


Abstract
This study reports that the affinity of HERG1 A for dofetilide is decreased from 0.125 ± 0.003 μM for wild-type (WT) channels to 15 ± 3 μM for F656V, a mutation in the COOH-terminal half of the S6. Similarly, the IC50 for quinidine was increased from 8 ± 4 μM for WT to 219 ± 65 μM for the F656V mutation, whereas affinity for external tetraethylammonium was similar for WT (51 ± 10 mM) and F656V (36 ± 10 mM, NS). Kinetics of onset of inactivation of F656V was similar to WT but kinetics of deactivation, activation, and recovery from inactivation differed from WT. However, mutations in nearby amino acids in the S6 more strikingly altered deactivation, activation, and recovery from inactivation but had little effect on affinity for dofetilide. To assess the effects of disruption of inactivation, the S631A mutation was made. The S631A mutation altered the IC50 for dofetilide to 20 ± 3 μM, but the IC50 for quinidine was unchanged at 8 ± 4 μM for WT and 10 ± 1 μM for S631A. To address whether the F656V mutation alters the IC50 for dofetilide in a channel that does not inactivate, the double mutation S631A/F656V was made. The IC50 for dofetilide of the double mutation was 32 ± 3 μM, which is not substantially different than that of S631A. These data support the notion that allosteric changes occurring during the process of inactivation are necessary for high-affinity dofetilide binding. In conclusion, the Phe-656 residue of HERG is a molecular determinant of high-affinity dofetilide binding.
HERG1is a member of the ether a-go-go (EAG) family of genes that encode voltage-gated potassium channels (Warmke and Ganetzky, 1994;Curran et al., 1995; Shi et al., 1997; Splawski et al., 1998). HERG1 A currents exhibit inward rectification due to rapid C-type inactivation (Sanguinetti et al., 1995; Smith et al., 1996; Spector et al., 1996b;Wang et al., 1997). The state-dependent transitions of HERG1 A have been modeled to consist of a series of closed states followed by slow activation to an open state followed by rapid inactivation to a nonconducting state (Wang et al., 1997). HERG1 A and Ikr are blocked by methanesulfonanilides, including dofetilide, which has an IC50 in the low nanomolar range (Jurkiewicz and Sanguinetti, 1993; Spector et al., 1996a; Wang and Duff, 1996; Ficker et al., 1998; Herzberg et al., 1998). Previous studies provide evidence that HERG channels are not blocked by dofetilide when the channel is kept closed and that opening of the activation gate is necessary for block (Kiehn et al., 1996;Snyders and Chaudhary, 1996).
Dofetilide is effective in the treatment of a range of cardiac arrhythmias in humans. The recently reported Diamond study (Diamond, 1996) provides evidence that dofetilide treatment may not be associated with the excess mortality in patients with structural heart disease that has plagued other antiarrhythmic drugs (Sword trial, 1996;Preliminary Report of CAST, 1989). An understanding of the molecular determinants of dofetilide binding to the HERG1 A K+ channel is relevant to structure-function analysis and for future design of drugs to block or open the HERG1 A channel. The purpose of this study was to identify molecular determinants for high-affinity binding of dofetilide to HERG1 A.
Previous site-directed mutation studies have identified amino acid residues that alter the affinity of HERG1 A for dofetilide (Ficker et al., 1998). The S620T mutation, putatively located in the pore helix near the external mouth of the channel, dramatically decreased the affinity of the HERG1 A channel for dofetilide (Ficker et al., 1998 ). The IC50 of dofetilide for HERG1 A was 0.32 ± 0.04 μM, whereas it was 248 ± 29 μM for S620T. Because the S620T mutation also disrupts inactivation, it was uncertain whether dofetilide interacted directly with this Ser-620 residue or whether the decreased affinity for dofetilide was related solely to the loss of inactivation (Ficker et al., 1998). All known mutations of HERG1 A that disrupt inactivation, such as S631A, also decreased the affinity of the channel for dofetilide (Ficker et al. 1998; Shi et al., 1998). Moreover, all wild-type(WT) EAG-related channels that do not inactivate also have a low (micromolar) affinity for dofetilide. In further experiments, Ficker et al. (1998) assessed a mutation equivalent to HERG S620T in the noninactivating bovine EAG (BEAG) channel. In those experiments the IC50 for dofetilide of BEAG T432S was 8 ± 1 μM compared with 32 ± 8 μM for BEAG WT. We hypothesized that another domain(s) of the channel is the molecular determinants of high (nanomolar)-affinity binding of dofetilide.
