


Abstract
γ-Aminobutyric acidA receptor γ-subunits are important for benzodiazepine (BZD) binding and modulation of the γ-aminobutyric acid-mediated Cl− current. Previously, by using γ2/α1 chimeric subunits, we identified two domains of the γ2-subunit, Lys-41-Trp-82 and Arg-114-Asp-161, that are, in conjunction, necessary and sufficient for high-affinity BZD binding. In this study, we generated additional γ2/α1 chimeric subunits and γ2 point mutants to identify specific residues within the γ2 Lys-41-Trp-82 region that contribute to BZD binding. Mutant γ2 and γ2/α1 chimeric subunits were expressed with wild-type α1 and β2 subunits in HEK 293 cells, and the binding of several BZDs was measured. We present evidence that the γ2 region Met-57-Ile-62 is important for flunitrazepam binding and that, in particular, γ2 Met-57 and γ2 Tyr-58 are essential determinants for conferring high-affinity binding. Furthermore, we identify an additional residue, γ2 Ala-79, that not only is important for high-affinity binding by flunitrazepam (a strong positive modulator) but also plays a crucial role in the binding of the imidazobenzodiazepines Ro15-1788 (a zero modulator) and Ro15-4513 (a weak negative modulator) in the BZD binding pocket. Results from site-directed mutagenesis of γ2 Ala-79 suggest that this residue may be part of a microdomain within the BZD binding site that is important for binding imidazobenzodiazepines. This separation of drug-specific microdomains for competitive BZD ligands lends insight into the structural determinants governing the divergent effects of these compounds.
γ-Aminobutyric acid (GABA) receptors are the major inhibitory neurotransmitter receptors in the mammalian brain. The native receptor is likely to be a heteropentameric protein (Nayeem et al., 1994), assembled from multiple subunit subtypes: 6 α, 4 β, 3 γ, 1 δ, 1 ε, 3 ρ, and 1 π (Barnard et al., 1998). The receptor contains an integral chloride-selective channel with specific binding sites for GABA and a variety of neuroactive drugs, including benzodiazepines (BZDs), barbiturates, neurosteroids, and anesthetics (Sieghart, 1995; Smith and Olsen, 1995). BZDs, clinically used for their anxiolytic, muscle relaxant, sedative, and antiepileptic actions, exert their therapeutic effects by allosterically modulating the activation of the GABAA receptor. Because of their clinical usefulness, a substantial effort has been made to understand the structural determinants of BZD binding in this receptor.
A variety of structurally diverse ligands bind with high affinity to the BZD binding site. These compounds include classic benzodiazepines, triazolopyridazines, imidazopyridines, cyclopyrrolones, pyrazoloquinolinones, and β-carbolines (Barnard et al., 1998). Depending on the ligand and the subunit composition of the GABAA receptor, the modulatory actions of these compounds range from full agonist (positive modulator) to inverse agonist (negative modulator). BZD positive modulators decrease the GABA concentration needed to elicit half-maximal channel activity (EC50), whereas BZD negative modulators increase the GABA EC50 value. BZD antagonists block the effect of both positive and negative modulators. Although all BZD binding site ligands appear to compete for a common binding site (McKernan et al., 1998), it is likely that different microdomains within the site interact with different subsets of BZD ligands (Davies et al., 1996).
The BZD binding site of the GABAA receptor has been proposed to lie at the interface between the α- and γ-subunits, with residues from each subunit contributing to the binding site (Smith and Olsen, 1995; Sigel and Buhr, 1997). In the α1 subunit, photoaffinity-labeling (Duncalfe et al., 1996) and mutagenesis (Wieland et al., 1992; Davies et al., 1998) experiments have identified histidine at position 101 (α1 H101) as forming part of the BZD binding site. Other α1 residues implicated in BZD binding include Tyr-159, Thr-162, Gly-200, Thr-206, Tyr-209, and Val-211 (Pritchett and Seeburg, 1991; Wieland and Luddens, 1994; Amin et al., 1997; Buhr et al., 1997b). In the γ2 subunit, only two residues have been identified as key determinants for BZD binding: γ2 Phe-77 (Buhr et al., 1997a; Wingrove et al., 1997) and γ2 Met-130 (Buhr and Sigel, 1997; Wingrove et al., 1997).
