


Abstract
In previous studies we showed that the wild-type histamine H2 receptor stably expressed in Chinese hamster ovary cells is constitutively active. Because constitutive activity of the H2 receptor is already found at low expression levels (300 fmol/mg protein) this receptor is a relatively unique member of the G-protein-coupled receptor (GPCR) family and a useful tool for studying GPCR activation. In this study the role of the highly conserved DRY motif in activation of the H2 receptor was investigated. Mutation of the aspartate 115 residue in this motif resulted in H2 receptors with high constitutive activity, increased agonist affinity, and increased signaling properties. In addition, the mutant receptors were shown to be highly structurally instable. Mutation of the arginine 116 residue in the DRY motif resulted also in a highly structurally instable receptor; expression of the receptor could only be detected after stabilization with either an agonist or inverse agonist. Moreover, the agonist affinity at the Arg-116 mutant receptors was increased, whereas the signal transduction properties of these receptors were decreased. We conclude that the Arg-116 mutant receptors can adopt an active conformation but have a decreased ability to couple to or activate the Gs-protein. This study examines the pivotal role of the aspartate and arginine residues of the DRY motif in GPCR function. Disruption of receptor stabilizing constraints by mutation in the DRY motif leads to the formation of active GPCR conformations, but concomitantly to GPCR instability.
G-protein-coupled receptors (GPCRs) form a large and functionally diverse superfamily of receptors that transduce signals across cell membranes. Agonist occupancy of GPCRs is believed to result in a conformational change in the receptor, leading to activation of G-proteins (Oliveira et al., 1994; Gudermann et al., 1997; Gether and Kobilka, 1998). Although much is known about structural features of GPCRs involved in ligand recognition and G-protein binding, the actual mechanism underlying ligand activation of GPCRs remains unclear. Studies with mutant GPCRs suggest that intracellular regions of the GPCRs, in particularly the second and third intracellular loops and sometimes the cytoplasmic tail, interact with G-proteins thereby mediating signal transduction (Moro et al., 1993; Zhu et al., 1994; Gudermann et al., 1995; Smit et al., 1996b,c). Recent studies suggest that the so-called DRY motif has a pivotal role in the signal transduction pathways of GPCRs (Oliveira et al., 1993; Zhu et al., 1994; Scheer et al., 1996, 1997; Ballesteros et al., 1998; Rasmussen et al., 1999). The DRY motif is a highly conserved triplet of amino acids, Asp-Arg-Tyr (Probst et al., 1992;Savarese and Fraser, 1992), located at the boundary of transmembrane helix 3 and the second intracellular loop (see Fig. 1).

Schematic representation of the wild-type and four mutant rat histamine H2 receptors. Part of the amino acid sequence is indicated by the single letter code. The arrows indicate the respective mutations as obtained by site-directed mutagenesis: Asp (D) at position 115 was replaced with Asn (N) or Ala (A), and Arg (R) at position 116 was replaced with Asn (N) or Ala (A).
The fully conserved arginine residue of the DRY motif is considered to be a key residue in signal transduction of GPCRs (Oliveira et al., 1994; Scheer et al., 1996; Ballesteros et al., 1998), because mutations in this residue generally result in receptors with impaired signal transduction (Oliveira et al., 1993; Zhu et al., 1994; Jones et al., 1995; Scheer et al., 1996). Moreover, some diseases are reported to be the result of naturally occurring mutations in the arginine residue leading to dysfunction of the receptor (Sung et al., 1991; Rosenthal et al., 1994).
The aspartate residue is also highly conserved and suggested to play an important role in receptor activation. Mutations of the aspartate residue are reported to result in constitutive GPCR activity for some GPCRs (Cohen et al., 1993; Scheer et al., 1996; Morin et al., 1998;Rasmussen et al., 1999). It has been suggested that receptor activation involves protonation of the aspartate residue (Scheer et al., 1996). Experiments with rhodopsin have shown that the corresponding glutamate residue is involved in the proton uptake resulting in the formation of the activated metarhodopsin II intermediate (Arnis et al., 1994). Computational studies have linked the increased basal GPCR activity on mutation of the aspartate residue to the disruption of intramolecular constraints that keep the receptor in an inactive conformation resulting in a reorientation of the arginine residue (Oliveira et al., 1994; Scheer et al., 1996; Ballesteros et al., 1998).
The histamine H2 receptor is a member of the large family of GPCRs. The gene encoding the histamine H2 receptor has been cloned in several species (Gantz et al., 1991a,b; Ruat et al., 1991; Traiffort et al., 1995). Compared with other GPCRs, the histamine H2receptor is unique in that the wild-type receptor possesses a remarkably high degree of constitutive activity. With a receptor density of 300 fmol/mg protein, constitutive H2receptor activity could be detected in Chinese hamster ovary cells (Smit et al., 1996a). An even higher constitutive activity was suggested for the wild-type human H2 receptor because this receptor is also structurally instable (Alewijnse et al., 1998). Structural instability is usually found for constitutively active mutant GPCRs, in which stabilizing intramolecular constraints that keep the GPCR in an inactive conformation are disrupted (Gether et al., 1997; Samama et al., 1997). Because of these unique characteristics, the histamine H2 receptor could be useful to study GPCR activation.