Studies on a range of voltage-gated K+ channels indicate that amino acids in the S6 domain are molecular determinants of block by 4-aminopyridine, quinidine, and intracellular application of tetraethylammonium (TEA) (Choi et al., 1993; Shieh and Kirsch, 1994;Yeola et al., 1996; Liu et al., 1997; Zhang et al., 1998). Moreover, amino acids in the S6 of cardiac sodium and L-type calcium channels also are molecular determinants for binding of local anesthetics and dihydropyridine calcium channel blockers, respectively (Ragsdale et al., 1994; Hockerman et al., 1995; McPhee et al., 1995; Peterson et al., 1996). Accordingly, we hypothesized that a high-affinity dofetilide binding site was located in the S6 domain of HERG1 A. The specific objectives of this study were to use site-directed mutagenesis to define amino acids in the S6 that constitute a molecular determinant of high-affinity dofetilide binding to HERG1 A, without grossly altering inactivation properties. In the present study, we identify an amino acid residue in the S6 of HERG1 A, Phe-656, which is a determinant of the high-affinity binding of dofetilide.
Materials and Methods
Expression in Xenopus Oocytes.
HERG1 A in pSP64 was obtained from M. T. Keating, University of Utah, Salt Lake City, UT (Curran et al., 1995). M. T. Site-directed mutagenesis was carried out by overlap extension with the polymerase chain reaction according to Ho et al. (1989). WT and mutated HERG1 A were transcribed in vitro from the SP6 promoter. The RNA transcripts were injected into Xenopus oocytes and the expressed currents were analyzed after 1 to 3 days with two-microelectrode voltage-clamp techniques (Lees-Miller et al., 1997).
Electrophysiologic Recordings.
The oocytes were perfused with modified frog Ringers solution at room temperature (21–22°C) containing 114 mmol/l NaCl, 2.5 mmol/l KCl, 1 mmol/l MgCl2, 1.8 mmol/l CaCl2, and 10 mmol/l HEPES, pH 7.2, adjusted with NaOH. Niflumic acid was included (0.15 mM) to block chloride currents. Glass microelectrodes were filled with 3 M KCl with tip resistances of 0.5 to 2 MΩ. Oocytes were clamped with a Geneclamp 500 amplifier and voltage-clamp protocols were generated with pClamp software (Axon Instruments, Foster City, CA), a Pentium computer, and a Digidata 1200 interface board (Axon Instruments). Currents were sampled at a rate of 2 KHz. Currents were filtered with a 4-pole Bessel filter. The oocyte membrane was held at −80 mV between pulses.
The current-voltage relationships for the time-dependent current were studied by applying voltage-clamp steps from a holding potential of −80 mV to different depolarizing levels. Each outward current was elicited by a 2-s depolarizing pulse from potentials of −70 to +30 mV, in 10-mV steps. To assess kinetics of deactivation, a double-pulse protocol was used. The first pulse was introduced from a holding potential of −80 mV to a potential of +50 mV for 2 s followed by a second pulse to a variety of test potentials ranging from −20 to −120 mV for 2.5 s. The deactivation process was fit to monoexponential or biexponential functions.
To assess the onset of fast inactivation, expressed channels were activated and inactivated with a 300-ms pulse to +40. A 25-ms interpulse to −110 mV was introduced to permit recovery from inactivation followed by a test pulse to variable potentials ranging from −70 to +30 mV. This protocol has been previously used to study inactivation (Smith et al., 1996). The decay of the current was fit to a mono- or biexponential function. Test pulses were applied every 15 s. To assess recovery from fast inactivation, expressed channels were activated with a 300-ms pulse to +40 mV followed by a test pulse to a variable potential ranging from −120 to −20 mV in 10-mV steps. At membrane potentials of less than −80 mV, tail currents were fit to biexponential functions to account for the rapid increase in current caused by recovery from channel inactivation and the much slower decrease in current by deactivation.
After control data were obtained, dofetilide at various concentrations was introduced. As has been previously reported (Spector et al., 1996a), the first depolarization yielded little or no reduction in current when the channel is held constantly at −80 mV during dofetilide superfusion. Accordingly during dofetilide treatments the oocytes were pulsed continuously from a holding potential of −80 mV to +20 mV for 2.5 s with an interpulse interval of 15 s. Because the development of dofetilide block during superfusion is slow, we monitored the currents every 5 min and did not obtain records of the extent of block of the current for 20 min after each change in dofetilide concentration. We also assessed the effects of potential “run down” of HERG1 A current during time-dependent evaluations of these pulse protocols. The extent of run down of HERG1 A during 90 min of superfusion with Normal Frog Tyrodes was 3.8 ± 1%.