Previously, by using γ2/α1 chimeric subunits, we identified two domains of the γ2 subunit, Lys-41-Trp-82 and Arg-114-Asp-161, that together are necessary for high-affinity BZD binding (Boileau et al., 1998). In this study, by using γ/α chimeric subunits and γ2 point mutations, we focused on identifying residues within the γ2 Lys-41-Trp-82 region that contribute to BZD binding. We identify three novel residues in the γ2 subunit, γ2 Met-57, Tyr-58, and Ala-79, that are important determinants for high-affinity BZD binding.
Materials and Methods
Chimera Nomenclature.
All γ2/α1chimeric constructs in this study contain γ2 amino acids from Arg-114 to Asp-161 because this region was found to be necessary for BZD binding (Boileau et al., 1998). For ease of reading, the chimeric constructs (χ) are named for the γ2 residue before the junction of the first γ2/α1 crossover in the mature rat protein sequence (Fig.1). For example, χ40 contains γ2 sequence from Gln-1 to Asn-40 and from Arg-114 to Asp-161, whereas χ82 contains γ2 sequence from Gln-1 to Trp-82 and from Arg-114 to Asp-161. Mutations produced in chimeric backgrounds were named for the α1 residue mutated, the aligned position in the mature γ2 subunit, and the γ2 residue introduced. For instance, χ56 V76I denotes that the α1 residue (Val) in the χ56 subunit was mutated to the aligned residue (Ile) at position 76 of the mature γ2 sequence. Mutations produced in the γ2 subunit were named for the targeted γ2 residue, the position in the mature γ2 subunit, and the mutant amino acid. For example, γ2 A79C denotes that the Ala at position 79 in γ2 was mutated to Cys.
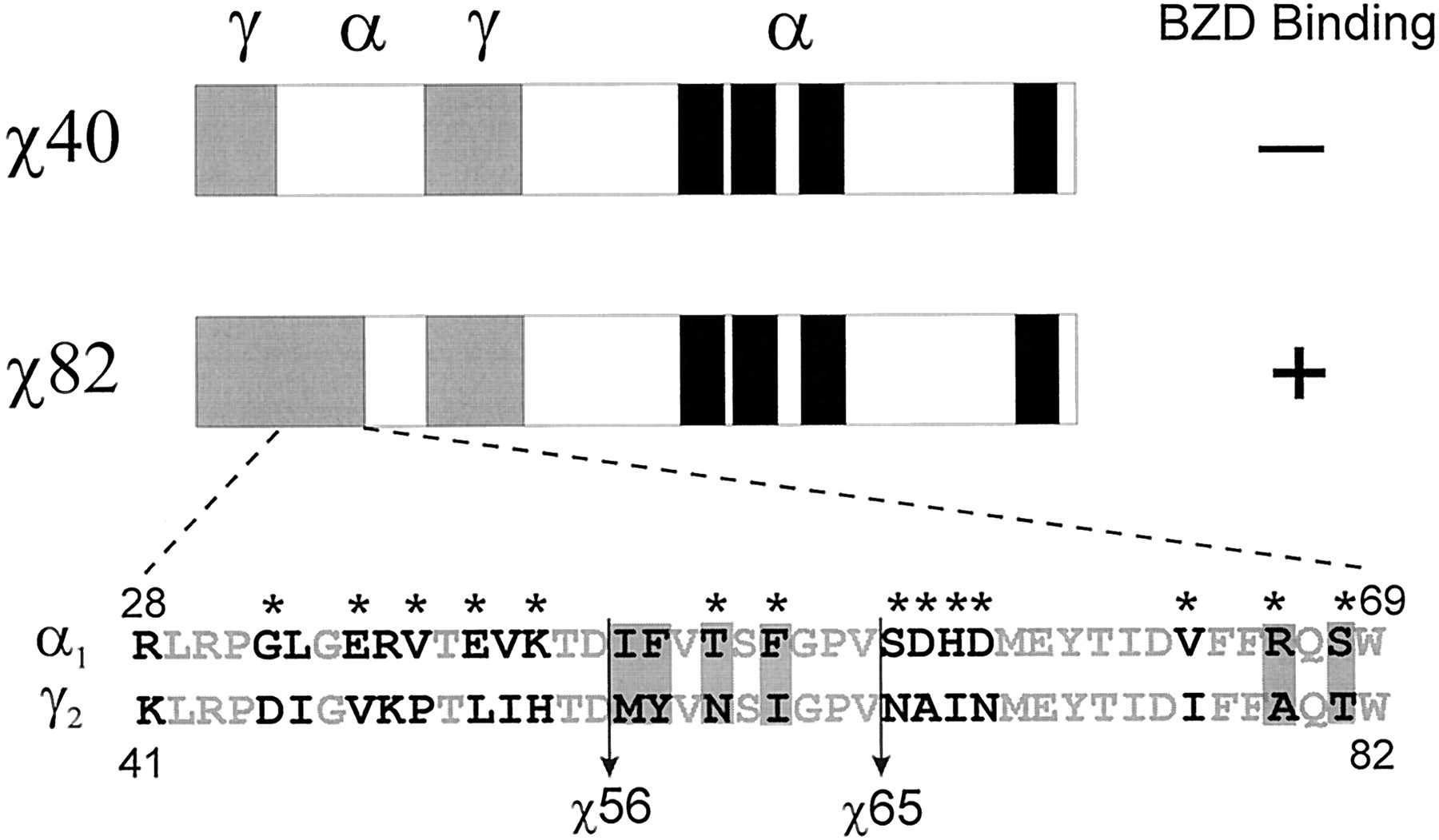
Constructed chimeric subunits and mutations used in the identification of γ2 residues important for BZD binding. γ2/α1 chimeras (χ) are named for the γ2 residue before the junction of the first γ2/α1 crossover. All χs contain additional amino acid residues from γ2 Arg-114 to Asp-161. Therefore, χ40 contains the γ2 sequence from Gln-1 to Asn-40 and from Arg-114 to Asp-161 (see Materials and Methods). The γ2 sequence is shown