Currently, information about the domains involved in G-protein coupling and receptor activation of the histamine H2receptor is limited. The importance of part of the C terminus and a conserved leucine residue in the second intracellular loop in Gs-protein coupling has been proposed (Smit et al., 1996b,c), but the role of the DRY motif in H2 receptor activation is not known. Investigation of the role of the aspartate residue of this motif in signal transduction might be of particular importance, because this residue is suggested to play an important role in keeping the GPCR in an inactive state (Scheer et al., 1996, 1997).
In this study the important role of the DRY motif in signal transduction of the histamine H2 receptor is demonstrated using mutant H2 receptors with point mutations in the DRY motif. Mutations of the aspartate residue of the DRY motif result in a constitutive activity and signaling of the H2 receptor mutants, whereas mutations of the arginine residue lead to reduced signal transduction. However, the arginine mutant receptors adopt an active conformation as the agonist affinity at these mutant receptors is increased.
Materials and Methods
Cell Culture.
HEK-293 cells and COS-7 cells were grown at 37°C in a humidified atmosphere with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM), containing 10% (HEK-293 cells) (v/v) or 5% (COS-7 cells) fetal calf serum supplemented with 2 mM l-glutamine, 50 I.U./ml penicillin, and 50 μg/ml streptomycin. HEK-293 cells were transiently transfected with 1 to 10 μg of DNA using calcium phosphate precipitation, whereas COS-7 cells were transiently transfected with 1 to 10 μg of DNA using DEAE-dextran (Brakenhoff et al., 1994).
Site-Directed Mutagenesis.
cDNAs encoding for the rat histamine H2 receptor with a mutation in the aspartate or arginine residue of the DRY motif were constructed using the polymerase chain reaction (Smit et al., 1996b) and verified by dideoxynucleotide sequencing.
Histamine H2 Receptor Binding.
Radioligand binding was assayed in cell homogenates prepared in phosphate-buffered saline (137 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, 1.8 mM KH2PO4) as described previously with the H2 antagonist [125I]iodoaminopotentidine ([125I]APT) (Leurs et al., 1994). Briefly, triplicate assays were performed in polyethylene tubes in 400 μl of 50 mM Na2/K phosphate buffer (pH 7.4) containing gelatin (0.1%), 0.3 nM [125I]APT, and 5 to 10 μg of cell homogenate in the absence or presence of 1 μM tiotidine. After 90 min at 30°C the incubations were stopped by rapid dilution with 3 ml of ice-cold 20 mM Na2/K phosphate buffer (pH 7.4) supplemented with 0.1% chicken egg albumin, and rapid filtration with a Brandel cell harvester (Semat, UK) through Whatman GF/C glass fiber filters (0.3% polyethyleneimine-treated). Filters were washed twice with 3 ml of buffer, and radioactivity retained on the filters was counted with an LKB-gamma counter at an efficiency of 63%. For measurements on receptor stability 10 to 20 μg of cell homogenate was incubated for different times at 37°C in 200 μl of 50 mM Na2/K phosphate buffer. Subsequently, the binding was measured as described before but with one important modification: to get a receptor occupation of 100%, unlabeled iodoaminopotentidine was added to all samples with a final concentration of 2.5 nM. For stabilization experiments, 10 to 20 μg of cell homogenate was incubated in the presence of a ligand for 2 h at 37°C in 200 μl of 50 mM Na2/K phosphate buffer. The samples were subsequently washed and centrifuged (10 min, 500g) three times to wash out the ligand. The binding was performed as described before. The binding data were evaluated by use of a GraphPad Prism. Changes in H2 receptor density were denoted as a percentage of the binding compared with that of nontreated control cells. Protein concentrations were determined according to Bradford using bovine serum albumin as a standard (Bradford, 1976).
Adrenergic α1B Receptor Binding.
Triplicate assays were performed in polyethylene tubes in 125 mM KCl, 25 mM HEPES, and 4.2 mM MgCl2, pH 7.4. For binding, 5 to 10 μg of cell homogenate and approximately 0.4 nM [3H]prazosin were incubated in the absence or presence of 1 μM prazosin in a total volume of 400 μl. After 90 min at 30°C, the incubations were stopped by rapid dilution with 3 ml of ice-cold buffer. The bound radioactivity was subsequently separated by filtration with a Brandel cell harvester (Semat) through Whatman GF/C glass fiber filters that had been treated with 0.3% polyethyleneimine. The radioactivity retained on the filters was measured by liquid scintillation counting. Experiments on the stability of the α1B-adrenergic receptor were performed as described for the histamine H2 receptor. Changes in α1B receptor density were denoted as a percentage of the binding compared with that of nontreated control cells.
Cyclic AMP Assay.
After 24 h, transiently transfected HEK-293 cells seeded in 12-well plates were washed twice with DMEM, supplemented with 50 mM HEPES (pH 7.4 at 37°C) and preincubated for 30 min at 37°C. Thereafter, the medium was aspirated, appropriate drugs in DMEM/HEPES, supplemented with a concentration (300 μM) of the phosphodiesterase inhibitor isobutylmethylxanthine (IBMX), were added, and the cells were incubated for 10 min at 37°C. The reaction was stopped by the rapid aspiration of the culture medium and the addition of 200 μl of 0.1 N cold HCl. The cells were kept on ice and disrupted by sonification (2 s, 40% output, Sonifier, Branson). The resulting homogenate was frozen at −20°C or directly neutralized with 1 N NaOH and assayed for the presence of cAMP.