The K d of binding was fit to the following model: Icontrol − Idofetilide/Icontrol =B max *C/[K d + C] with GraphPad Prism Software that involves nonlinear regression modeling where C indicates the concentration of drug,B max indicates the maximum level of block of the channel, and Icontrol is the tail current amplitude at +30 mV; similarly, Idofetilide is the amplitude of the tail current at +30 mV recorded during dofetilide treatment.
Rationale for Choice of Mutations.
We created four mutations in the S6 of HERG1 A: M651T, S654L, F656V, and N658V. Table1 shows a sequence alignment of homologous regions from the S6 of Shaker B, Kv1.5, Kv2.1, HERG1 A, BEAG, ELK1, and KcsA. Amino acids that have been reported to alter affinity for 4-aminopyridine (4-AP), TEA, and quinidine are indicated in bold (Choi et al., 1993; Shieh and Kirsch, 1994; Yeola et al., 1996;Liu et al., 1997). Amino acids whose accessibility to intracellular methanethiosulfonate reagents was altered in the open versus closed state of the channel are shown in italics and underlined in Table 1 (Liu et al., 1997).
Sequence alignment of homologous regions from the S6 of Shaker B, Kv1.5, Kv2.1, HERG1 A, BEAG, ELK1, and KcsA.
Previous studies have assessed the structural domains of Kv1.5 necessary for quinidine binding. The T505I or T505V mutations in the S6 of Kv1.5 produced an ∼10-fold increase in affinity for quinidine from ∼6 to 0.7 μM (Yeola et al., 1996). The T505 residue is equivalent to the T469 site of Shaker (Yeola et al., 1996). In Shaker, the T469S mutation reduced the affinity for internal TEA by ∼2-fold but substitution with a hydrophobic residue, T469V, produced little effect (Choi et al., 1993). The M651 residue in HERG1 A is equivalent to T505 in Kv1.5 and accordingly we made the M651T mutation.
The rationale for designing the F656V mutation relates to studies ofLiu et al. (1997) who identified pore sites in Shaker that were only accessible during channel opening. The V474C mutation, shown in italics in Table 1, showed the greatest difference in modification rate between the open and closed states of the channel. Thus, access to this amino acid was strikingly dependent on gating. Moreover, the V474C Shaker current was blocked by Cd2+ but only when the channels were opened. Similarly, Cd2+ could only be released from the channel when it was opened. A similar phenomena of block and release of dofetilide from IKr and HERG1 A occurs when the channel is opened (Carmeliet, 1992; Spector et al., 1996a). The Val-474 residue of Shaker is equivalent to Phe-656 of HERG1 A. Moreover Shieh and Kirsch (1994) reported that the I405V mutation of Kv2.1 increased 4-AP affinity, independent of changes in gating. From these data, the authors concluded that this site and another site in the S5 would be amino acid residues critical for 4-AP binding. The Ile-405 residue of Kv2.1 is equivalent to Phe-656 of HERG1 A. Accordingly, we created the F656V mutation.
The structural determinants of quinidine affinity for the sodium channel also have been examined. Residues Ile-764 and Ile-771 of the IV S6 are important determinants of molecular binding of quinidine to the sodium channel (Ragsdale et al., 1994; McPhee et al., 1995). According to the proposed alignment of this sodium channel with Kv1.5, these residues are equivalent to Gly-648 and Ser-654 of HERG1 A (Yeola et al., 1996). Moreover, Shieh and Kirsch (1994) reported that the mutation L403M of Kv2.1, a site equivalent to Ser-654 of HERG1 A, changed the affinity of Kv2.1 for internal TEA. Accordingly, we also created the S654L mutation of HERG1 A.
Results
F656V Alters Affinity of HERG1 A for Dofetilide and Quinidine but not External TEA.
A number of compounds block HERG1 A and IKr, including dofetilide; external TEA (TEAe), which interacts with the inactivation gate at Ser-631; and other nonspecific K+ channel blockers, such as quinidine. Figure 1shows the extent of block of WT and the F656V mutation by dofetilide (A), quinidine (B), and TEAe (C). The F656V mutation increased the IC50 for dofetilide by two orders of magnitude from 0.125 ± 0.003 μM for WT to 15 ± 3 μM. Similarly, the IC50 for quinidine was increased from 8 ± 4 for WT to 219 ± 65 μM for the F656V mutation. In contrast, the IC50 for TEAe was similar for WT (51 ± 10 mM) and F656V (36 ±10 mM, NS). The dose-response relationships for TEAe block did not fit a first-order binding isotherm because of the low affinity of HERG1 A for TEAe. Similar dose-response relationships of TEAe for HERG1 A have been previously reported bySmith et al. (1996). Because saturation was not observed, the measurement of affinity of HERG1 A and F656V for TEAe are considered estimates. However, a review of the raw data indicates that the extent of block by TEAe for WT and F656V shows little, if any difference. These data indicate that F656V is a molecular determinant for high-affinity dofetilide binding. Moreover, the F656V mutation alters affinity of the channel for dofetilide and quinidine but not for TEAe. In contrast, previous studies have reported that mutation of Ser-631 alters affinity of the channel for dofetilide and TEAe (Smith et al., 1996; Ficker et al., 1998) but we find that S631A does not alter the affinity for quinidine (see below). Because the F656V mutation did not change response to TEAe, it seemed likely that inactivation was intact. Accordingly, we assessed the inactivation and other gating characteristics of the channel.