in gray, the α1 sequence is shown in white, and the transmembrane segments M1 through M4 are shown in black. α1β2χ82 receptors specifically bind BZDs, whereas α1β2χ40 receptors do not (Boileau et al., 1998). α1 and γ2 sequence homology in the region from γ2 Lys-41 to Trp-82 is shown in expanded form. Identical amino acid residues are shown in light gray. Residues indicated with an asterisk were targeted for mutation. Vertical lines indicate crossover transitions for χ56 and χ65. Residues that were found to influence BZD binding are boxed.
Molecular Cloning.
Rat cDNA clones for the α1, β2, and γ2 GABAA receptor subunits were used for all molecular cloning. χ40 and χ82 were produced as previously described (Boileau et al., 1998). Point mutations were made in χ40 using either the MORPH Site-Specific Plasmid DNA Mutagenesis kit (5 Prime-3 Prime, Boulder, CO) or a modified form of recombinant polymerase chain reaction (PCR). In this method, a forward mutagenic oligonucleotide was paired with a reverse template-specific oligonucleotide and amplified by PCR using an appropriate template. The resulting product was purified to remove excess oligonucleotides using the High Pure PCR Product Purification kit (Boehringer-Mannheim Biochemicals, Indianapolis, IN). Using the same template, the purified primary product, now acting as a reverse primer, was paired with an upstream vector-specific oligonucleotide and amplified. After the secondary amplification, the final product was purified as earlier and subcloned into the appropriate background.
χ56 and χ65 were produced by recombinant PCR. Then, α1-to-γ2 point mutations between χ65 and χ82 (S67N/D68A/H69I/D70N, V76I, R79A, and S81T) were made in χ65 using the modified recombinant PCR method. Single, double, and triple α1-to-γ2 mutations between χ56 and χ65 (I57M, F58Y, T60N, F62I, I57M/F58Y, T60N/F62I, I57M/F58Y/T60N, and F58Y/T60N/F62I) were made in χ56 using the modified method. Point mutations in the γ2 subunit (A79C, A79R, A79Q, A79Y, T81A, T81C, and T81S) were mutated using recombinant PCR withmyc epitope-tagged γ2 as template.