The amount of cAMP in transiently transfected HEK-293 cells was determined using a competitive protein kinase A binding assay according to Nordstedt and Fredholm (1990), with some minor modifications (Smit et al., 1996a). Briefly, 200 μl of protein kinase A was mixed with 25 to 200 μl of the HEK-293 homogenate or cAMP standards and 30,000 dpm [3H]cAMP. After incubation for 150 min at 4°C the mixture was rapidly diluted with 3 ml of ice-cold 50 mM Tris/HCl (pH 7.4 at 4°C) and filtered through Whatman GF/B filters using a Brandel cell-harvester (Semat). The radioactivity retained on the filters was measured by liquid scintillation counting.
Chemicals.
Histamine dihydrochloride, IBMX, forskolin, cAMP, gelatin, polyethyleneimine, l-phenylephrine, and chicken egg albumin were obtained from Sigma. [5′,8-3H]cAMP (30–60 Ci/mmol) and [7-methoxy-3H]prazosin (65–85 Ci/mmol) were obtained from Amersham. Aminopotentidine and iodoaminopotentidine were taken from laboratory stock. Gifts of cimetidine and burimamide (SmithKline Beecham, United Kingdom), ranitidine dihydrochloride (Glaxo, United Kingdom), tiotidine (Imperial Chemical Industries, United Kingdom), and famotidine (Merck Sharp Dohme, The Netherlands) are greatly acknowledged. Prazosin hydrochloride was obtained from Pfizer. Corynanthine hydrochloride was obtained from Roth (Karlsruhe, Germany).
Statistical Analysis.
All data shown are expressed as mean ± S.E. of at least three independent experiments. Statistical analysis was carried out using Student's t test and expressed as P < .05 (*), P < .01 (**), and P < .001 (***).
Results
Functional Analysis of the D115A, D115N, R116A, and R116N Mutant H2 Receptors.
Using the polymerase chain reaction aspartate115 or arginine116 of the highly conserved DRY motif located in the second intracellular loop of the rat histamine H2 receptor were mutated into an alanine or asparagine residue (Fig. 1). The mutant receptor cDNAs were transiently transfected into HEK-293 cells for additional analysis. Remarkably, all mutant receptors were expressed at significantly lower levels than the wild-type receptor (Table1). Expression of the R116A and R116N mutant receptors was too low to allow further characterization.
Binding characteristics of wild-type or mutant H2 receptors after transient expression in HEK-293 cells
For the D115A and D115N mutant receptors, no major differences in the binding of H2 antagonists compared with that of the wild-type receptor were found. The affinity of [125I]APT (Table 1) was only slightly affected by the introduction of the mutation. In contrast, the agonist binding properties were significantly affected by the mutation of the aspartate residue (Table 1). The affinity of histamine for either the D115A or D115N mutant receptors was significantly increased compared with the wild-type receptor (Table 1, Fig. 2). For the wild-type H2 receptor, a single, low affinity binding site for histamine was observed. In contrast, a high- and low-affinity site could be distinguished for the D115N mutant receptor and the D115A mutant receptor (Table 1, Fig. 2). Addition of 10 μM guanosine 5′-3-O-(thio)triphosphate (GTPγS) did not shift the histamine displacement curve of both mutant receptors back to the affinity detected at the wild-type receptor (data not shown).

Displacement by histamine of the [125I]APT binding to the D115N (closed circles) and the D115A (closed squares) mutant H2 receptors and the wild-type (open circles) H2 receptor transiently expressed in HEK-293 cells. Cell homogenates were incubated with 0.3 nM [125I]APT for 90 min at 30°C in the presence of increasing concentrations of histamine. Data presented are from one typical experiment out of three experiments performed in triplicate.
Subsequently, the effect of the mutation of Asp-115 into Ala or Asn on the histamine-induced cAMP production was investigated. Because expression of both mutant receptors was about 10-fold lower than that of the wild-type receptor, the effect of receptor expression on the histamine-induced cAMP production was first studied. By transfecting different amounts of the expression vector pcDNA3-rH2 the expression level of the wild-type histamine H2 receptor was varied. Increasing the expression of rH2 receptors in HEK-293 cells from 341 fmol to approximately 1.5 pmol did not affect the EC50 value of the histamine-induced cAMP response (Table 2). The maximum histamine-induced response, however, was found to be increased from 12 ± 2 pmol/well at low receptor density to 42 ± 6 pmol/well at the highest receptor density (Table 2) as expected. Subsequently, the effect of the mutation of Asp-115 into Ala or Asn on the histamine-induced cAMP response was investigated at comparable densities of wild-type or mutant receptors. The EC50 value of the histamine-induced cAMP response in HEK-293 cells expressing either the D115A or the D115N mutant receptor was not significantly different from the EC50 value determined in HEK-293 cells expressing the wild-type H2 receptor (Table 2). Both mutant receptors, D115A and D115N, however, showed a dramatic increase in the maximum histamine-induced response compared with the wild-type receptor at a comparable density (Table 2).
