in the absence of ligand exists primarily in the R form (i.e.,J = 0.0001) and a set of mutations that selectively enhance the value of J. The shifts in affinity induced for ligands of varying efficacy (computed from 4b and expressed relative to the wild-type value) is related to the initial (wild-type) intrinsic activity as shown in Fig. 5B. This relation is close to linear for small changes of J, but it is warped into a parabolic shape as J grows larger, until saturation occurs. Note that such a theoretically predicted pattern is quite close to that observed experimentally in this study (Fig. 4).
The linkage between shift in affinity and intrinsic activity of ligands is thus a major diagnostic tool to uncover mutations that affect the intrinsic equilibria in the receptor. However, if a mutation can shiftJ and γ into opposite directions, then the six-cycle model predicts opposite effects on affinity and intrinsic activity of ligands. This situation is simulated in Fig.6, where identical enhancements ofJ are imposed on a “wildtype” receptor either in the absence (Fig. 6, left) or presence (Fig. 6, right) of a concomitant decrease of γ. In both cases, agonist binding curves (Fig. 6, bottom) are shifted to the left as J increases, indicating enhanced affinity. However, the fraction of G-coupled R* (i.e., the “biological response”) goes up with the increase of J in the first case (Fig. 6, top left), and diminishes in the second (Fig.6, top right). Additional simulations using different values for the parameters αβδ (not shown) indicated that in either cases the shifts in affinity are related to the “wild-type” intrinsic activity of ligands as illustrated using the simplified equations (2b and 4b) plotted in Fig. 5B. Therefore, both mutations seem to enhance binding affinity of ligands in an efficacy-dependent mariner. However, whereas the first also enhances “basal” activity and apparent “coupling” of the receptor, the second does the opposite. We may conclude from this analysis that an increase of J with a parallel decrease of γ explains the difference in properties between the defective R143E mutant and the constitutively active mutants compared in this study.
Footnotes
-
Send reprint requests to: Susanna Cotecchia, M.D., Institut de Pharmacologie et de Toxicologie. 27, Rue du Bugnon. Faculté de Médecine, 1005 Lausanne, Switzerland. E-mail:susanna.cotecchia{at}ipharm.unil.ch
-
This work was supported by the Fonds National Suisse de la Recherche Scientifique (Grant 31-51043.97) and by the European Community (Grants BMH4-CT97-2152 and BMH4-CT98-3566).
- Abbreviations:
- AR
- adrenergic receptor(s)
- GPCR
- G protein-coupled receptor
- IP
- inositol phosphate
- TM
- transmembrane
- E/DRY
- Glu/Asp-Arg-Tyr
- MD
- molecular dynamics
- DMEM
- Dulbecco's modified Eagle's medium
- HEAT
- [β-(4-hydroxyphenyl)-ethylaminomethyl]tetralone
- i3
- third intracellular loop
- i2
- second intracellular loop
- GnRH-R
- gonadotropin-releasing hormone receptor
- Received February 15, 1999.
- Accepted October 21, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}