All point mutations and chimeras were subcloned into pCEP4 (InVitrogen, Carlsbad, CA) for transient expression in HEK 293 cells (ATCC CRL 1573) and were verified by restriction enzyme analysis and double-stranded DNA sequencing.
Transient Expression in HEK 293 Cells.
Cells were grown in 100-mm tissue culture dishes in minimum essential medium with Earle's salts (Life Technologies, Inc., Gaithersburg, MD) containing 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) in a 37°C incubator under a 5% CO2 atmosphere. Cells were cotransfected at 50 to 60% confluency with α1-pCEP4, β2-pCEP4, and either γ2-pCEP4 or χ-pCEP4 using a standard CaHPO4precipitation method (Graham and van der Eb, 1973). The vector pAdVAntage (Promega, Madison, WI) was added to enhance expression levels (4 μg of each subunit and 12 μg of pAdVAntage/100-mm plate). Cells were harvested and membrane homogenates were prepared as previously described (Boileau et al., 1998).
Binding Assays.
Competition binding experiments with various BZD-site ligands were performed as previously described (Boileau et al., 1998). In brief, membrane homogenates (100 μg) were incubated at room temperature with [3H]flunitrazepam (85 Ci/mmol; DuPont-New England Nuclear, Boston, MA) or [3H]Ro15-4513 (21.7 Ci/mmol; DuPont-New England Nuclear) at a concentration slightly lower thanKI and 7 to 10 concentrations of unlabeled competing ligand in a final volume of 250 μl. The unlabeled BZDs, flunitrazepam, Ro15-1788, and Ro15-4513 were generously supplied by Dr. Sepinwall (Hoffman-La Roche, Nutley, NJ). Data were fit by using the equation y =Bmax/[1 + (x/IC50)], where y is the specifically bound dpm, Bmax is the maximal binding, and x is the concentration of displacing drug (Prism; GraphPad Software, San Diego, CA).KI was calculated according to the Cheng-Prusoff/Chou equation (Cheng and Prusoff, 1973; Chou, 1974).
Immunofluorescence.
χ40, χ56, χ82, and γ2 were tagged between the third and fourth residues of the mature subunit with the myc 9E10 epitope (EQKLISEEDL) using recombinant PCR and subcloned back into each respective template. The mycepitope tag had no effect on the function or expression of the subunits. Cells were grown and transfected in 12-well dishes on poly(d)-lysine (Sigma-Aldrich)-coated 12-mm glass coverslips. Forty-eight hours after the transfection, cells were washed and fixed in 2% paraformaldehyde. Nonspecific immunoreactivity was reduced by blocking cells with 2% BSA in PBS containing: 2.7 mM KCl, 1.5 mM KH2PO4, 0.5 mM MgCl2, 137 mM NaCl, and 14 mM Na2HPO4, pH 7.1. Antibodies were diluted in the corresponding blocking buffer. In some cases, the cells were permeabilized using PBS plus 0.1% Triton X-100 before the antibody addition. The primary antibody was an anti-myc 9E10 antibody, generously supplied by Dr. Johannes Hell (University of Wisconsin-Madison), diluted at 1:500; the secondary antibody, biotin-SP goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA), was diluted to 4.4 μg/ml. The final incubation was in Texas Red-conjugated streptavidin (Jackson ImmunoResearch), diluted to 4.2 μg/ml. After several washes, the coverslips were mounted onto slides and visualized. Fluorescent images of cells were acquired with a Zeiss 35 M inverted microscope (Carl Zeiss, Thornwood, NY), 63×/1.4 NA Plan-Apochromatic objective, Texas Red filter set (Chroma Technology, Brattleboro, VT) and a Princeton Instruments MicroMax cooled CCD digital camera (Princeton Instruments, Trenton, NJ). All images were acquired at full chip resolution (Kodak KAF-1400 chip, 1035 × 1317 pixels, 6.8-μm pixel size) within the dynamic range of the camera (12-bit, 4096 gray levels) using Metamorph 4.1 imaging software (Universal Imaging, West Chester, PA). Images were scaled appropriately, converted to 8-bit images, and imported into Adobe Photoshop (ADOBE Systems, Mountview, CA).