Histamine-induced cAMP production in HEK-293 cells transiently expressing the D115A or D115N mutant H2 receptor or different levels of the wild-type H2 receptor
Because the increased agonist affinity and responsiveness are typical characteristics of constitutively active GPCRs (Lefkowitz et al., 1993), we carefully examined the basal signaling of the mutant H2 receptors. Comparing the basal cAMP production of wild-type and mutant receptors at similar receptor densities revealed that the basal cAMP production for both mutant receptors was significantly increased over that of the wild-type receptor (Fig.3A). Only at much higher H2 receptor densities, 5.2 pmol/mg protein, the basal cAMP production of the wild-type receptor is comparable with that of the mutant receptors at low receptor density (Fig. 3A). In addition, the cAMP production induced by 1 μM forskolin was also significantly higher at both mutant receptors than that of the wild-type receptor at a comparable receptor density (Fig. 3B). At high H2 receptor density (5.2 pmol/mg protein) the forskolin-induced cAMP production is again comparable with that of both mutant receptors at low receptor density. By incubation with the inverse agonist ranitidine (100 μM), the increase in both basal and forskolin-induced cAMP production was reduced to basal and forskolin-induced cAMP levels of mock-transfected cells (Fig. 3, A and B). Ranitidine did not affect basal or forskolin-induced cAMP production in mock-transfected cells (Fig. 3, A and B).

A, basal cAMP production in the absence (gray bars) or presence of 100 μM ranitidine (white bars) in HEK-293 cells transiently transfected with the wild-type H2 receptor at high (WT5168) and low (WT596) density or the D115N or D115A mutant H2 receptor. B, forskolin (1 μM)-induced cAMP production in the absence (gray bars) or presence of 100 μM ranitidine (white bars) in HEK-293 cells transiently transfected with the wild-type H2 receptor at high (WT5168) and low (WT596) density or the D115N or D115A mutant H2 receptors.Bmax values: wild-type receptor low density, 596 ± 190 fmol/mg; wild-type receptor high density, 5168 ± 1144 fmol/mg; D115A mutant receptor, 280 ± 191 fmol/mg; and D115N mutant receptor, 163 ± 80 fmol/mg. Cells were incubated in the absence or presence of the indicated drugs for 10 min at 37°C in DMEM containing 300 μM IBMX and 25 mM HEPES, pH 7.4. The data shown represent the mean ± S.E. of at least three independent experiments performed in duplicate.
Regulation of Wild-Type and Mutant H2 Receptors by Prolonged Treatment with Various Ligands.
To determine the effect of an agonist or inverse agonist on receptor expression after 24-h incubation, the ligands were added to transiently transfected HEK-293 cells directly after the transfection. The experiments on receptor regulation were only done for the mutant receptors in which the aspartate or the arginine residue were mutated to an alanine as the mutants with a mutation to an asparagine behaved similarly. In this set of experiments HEK-293 cells expressing our mutant H2 receptors (Bmax: D115A 163 ± 98 fmol/mg and R116A mutant receptor 140 ± 107 fmol/mg) were compared with HEK-293 cells expressing a high density of wild-type H2 receptors (4.3 ± 0.8 pmol/mg) to obtain similar basal cAMP levels. Prolonged treatment with a concentration (1 mM) of the agonist histamine induced a small but significant down-regulation in HEK-293 cells expressing the wild-type receptor (Table 3). However, it induced a significant up-regulation in HEK-293 cells expressing the D115A mutant receptor. Interestingly, after 24-h incubation with 1 mM histamine, HEK-293 cells transfected with the R116A receptor showed a 10-fold increase in receptor expression to a receptor density of 1.5 ± 0.6 pmol/mg protein (Table 3). Prolonged treatment with the inverse agonist ranitidine (100 μM) resulted in an increase in receptor expression in HEK-293 cells expressing either the wild-type or mutant receptors (Table 3). The increase in receptor expression of the D115A or R116A receptor was, however, much higher than the increase in expression of the wild-type receptor (Table 3).
Regulation of the expression of wild-type and mutant H2receptors after prolonged treatment of transfected HEK-293 cells with an agonist or inverse agonist
Functional Analysis of the R116A and the R116N Mutant Receptor after Pretreatment with Ranitidine.
Under normal conditions, expression of both arginine mutant receptors was too low for pharmacological characterization (Table 1). However, as receptor densities of both mutant receptors were increased by 100 μM ranitidine pretreatment, pharmacological characterization was allowed. Pretreatment itself did not significantly affect the binding properties (Table 4) or the histamine or forskolin-induced cAMP production (Fig.4, A and B) of the wild-type receptor. Basal cAMP levels of the wild-type receptor were, however, increased on ranitidine treatment (Fig. 4A). The affinity of [125I]APT for the R116A and R116N mutant receptor was comparable with the affinity determined at the wild-type H2 receptor (Table 4). However, the affinity of histamine for either the R116A or R116N mutant receptor was significantly higher compared with the wild-type receptor (Table 4). The maximum histamine-induced response at the R116A and R116N mutant receptor was significantly lower than the response at the wild-type receptor (Fig. 4B). Furthermore, the EC50 value of the histamine-induced cAMP response was increased from 101 ± 69 nM at the wild-type H2 receptor to 1408 ± 441 nM at R116A and 761 ± 102 nM at the R116N mutant receptor. Ranitidine pretreatment itself did not affect the EC50 and the Emaxvalues for the histamine-induced cAMP production at the wild-type H2 receptor. Comparing the basal cAMP production of wild-type and mutant receptors at similar receptor densities revealed that the basal cAMP production for both mutant receptors was comparable, but significantly decreased compared with the wild-type receptor (Fig. 4A). The basal cAMP levels at both mutant receptors were comparable to the cAMP production of the wild-type receptor after inhibition by 100 μM ranitidine, and, consequently, at the mutant receptors no effect of ranitidine was observed (Fig. 4A). Consistent with these findings, the forskolin-induced cAMP production was also significantly decreased at both mutant receptors compared with the wild-type receptor (Fig. 4B).