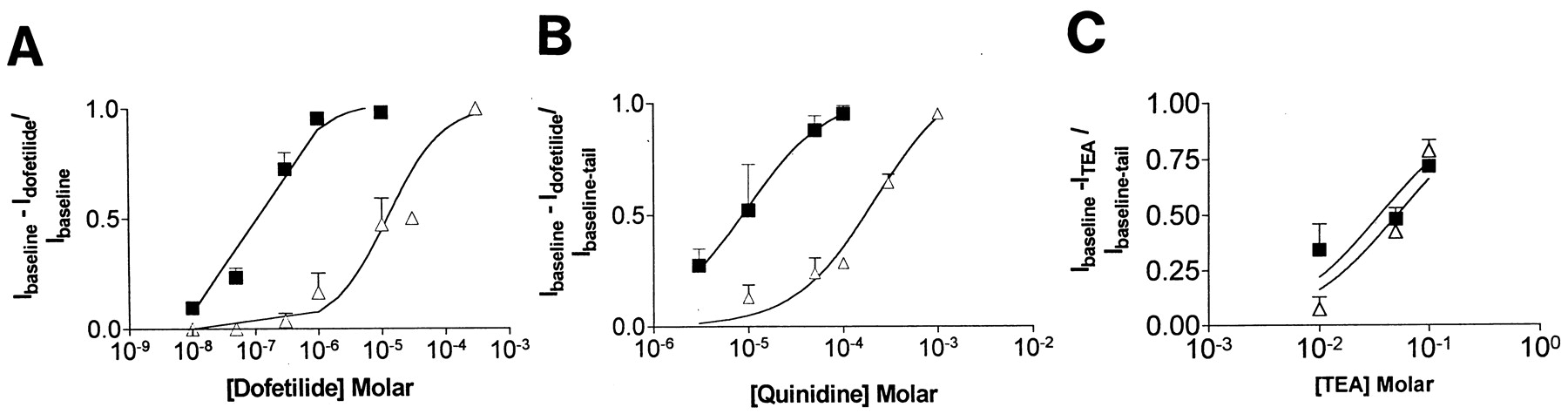
The extent of block of HERG1 A and of the F656V mutation by dofetilide, quinidine, and TEAe. The IC50 of dofetilide was 0.125 ± 0.003 μM for HERG1 A and 15 ± 3 μM for F656V. The IC50 of quinidine was 8 ± 4 for WT and 219 ± 65 μM for F656V. Because the F656V mutation does not change the inactivation process (see below), we predicted that the affinity for TEAe would be similar in HERG1 A and F656V. The estimate of IC50 for TEAe was 50 ± 10 mM for HERG1 A and 36 ± 10 mM for F56V.
F656V Is Inwardly Rectified with Inactivation Characteristics Similar to HERG1 A.
If F656V inactivates in a manner similar to HERG1 A, we expect that the time-dependent currents would be inwardly rectified. Figure 2 shows typical currents of WT in the left column and F656V in the right column. From a holding potential of −80 mV, currents were evoked by a series depolarizing potentials, P1, for 2500 ms to potentials between −70 and +50 mV followed by the P2 pulse to a holding potential of −60 mV. The current measured at the end of P1 was related to the depolarizing potentials. These mean current-voltage data are shown in Fig. 2, E and F. Inward rectification was evident for both WT and F656V. From these data, we concluded that the characteristic of inward rectification is common to WT and F656V.

The character of the F656V currents in comparison to HERG1 A when 3 ng of mRNA was injected. HERG1 A (A) and F656V (B) show families of activating and tail currents elicited from a holding potential of −80 mV by a series of depolarizing pulses to −30, −10 and +20 mV, P1, for 2500 ms followed by a second pulse, P2, to a potential of −60 mV. The characters of the currents are similar. C and D, deactivating tail currents elicited by the protocol shown in the inset. A double pulse protocol was used. The first pulse was introduced from a holding potential of −80 mV to a potential of +50 mV for 2 s followed by a second pulse to a variety of test potentials ranging from −20 to −120 mV for 2.5 s. The deactivation process is more rapid for F656V than WT. E and F, current-voltage relationship of the time-dependent activating current (●) and the tail currents (▪) for HERG1 A in E and F656V in F. Both the time-dependent currents and the tail currents show inward rectification.