Statistical Analysis.
We compared the effects of the mutations with the use of one-way ANOVA, applying Dunnett's post-test for significance of differences (Prism; GraphPad Software).
Results
Identification of γ2 Residues Essential for Flunitrazepam Binding.
The focus of the current study was to identify specific amino acid residues within the γ2 Lys-41-Trp-82 region that contribute to BZD binding. Previously, by comparing a γ2/α1 chimera that bound BZDs with high affinity (χ82) with one that did not (χ40), we determined that the γ2 Lys-41-Trp-82 region was important for BZD binding (Boileau et al., 1998). An amino acid alignment of the γ2 Lys-41-Trp-82 region with the equivalent α1 region (Arg-28-Trp-69) reveals 22 identical residues, 8 conservative substitutions, and 12 nonconservative substitutions (Fig. 1). In an attempt to identify single γ2 amino acid residues that contribute to BZD binding, 14 of the 20 nonidentical α1 residues in χ40 were individually mutated to the aligned γ2 residues (Fig. 1, asterisks). When the χ40 mutants were expressed with wild-type α1 and β2 subunits in HEK 293 cells, no specific [3H]flunitrazepam binding was detected (results not shown). Because no single α1-to-γ2 amino acid residue substitution tested restored high-affinity binding, it is likely that more than one γ2 residue in the Lys-41-to-Trp-82 region is important for [3H]flunitrazepam binding.
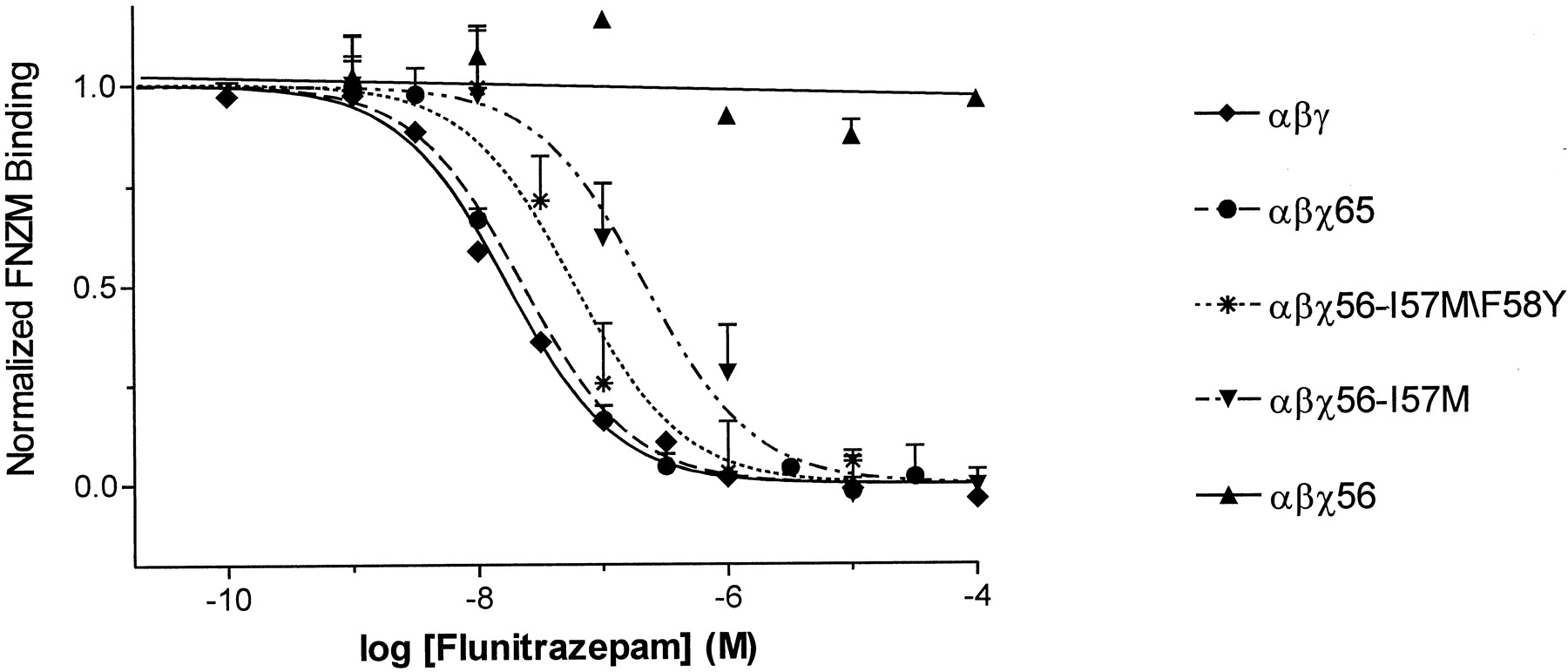
In an effort to identify multiple residues that might be required for BZD binding, two additional chimeric subunits, χ56 and χ65 (Fig.1), were constructed that contain a longer γ2 amino-terminal sequence. The new chimeric subunits were individually expressed with wild-type α1 and β2 subunits in HEK 293 cells, and the binding of [3H]flunitrazepam was measured. [3H]Flunitrazepam binding to α1β2χ56 receptors was not displaceable by concentrations of unlabeled drug up to 100 μM (Fig. 2, Table1), whereas α1β2χ65 receptors specifically bound [3H]flunitrazepam with an apparent affinity (KI = 24 nM) only 2-fold lower than that for wild-type α1β2γ2 receptors (Fig. 2, Table 1). Therefore, residues within γ2 Met-57-Val-65 are essential for high-affinity [3H]flunitrazepam binding.