Binding characteristics of the wild-type or mutant rat H2receptors after transient expression in HEK-293 cells

A, basal cAMP production in the absence (gray bars) or presence of 100 μM ranitidine (white bars) in HEK-293 cells transiently expressing either the R116N, the R116A, or the wild-type (WT) H2 receptor. To increase expression of the mutant receptors, the cells were pretreated with 100 μM ranitidine directly after the transfection. B, basal (black bars), 100 μM histamine (white bars), and 1 μM forskolin-induced cAMP production (gray bars) in HEK-293 cells transiently expressing either the R116N, the R116A, or the wild-type (WT) H2 receptor. To increase expression of the mutant receptors, the cells were pretreated with 100 μM ranitidine directly after the transfection.Bmax values: untreated wild-type receptor, 1.8 ± 1.5 pmol/mg; pretreated wild-type receptor, 2.4 ± 1.7 pmol/mg; pretreated R116A, 0.7 ± 0.4 pmol/mg; and pretreated R116N, 1.2 ± 0.9 pmol/mg. Transiently transfected HEK-293 cells were incubated in the absence or presence of the indicated drugs for 10 min at 37°C in DMEM containing 300 μM IBMX and 25 mM HEPES, pH 7.4. The data shown represent the mean ± S.E. of at least three independent experiments.
Investigation of the Effect of the Mutation of Asp-115 or Arg-116 into Ala on the Stability of the Histamine H2Receptor.
The effect of the mutation of Asp-115 or Arg-116 into Ala on the stability of the receptor protein was subsequently investigated. The R116A mutant receptor was again pretreated with 100 μM ranitidine directly after the transfection. For proper comparison, the wild-type and D115A mutant H2 receptors were also pretreated with 100 μM ranitidine. Pretreatment of the transfected cells with 100 μM ranitidine did not affect the stability of the wild-type receptor (data not shown). As can be seen in Fig.5A, incubation of the wild-type receptor at 37°C in phosphate buffer resulted in a decrease in [125I]APT binding over time. After 24-h incubation at 37°C, the [125I]APT binding to the H2 receptor was decreased to 49 ± 6% of control incubations at 4°C. [125I]APT binding to the D115A receptor was also decreased over time but at a much higher rate than for the wild-type H2receptor (Fig. 5A). After 24-h incubation at 37°C, only 22 ± 3% of the original [125I]APT binding was left for the D115A mutant receptor. Similarly, at 37°C the [125I]APT binding to the R116A mutant receptor rapidly decreased over time (Fig. 5A). The decrease in [125I]APT binding over time for the D115A and the R116A mutant receptors was comparable and significantly higher than for the wild-type H2 receptor.

A, [125I]APT binding to HEK-293 cells transiently expressing either the D115N (open circles), the R116A (open squares), or the wild-type (closed circles) H2 receptor after incubation at 37°C for different time points. The [125I]APT binding after incubation is expressed as the percentage of [125I]APT binding of a control incubation at 4°C. The data shown represent the mean ± S.E. of at least three independent experiments performed in duplicate. The asterisks indicate a significant difference from control cells. B, [125I]APT binding to HEK-293 cells transiently expressing either the D115N, the R116A, or the wild-type (WT) H2receptor after 2-h incubation at 37°C in the presence of 100 μM ranitidine (striped bars) or 100 μM histamine (white bars) or in the absence of a ligand (gray bars). After incubation the cells were washed extensively (three times) with buffer to wash out the ligand and then used in a binding experiment. The [125I]APT binding after treatment is expressed as the percentage of [125I]APT binding of a 2-h control incubation at 4°C. The data shown represent the mean ± S.E. of at least three independent experiments performed in duplicate.
The loss of [125I]APT binding at 37°C was blocked in the presence of either an H2 receptor agonist or an antagonist (Fig. 5B). A significant decrease in [125I]APT binding to all receptors was found after 2-h incubation at 37°C in the absence of any ligand (D115A, 65 ± 6%; R116A, 55 ± 9%; wild-type receptor, 15 ± 9%). In the presence of either 1 mM histamine or 100 μM ranitidine, however, the decrease in [125I]APT binding after 2-h incubation at 37°C was almost completely prevented (Fig.5B).
Investigation of the Stability of the α1B-Adrenergic Receptor and the R143A Mutant α1B-Adrenergic Receptor.