Inward rectification suggested that both WT and F656V would have grossly similar inactivation characteristics. Accordingly, the character of the onset of inactivation of WT and F656V were compared. Representative examples of the inactivation time course are shown in Fig. 3 for WT (A) and F656V (B) with a protocol similar to that of Smith et al. (1996) and Spector et al. (1996b). No substantial difference in the time course of onset of inactivation was noted. Data were best fit to a monoexponential function. The mean time constants for the onset of inactivation are plotted against voltage in Fig. 3C and are similar for WT and F656V. For example, the mean time constants at −20 mV were 13 ± 1 ms and 11 ± 1 ms for WT and F656V, respectively, and at +20 mV were 9 ± 1 ms for WT and 8 ± 0.2 ms for F656V. These data indicate that the characteristics of onset of inactivation are similar in WT and F656V.

shows the onset of inactivation of HERG1 A and F656V. The hyperpolarized interpulse to −110 mV was applied for 25 ms for WT and 12.5 ms for F656V, a protocol similar to that used by Spector et al. (1996b). Recovery from inactivation at −110 mV proceeds with a time constant of 4 ms for WT and 2 ms for F656V. Therefore, the duration of the interpulse intervals was of sufficient time to allow an ∼90% recovery from inactivation but too short for substantial deactivation. Representative examples of the time course of onset of inactivation for HERG1 A (A) and F656V (B) are shown. The dashed lines show the fits to monoexponential functions in each case. C, mean τ values of onset of inactivation comparing HERG1 A (■), F656V (▿), S654L (▴), N658V (●), and M651T (♦). There was no significant difference in the onset of inactivation comparing HERG1 A and F656V. The voltage-τ relationship for onset of inactivation of M651T and F656V are similar, as are those of N658V and the S654L.
To assess whether the F656V mutation altered the process of recovery from inactivation, a protocol similar to that of Spector et al. (1996b)was used. Representative examples of the time course of recovery from inactivation and its fitting to a monoexponetial process are shown in Fig. 4. The general character of recovery from inactivation was similar in WT and F656V. The mean time constants for recovery from inactivation are plotted against voltage for WT and F656V in Fig. 4C. The time constants of recovery from inactivation were significantly slower for WT than for F656V. For example, the mean time constant at −20 mV was 13 ± 1 ms for WT and 8 ± 0.3 ms for F656V (P < .01, Student's t test). These data indicate that F656V alters the time course of recovery from inactivation.

shows representative examples of the recovery from inactivation for HERG1 A (Panel A) and for F656V (Panel B). The time scale is identical in these Panels. Recovery from inactivation was similar in HERG1 A and the F656V mutation. Panel C shows the mean τ values for recovery from inactivation for HERG1 A (■), F656V (▿), S654L (▴), N658V (●), and M651T (♦) (n > 6 for each). The voltage-τ relationship for recovery from inactivation of M651T and F656V are similar.
Activation Characteristics of F656V Compared with WT.
To assess whether the F656V mutation had altered the process of activation, the peak of the tail currents at the onset of the P2 pulse was related to the P1 potentials and presented for WT in Fig. 2E and F656V in Fig. 2F. The voltage-dependence of the activated tail currents of WT were well fit to a Boltzman function with V1/2for activation of −26 ± 7 mV (slope 9 ± 6), whereas the V1/2 for F656V was −9 ± 6 mV (slope 15 ± 1 mV). These data indicate that the F656V mutation significantly shifts the voltage dependence of activation to more depolarized potentials.
Deactivation.
To assess whether the F656V mutation had altered the process of deactivation, the time constants of the decay of the tail currents at the onset of the P2 pulse were assessed. Representative examples of the deactivation currents are shown for WT in Fig. 2C and for F656V in Fig. 2D. At −80 mV, the τ value of the fast component of deactivation for WT (173 ± 9 ms) was slower than for F656V (75± 5 ms), whereas at −40 mV the τ values were 1088 ± 240 and 316 ± 9 ms, respectively (P< .01).
Other S6 Mutations Tested Have Little or Modest Effects on Affinity for Dofetilide.
A number of other S6 mutations were studied, including M651T, S654L, and N658V. The IC50 of these mutant channels for dofetilide were 0.120 ± 0.08, 0.3 ± 0.1, and 0.119 ± 0.7 μM, respectively.