γ2 Met-57 and Tyr-58 are essential for high-affinity flunitrazepam binding. [3H]Flunitrazepam affinity was measured by homologous competition radioligand binding assays on membranes prepared from HEK 293 cells transfected with αβγ, αβχ56, αβχ65, αβχ56-I57M/F58Y, and αβχ56-I57M receptors. αβχ56 data were fit by linear regression analysis, and the total binding of [3H]flunitrazepam was normalized to 1.0 for this receptor combination. The slope did not significantly deviate from zero. Data for the other receptors were normalized to specific binding and fit by nonlinear regression analysis. Data shown are single representative experiments; points are mean ± S.E. of triplicate determinations.KI values are summarized in Table 1.
Binding affinities of BZD site ligands for wild-type, chimeric, and mutant chimeric GABAA receptors
Only four residues in the γ2 Met-57-Val-65 region, γ2 Met-57, Tyr-58, Asn-60, and Ile-62, are different than the aligned α1 residues (Fig. 1). In a χ56 background, the α1 residues aligned with these four γ2 residues were individually and in combination mutated to the corresponding γ2 amino acid residues. The effects of the mutations were examined in a χ56 background to identify gain of function mutations. The mutant subunits (χ56*) were expressed with wild-type α1 and β2 subunits in HEK 293 cells, and the specific binding of [3H]flunitrazepam was measured. No specific [3H]flunitrazepam binding was detected for α1β2χ56* receptors containing the individual F58Y, T60N, or F62I mutations (results not shown). α1β2χ56* receptors containing the single I57M mutation, the double (I57M/F58Y; T60N/F62I) mutations, and the triple (I57M/F58Y/T60N; F58Y/T60N/F62I) mutant combinations specifically bound [3H]flunitrazepam (Fig. 2, Table 1). The [3H]flunitrazepam binding affinities for α1β2χ56* receptors containing the single I57M mutation (KI = 232 nM) and the double T60N/F62I mutation (KI = 219 nM) were approximately 10-fold lower than that for α1β2χ65 receptors (KI = 24 nM), whereas the α1β2χ56* receptors containing the double I57M/F58Y mutation (KI = 56 nM) and the triple I57M/F58Y/T60N or F58Y/T60N/F62I mutations (KI = 46 and 82 nM, respectively) were only 2.4-, 1.9-, and 3.5-fold lower, respectively. The results demonstrate that within the γ2 Met-57-Ile-62 region, γ2 Met-57 and γ2 Tyr-58 play a major role in conferring high-affinity flunitrazepam binding.
Surface Expression of α1β2χ GABAA Receptors.