Scheer et al. (1996) showed that a mutation in the arginine residue of the DRY motif of the α1B-adrenergic receptor expressed in COS-7 cells also resulted in the inactivity of this receptor. As found for the arginine mutants of the H2 receptor the agonist affinity of the R143A mutant receptor was significantly increased, whereas no constitutive activity was observed (Scheer et al., 1996). Our present studies on receptor regulation show that expression of the wild-type and R143A α1B-adrenergic receptor was significantly increased after long term treatment with the antagonist corynanthine (10 μM) (Fig. 6A). Expression of the R143A mutant receptor was also significantly increased after long term treatment with the agonistl-phenylephrine, thus suggesting a structural instability of the mutant receptor (Fig. 6A). l-Phenylephrine did not affect the expression of the wild-type α1B-adrenergic receptor. In experiments on the receptor stability, incubation of the wild-type α1B-adrenergic receptor at 37°C did not affect [3H]prazosin binding (Fig. 6B). [3H]Prazosin binding to COS-7 cells expressing the α1BR143A mutant receptor, however, significantly decreased over time on incubation at 37°C (Fig. 6B).
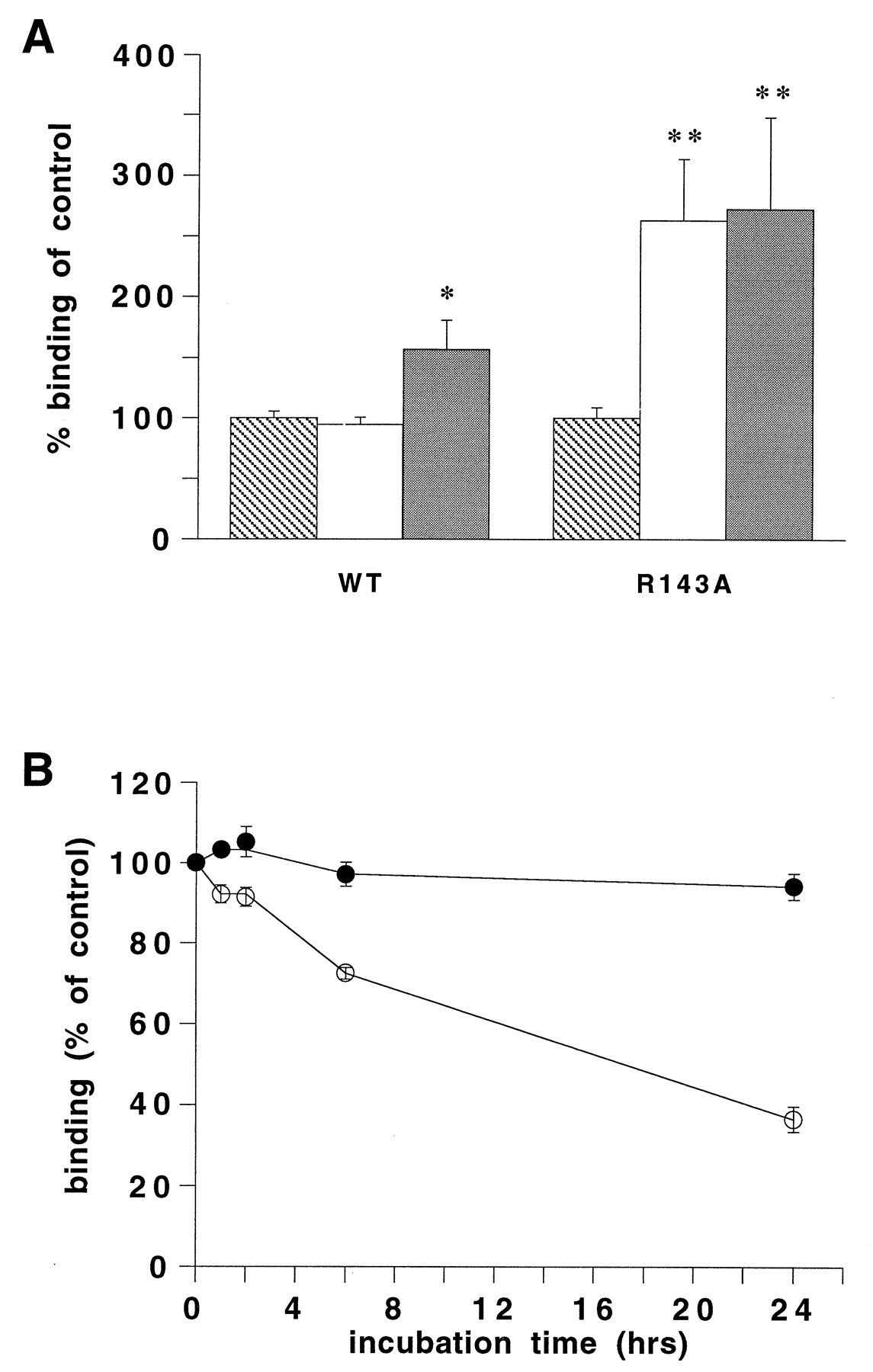
A, [3H]prazosin binding to COS-7 cells transiently expressing the R143A or wild-type α1B-adrenergic (WT) receptor after 48-h treatment with 100 μM l-phenylephrine (white bars) or 10 μM corynanthine (gray bars) or in the absence of a ligand (striped bars). The [3H]prazosin binding after treatment is expressed as the percentage of [3H]prazosin binding of untreated cells. The data shown represent the mean ± S.E. of at least three independent experiments performed in duplicate. The asterisks indicate a significant difference from untreated cells. B, [3H]prazosin binding to COS-7 cells transiently expressing the R143A (open circles) or wild-type α1B-adrenergic receptor (closed circles) after incubation for different time points at 37°C. The [3H]prazosin binding after incubation is expressed as the percentage of [3H]prazosin binding of a control incubation at 4°C. The data shown represent the mean ± S.E. of at least three independent experiments performed in duplicate.