Figure 5 shows the character of activating currents of these mutations. The deactivation time constant of M651T is markedly prolonged, whereas in the N658V mutation it is abbreviated. The mean τ values of the rapid component of deactivation at −90 mV for WT, M651T, and N658V were 116 ± 7, 508 ± 205, and 16 ± 6 ms, respectively. Thus, the deactivation τ can be altered by 50-fold (from 16 to 508 ms) and yet the IC50 for dofetilide of M651T and N658V are the same as WT. The V1/2 for activation as measured from the tail currents were shifted to −32 ± 6 mV for M651T, to −18 ± 2 mV for N658V, and to −6 ± 5 mV for S654L compared with −26 ± 7 mV for WT, but their IC50values were similar. Thus, shortening or prolonging deactivation or shifting the voltage dependence of activation did not correlate with a change in the affinity of the channel for dofetilide.

Phenotypic features of the other mutations made in the S6 that had little or only modest effects on the IC50for dofetilide. A–C, phenotypes for S654L, M651T, and N658V, respectively. The M651T mutation substantially slows deactivation, whereas the deactivation of the N658V mutation is much more rapid than WT. D, phenotype of the double mutation S631A/F656V. The double mutation interferes with inactivation. The IC50 for block by dofetilide was 32 ± 3 μM, which was not grossly different than is observed for the individual mutations [S631A (20 ± 3 μM) or for F656V (15 ± 3 μM)].
Figure 3C also contrasts the kinetics of the onset of inactivation of M651T, S654L, N658V, F656V, and HERG1 A WT. The M651T mutation has characteristics of inactivation that are similar to F656V. Indeed, at −20 mV the τ values of onset of inactivation are identical even though the IC50 for dofetilide differs by two orders of magnitude (0.120 ± 0.08 μM/l for M651T and 15 μM for F656V). Moreover, shifts in voltage dependence of inactivation also do not appear to correlate with affinity for dofetilide. For example, the S654L mutation has a shifted voltage dependence of onset of inactivation and a shifted IC50 for dofetilide; however, the N658V mutation has a very similar shift in the onset of inactivation but the IC50 for dofetilide is similar to that of WT, 0.119 ± 0.7 μM. Thus, for these mutations, there is no correlation between alteration in the character of onset of inactivation and the IC50 for dofetilide.
Figure 4C compares the kinetics of recovery from inactivation of mutant and WT HERG1 A channels. The M651T mutation has a voltage-dependent recovery from inactivation, which is similar to F656V over voltages from −90 to −50 mV. At −70 mV, the τ of recovery from inactivation is 6 ± 0.2 for F656V and 6 ± 0.5 ms for M651T, even though the IC50 for F656V is two orders of magnitude different than M651T. In addition shifts of the voltage dependence of recovery from inactivation to the right, as seen with N658V do not correlate with a change in the IC50 for dofetilide. Thus, there is no correlation between alteration in the character of recovery from inactivation and the IC50 for dofetilide.
Pharmacologic Responses to S631A and to Double Mutant F656V/S631A.
To assess the effects of disrupting inactivation, the S631A mutation was made. The IC50 values of TEAe, a drug known to interact with the S631 site (Smith et al., 1996); dofetilide, a drug also known to have its IC50 altered by S631A (Ficker et al., 1998); and quinidine, a nonspecific potassium channel blocker were assessed in HERG1 A and in the S631A and F656V mutants. In this study, the S631A mutation decreased the affinity for dofetilide to 21 ± 4 μM, a result similar to that previously reported (Ficker et al., 1998). However, the IC50 for quinidine is not affected by the S631A mutation (8 ± 2 μM for WT and 10 ± 1 μM for S631A). In contrast to the S631A mutation, the F656V mutation, as reported above, decreases the affinity for quinidine and dofetilide but not for TEAe. Moreover, affinity for TEAe is altered by S631A, affinity for quinidine is altered by F656V, and affinity for dofetilide is altered by mutations in either sites. These data indicate that these different drugs have different responses to the S631A and the F656V mutations.
To address whether the F656V mutation alters the IC50 for dofetilide in a channel that does not inactivate (S631A), the double mutation S631A/F656V was made (Fig. 5D). The IC50 for dofetilide of the double mutant S631A/F656V is 32 ± 3 μM, which was not grossly different than is observed for the individual mutations S631A (20 ± 3 μM) or for F656V (15 ± 3 μM). These data support the notion that allosteric changes occurring during the process of inactivation are necessary for high-affinity dofetilide binding.
Discussion
New Information Provided by This Study.