Discussion
The aspartic acid of the highly conserved DRY motif (Probst et al., 1992; Oliveira et al., 1993) is found to be involved in stabilizing intramolecular interactions, probably with the neighboring arginine residue, constraining GPCRs in an inactive conformation (Scheer et al., 1996, 1997; Ballesteros et al., 1998). Mutations at the aspartate residue are thought to disrupt the constraint, thereby leading to a conformational change of the receptor into an active conformation (Scheer et al., 1996, 1997; Ballesteros et al., 1998). The change in conformation is hypothesized to result in a shift of the arginine residue of the DRY motif out of the polar pocket (Oliveira et al., 1993; Scheer et al., 1996, 1997; Ballesteros et al., 1998). In addition, disruption of the constraint by the introduction of a mutation might also result in structural instability of GPCRs as shown for some constitutively active mutant receptors (Gether et al., 1997;Samama et al., 1997).
In this study the role of the aspartate residue and the arginine residue of the DRY motif in signal transduction of the histamine H2 receptor was investigated. Pharmacological characterization of the mutant receptors in HEK-293 cells revealed a significantly lower expression level of all the mutant receptors than for that of the wild-type H2 receptor. Expression of the arginine mutants was even too low to allow further characterization. The decreased expression levels found for the mutant receptors could be explained by an increased basal receptor down-regulation as a consequence of constitutive receptor activation (Heinflink et al., 1995; MacEwan and Milligan, 1996; Smit et al., 1996a; Lee et al., 1997; Milligan and Bond, 1997; Alewijnse et al., 1998; Leurs et al., 1998). Another possibility is that the low expression is the result of a structural instability of the receptor (Gether et al., 1997; Samama et al., 1997; Alewijnse et al., 1998).
Investigation of both aspartate mutants of the rat H2 receptor showed a high constitutive receptor activity of these mutant receptors. Studies of the functionality of the mutant receptors were performed at comparable receptor densities, because it was shown for the wild-type H2receptor that in HEK-293 cells histamine-induced cAMP production was dependent on receptor expression (Table 2). At comparable receptor density, basal cAMP production at both D115N and D115A mutant receptors was significantly increased compared with the wild-type receptor (Fig.3A). Only at a 10-fold higher expression of the wild-type H2 receptor did basal cAMP production of the wild-type and mutant H2 receptors became comparable (Fig. 3A). Because constitutive receptor activity also affects forskolin-induced cAMP production (Alewijnse et al., 1997), the increased forskolin-induced cAMP production in HEK-293 cells, expressing the D115A or D115N mutant H2 receptor, confirmed a high constitutive activity of the mutant receptors (Fig.3B). Studies of the maximal histamine-induced cAMP production also suggest a high constitutive activity of the aspartate mutant receptors, because the maximal histamine-induced cAMP production at both mutant receptors was significantly increased over that of the wild-type receptor expressed at a comparable receptor density (Table 2).
As found for constitutively active mutant GPCRs, the agonist affinity at the D115A or D115N H2 receptors was significantly increased over that of the wild-type H2 receptor. GTPγS does not shift the histamine competition curve at both mutant receptors back to the wild-type affinity, suggesting that the increased histamine affinity is not a result of better G-protein coupling. Because the experiments were performed in cell homogenates, the resistance to GTPγS might be explained by the presence of endogenous guanine nucleotides. Yet, studies with extensively washed membrane preparations resulted in comparable data (unpublished observations).
The aspartate mutants of the H2 receptor thus show all the characteristics typical for constitutively active GPCRs (Lefkowitz et al., 1993). Mutations of the aspartate residue in the DRY motif of the α1b-adrenergic, β2-adrenergic, and V2vasopressin receptor also result in constitutive GPCR activity (Cohen et al., 1993; Scheer et al., 1996; Morin et al., 1998; Rasmussen et al., 1999). Moreover, mutation of the homologous Glu-134 of rhodopsin into glutamine results in a small but significant, light-independent activation (Cohen et al., 1993). For the GnRH receptor, the aspartate mutation increases the maximal agonist response, but the effect on the basal response has not been investigated (Arora et al., 1997). In contrast, for some adrenergic (Fraser et al., 1988) and muscarinic receptors (Fraser et al., 1988; Savarese and Fraser, 1992; Lu et al., 1997), mutations in the aspartate residue of the DRY motif did not result in constitutive GPCR activity. It should be noted, however, that for most of these mutant receptors the expression was significantly lower than the expression of the wild-type receptor (Arora et al., 1997; Lu et al., 1997). Consequently, the studies on signal transduction properties of these mutant GPCRs are not conclusive.