Previous studies have identified mutations such as S631A and S620T that alter the affinity of HERG1 A for dofetilide, but also disrupt inactivation (Ficker et al., 1998; Zou et al., 1998). The decreased affinity of the channel for dofetilide produced by the F656V mutation was not associated with disrupted inactivation. The F656V mutation alters affinity of the channel for dofetilide and quinidine but not TEAe, whereas the S631A mutation alters affinity of the channel for dofetilide and TEAe (Smith et al., 1996; Ficker et al., 1998) but not for quinidine. To address whether the F656V mutation alters the IC50 for dofetilide in a channel that does not inactivate (S631A), the double mutation S631A/F656V was made, and the IC50 for dofetilide of the double mutant was not grossly different than is observed for the individual mutations. These data support the notion that allosteric changes occurring during the process of inactivation appear to be necessary for high-affinity dofetilide binding.
F656V Alters Affinity for Dofetilide Unrelated to Changes in Channel Gating.
Previous studies have demonstrated that mutation of S620 to T dramatically decreases HERG1 A's affinity for dofetilide (Ficker et al., 1998). However, the S620T mutation also disrupts inactivation (Ficker et al., 1998), therefore, it was uncertain whether dofetilide interacted directly with this S620 residue or whether the decreased affinity for dofetilide was related solely to the loss of inactivation. In contrast, the F656V mutation decreases the affinity of HERG1 A without a disruption of the inactivation characteristics. Moreover, it is unlikely that quantitative differences in activation, deactivation or onset and recovery from inactivation are responsible for the reduced affinity of F656V for dofetilide because mutations of nearby amino acids that either prolong or shorten these gating kinetics have no effect on the IC50 of dofetilide.
It might be argued that the mutation of F656V has changed the entire topology of the internal mouth of the channel and thus this residue is not the real binding site. This argument seems unlikely. F656V is a conservative substitution of one hydrophobic residue for another. In addition, the channel continues to function in a manner qualitatively similar to WT despite the mutation.
Phe-656 Is Necessary but not Sufficient for High-Affinity Dofetilide Binding: Contribution of Inactivation.
The structure of the KcsA K+ channel has been determined by X-ray crystallography (Doyle et al., 1998). The apparent structure of IRK1, as determined by mutagenesis, differs significantly from KcsA (Minor et al., 1999). However, KcsA is more similar in its primary structure to voltage-gated channels than to IRK1, suggesting that the topologic features of KcsA may provide a structural framework for considering the molecular determinants of interaction of dofetilide with HERG Phe-656. Thr-107 in KcsA aligns with Phe-656 of HERG1 A. Thr-107 is located near the intracellular pore mouth and faces the pore stream and is at the apex of the “inverted teepee tent” configuration of the KcsA potassium channel, placing it at or near the narrowest portion of the intracellular mouth of the channel. Therefore, binding of a large organic compound at this site might be expected to occlude the channel. The presence of a phenylalanine at this site in HERG1 A appears to be important in creating a binding site for dofetilide and quinidine. Dofetilide consists of a long aliphatic chain with substituted benzene rings at each end. Benzene rings are known to stack via pi bonds and it is possible that the benzene head groups of dofetilide could interact with the benzene ring of phenylalanine. Interestingly, none of the Shaker, Shab, Shaw, Shal family of K+ channels has a phenylalanine at a position equivalent to Phe-656. This may account for the relative specificity of dofetilide for HERG1 A. However, the noninactivating BEAG and ELK1 channels, which also have an F in positions equivalent to Phe-656 (Table 1), are relatively insensitive to methanesulphonanilides. Therefore, the presence of Phe-656 is necessary for high-affinity dofetilide binding, but other characteristics of the channel also must contribute to differences in dofetilide binding. Because ELK1 and BEAG also do not inactivate, these data suggest that there are two characteristics that are determinants of high-affinity dofetilide binding, the Phe-656 amino acid and allosteric changes that take place during the process of inactivation.
The relative contribution of the Phe-656 site and the allosteric changes associated with inactivation to the affinity for dofetilide was assessed by evaluating whether the F656V mutation alters the IC50 for dofetilide in the setting of a channel that does not inactivate. Accordingly, a double mutation was created, including both S631A, a mutation that disrupts the inactivation process, and F656V, a site that alters binding of dofetilide to its site in the S6. We predicted that if inactivation was disrupted, dofetilide could not bind efficiently to the S6, no matter what amino acid is present at the Phe-656 position and therefore the IC50 of the double mutations should be generally similar to that seen with S631A. Our data indicate that the double mutation has an IC50 of 32 μM, a value in the same range as that seen with the individual S631A or F656V mutations. These data support the notion that the process of inactivation is necessary for high-affinity dofetilide binding.