Besides being highly constitutively active, the D115A H2 receptor was also shown to be highly structurally instable (Fig. 5A), suggesting that the mutation disrupts stabilizing intramolecular constraints that keep the receptor in the inactive conformation. Interaction with an H2ligand, independent of the nature of the ligand, resulted in a stabilization of the receptor (Fig. 5B). These findings perfectly explain the increased expression of both D115A and D115N H2 receptors on long term treatment of the receptor with either an agonist or an inverse agonist (Table 3). In agreement with our findings, the constitutively active aspartate mutants of the β2-adrenergic receptor were also shown to be structurally instable (Rasmussen et al., 1999). Structural instability is a relatively new phenomenon first described for a constitutively active mutant of the β2-adrenergic receptor either overexpressed in Sf9 cells (Gether et al., 1997) or in vivo in the heart of transgenic mice (Samama et al., 1997). At the mutant receptor, an increase in vitro structural instability was observed leading to a loss of ligand binding (Gether et al., 1997). Expression of the mutant receptor was up-regulated in vivo by inverse agonists, neutral antagonists, and partial agonists (Samama et al., 1997). Full agonists did not up-regulate expression of the mutant receptor, because these compounds, in addition to stabilizing the receptor, also induce receptor down-regulation.
Under normal conditions, expression of the R116A and R116N mutants of the H2 receptor was too low to allow characterization. The extremely low expression of the arginine mutant receptors might be due to a high structural instability of these mutant receptors. As expected for highly structurally instable receptors, the expression of the R116A and R116N H2 receptors was dramatically increased after long term incubation with either an agonist or an inverse agonist (Table 3). Expression levels of both arginine mutant receptors after prolonged treatment with a ligand were high enough to allow additional investigation of these mutant receptors. To confirm the suggested high structural instability of the R116A receptor, experiments on receptor stability were performed. As found for the D115A mutant H2 receptor, the decrease over time in [125I]APT binding at 37°C was indeed significantly higher for the R116A H2 receptor than for the wild-type H2 receptor (Fig. 5A).
Pharmacological characterization of both arginine mutant H2 receptors revealed that, as seen for constitutively active GPCRs, the affinity of histamine for the mutant receptors was significantly increased over that of the wild-type H2 receptor. The increase in agonist affinity was, however, not accompanied by an increase in constitutive receptor activity. In contrast, basal cAMP levels of the R116A and R116N H2 receptors were significantly decreased compared with the wild-type H2 receptor, and the inverse agonist ranitidine no longer affected the basal cAMP levels (Fig. 4A). Furthermore, the maximal histamine-induced cAMP production was also significantly lower at the R116A and R116N H2 receptors than it was at the wild-type receptor (Fig. 4B). Consistent with these findings, the EC50 value of the histamine-induced cAMP production was increased at both mutant receptors. We therefore conclude that the arginine mutants of the H2receptors are in a high-affinity state but their ability to couple or activate the Gs-protein is decreased. With respect to the two-state model (Kenakin, 1996), this means that the equilibrium constant of the active (R*) and the inactive (R) state of the receptor (J parameter) and the affinity of the R* for its G-protein (M parameter) are changed simultaneously in opposite directions. In agreement with our findings at the histamine H2receptor, mutations of the arginine residue of the DRY motif have been shown to result in receptor inactivity for other GPCRs (Oliveira et al., 1993; Zhu et al., 1994; Jones et al., 1995; Scheer et al., 1996). The impaired signal transduction is, however, for most receptors not accompanied by an increase in agonist affinity except for the arginine mutant of the α1B-adrenergic receptor (Scheer et al., 1996). Our investigation of this mutant receptor revealed that this receptor is also highly structurally instable, as found for the arginine mutant of the histamine H2 receptor.
In conclusion, this study emphasizes the importance of the conserved DRY motif in signal transduction of GPCRs. Mutations of the aspartate residue resulted in a receptor with increased agonist affinity and high constitutive activity. Mutations of the arginine residue also resulted in a mutant receptor with increased agonist affinity, but the level of second messenger production is decreased. This mutant receptor seems to be in an active conformation, but the ability to couple to or activate the G-protein is decreased. We hypothesize that the disruption of an intramolecular constraint that stabilizes the receptor in an inactive conformation is responsible for the generation of an R* state of the receptor resulting in high agonist affinity. Because the arginine mutants display an impaired interaction with the G-protein, in those cases disruption of this constraint does not lead to constitutive signal transduction, despite the formation of R*. Moreover, the disruption of the stabilizing constraints also lead to GPCR instability. Our findings seem to be relevant for the whole family of GPCRs, because we show similar findings for an arginine mutant of the α1B-adrenergic receptor.
Footnotes
-
Send reprint requests to: Rob Leurs, De Boelelaan 1083, 1081 HV Amsterdam, the Netherlands. E-mail: leurs{at}chem.vu.nl
-
The authors acknowledge support from the EU BIOMED2 program “Inverse Agonism: Implications for Drug Design.”
- Abbreviations:
- GPCR
- G-protein-coupled receptor
- DMEM
- Dulbecco's modified Eagle's medium
- [125I]APT
- [125I]iodoaminopotentidine
- IBMX
- isobutylmethylxanthine
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- Received August 16, 1999.
- Accepted January 25, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}