Evidence against the concept that inactivation is a determinant of dofetilide binding is the observation that depolarization to extremely high voltages (+80 mV) appears to decrease the extent of dofetilide block compared with that seen at +10 mV (Snyders and Chaudhary, 1996;Kiehn et al., 1996). Ficker et al. (1998) have previously considered whether inactivation could contribute to dofetilide block or whether all of the characteristics of dofetilide block can be explained by open-channel block. Evidence against open-channel block was that dofetilide did not produce significant changes in open-channel time (Kiehn et al., 1996). To account for these observations, Ficker et al. (1998), proposed that dofetilide binds to a preinactivated state of the channel, which is consistent with the single channel-bursting behavior of the channel. The presence of a preinactivated state as a determinant of binding of dofetilide is consistent with our findings.
Proposed Model of Dofetilide Binding to HERG1 A.
Although a preinactivated state of the channel may be a determinant of dofetilide binding, we present an alternative model that proposes that sequential allosteric changes, including activation and subsequent inactivation (or preinactivation) are necessary for high-affinity dofetilide binding. In this model, opening of the activation gate allows access of dofetilide to the inner vestibule of the channel. Thereafter, allosteric changes associated with inactivation allow association of dofetilide with its high-affinity binding site. Following repolarization, the channels reactivate and close allowing dissociation of dofetilide from the high-affinity site, but the activation gate must reopen to allow dofetilide to leave the inner vestibule of the channel pore. In this model, the activation gate determines access of dofetilide to the inner vestibule of HERG1 A and as such is a critical determinant of association with and dissociation from the pore, whereas the process of inactivation appears to be necessary for association of dofetilide with its high-affinity binding site in the S6. Consistent with this model are the observations that an opening of the activation gate is necessary for dofetilide block and unblock (Kiehn et al., 1996;Snyders and Chaudhary, 1996). The evidence that allosteric changes associated with the inactivation process are necessary for block is that all known mutations of HERG1 A that disrupt inactivation also decrease the affinity of the channel for dofetilide independent of whether or not they face the pore stream. Moreover, all WT EAG-related channels that do not inactivate but have an Phe at positions equivalent to Phe-656 also have a low (micromolar) affinity for dofetilide, whereas the inactivating channels ERG2 and ERG3 are sensitive to nanomolar concentrations of methanesulfonanilides (Shi et al., 1997,1998). Our model does not define whether the inactivation gate directly alters access to the receptor at Phe-656 or whether allosteric changes in the entire pore that occur during inactivation alter access of dofetilide to its high-affinity receptor. An alternative explanation of how inactivation could alter dofetilide binding to the S6 is that inactivation stops potassium flux across the pore. Ongoing potassium flux could inhibit binding. In summary, in this model conformational changes in the activation gate determine access of dofetilide to the inner vestibule, whereas a transition state involved in inactivation appears necessary for binding of dofetilide to the high-affinity site in the S6.
The sequential binding model also can account for the observation that extreme inactivation produced by depolarizing to +80 mV appears to decrease the extent of dofetilide block compared with that seen at +10 mV (Kiehn et al., 1996; Snyders et al., 1996). Our model requires two allosteric steps to achieve dofetilide block; first the channel must open to allow access of dofetilide to the inner vestibule, and then the channel must transition to an inactivated state. At +80 mV, the current-voltage relationship of HERG indicates that virtually all channels will be in the inactivated state (Sanguinetti et al., 1995), with only rare openings (a state necessary for dofetilide binding). In contrast, at +10-mV channels are in equilibrium between open and inactivated states and so dofetilide can block the channel. In conclusion, the Phe-656 residue of HERG1 A is a molecular determinant of high-affinity dofetilide binding.
Acknowledgments
We are appreciative of the helpful suggestions of Drs R. S. Clark, A. M. Gillis, and R. S. Sheldon and the technical assistance of Lilong Tang and Kevin Schade.
Footnotes
-
Send reprint requests to: H. J. Duff, M.D., F.R.C.P.(C), Department of Medicine, University of Calgary, 3330 Hospital Dr. NW, Calgary, Alberta, Canada T2N 4N1. E-mail:hduff{at}ucalgary.ca
-
This work was supported by Medical Research Council of Canada, the Heart and Stroke Foundation of Alberta, and the Andrews Family Professorship in Cardiovascular Medicine.
- Abbreviations:
- EAG
- ether a-go-go
- WT
- wild type
- TEA
- tetraethylammonium
- TEAe
- external TEA
- 4-AP
- 4-aminopyridine
- TEAe
- tetraethylammonium external
- Received July 28, 1999.
- Accepted November 9, 1999.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}