Although the ability of γ2 Met-57 and γ2 Tyr-58 to restore high-affinity flunitrazepam binding to α1β2χ56 receptors may indicate that these residues are directly involved in flunitrazepam binding, it is also possible that these residues are required for the proper assembly and cell surface expression of α1β2χ56 receptors. To distinguish between these possibilities, we examined the assembly and cell surface expression of the γ2/α1 chimeric subunits through the use of immunohistochemistry. Because nonpentameric GABAAreceptors and unassembled α1, β2, and γ2 subunits are not expressed on the cell surface (Connolly et al., 1996; Tretter et al., 1997), cell surface expression of the γ2/α1 chimeric subunits provides strong evidence that they are assembling into mature, pentameric receptors.
The amino termini of γ2, χ40, χ56, and χ82 subunits were tagged with the myc-9E10 epitope, and the expression of the tagged subunits was assayed using anti-myc antibodies and Texas Red fluorescence. As seen in Fig. 3, nonpermeabilized HEK 293 cells expressing myc-tagged α1β2γ2, α1β2χ40, α1β2χ56, and α1β2χ82 receptors display clear surface labeling, indicating that the γ2/α1 chimeric subunits are expressed on the cell surface. Thus, the inability of χ40- and χ56-containing receptors to bind BZDs is not due to an inability of the chimeric subunits to assemble in cell-surface GABAA receptors but most likely reflects a lack of essential γ2 residues required for BZD binding. In these experiments, we assume the γ/α chimeras are assembling as γ-subunits and not as α-subunits.

Immunofluorescence of GABAA receptors expressed in HEK 293 cells demonstrates that chimeric subunits reach the cell surface. Cells transfected with α1, β2, andmyc-tagged γ2 or myc-tagged γ2/α1 chimeric subunits were either surface labeled (−) with an anti-myc 9E10 antibody or permeabilized with Triton X-100 before labeling (+) as described in Materials and Methods.
Ro15-4513 and Ro15-1788 Binding to α1β2χ GABAA Receptors.
The apparent affinities (KI values) of α1β2χ65, α1β2χ82, and α1β2γ2 receptors for [3H]flunitrazepam (BZD agonist), Ro15-1788 (BZD antagonist), and Ro15-4513 (BZD inverse agonist) were measured and compared. The affinity of α1β2χ65 receptors for [3H]flunitrazepam was not significantly different from that of α1β2χ82 receptors and was only 2-fold lower than that of α1β2γ2 receptors (Fig.4, Table 1). In contrast, the affinities of α1β2χ65 receptors for Ro15-4513 and Ro15-1788 were significantly lower than those of α1β2χ82 receptors (approximately 3-fold) and were 25- and 10-fold lower than those of the wild-type receptors, respectively (Fig. 4, Table 1). These results suggest that amino acid residues within the γ2 Asn-66-Trp-82 region preferentially influence Ro15-4513 and Ro15-1788 binding.

Displacement of [3H]flunitrazepam binding from membranes prepared from HEK 293 cells transfected with αβγ (▴), αβχ82 (▪), and αβχ65 (●). Competition of [3H]flunitrazepam binding by flunitrazepam (top), Ro15-4513 (middle), and Ro15-1788 (bottom) is shown. Data are from single representative experiments; points are mean ± S.E. of triplicate determinations. Data were fit by nonlinear regression analysis. The KI values are summarized in Table 1.
In χ65, a quadruple mutant was constructed that replaced the α1 residues S53, D54, H55, and D56 with the corresponding γ2 residues (Asn-66, Ala-67, Ile-68, and Asn-69, respectively, Fig. 1). The quadruple mutant was expressed with wild-type α1 and β2 subunits in HEK 293 cells, and the binding affinities for Ro15-1788 and Ro15-4513 were measured and compared with α1β2χ82 receptor values (Table1). The χ65 quadruple mutation (S66N/D67A/H68I/D69N) did not increase the binding affinities for Ro15-4513 or Ro15-1788 to α1β2χ82 receptor values.
Only three other residues in the γ2 Asn-66-Trp-82 region, γ2 Ile-76, Ala-79, and Thr-81, are different from the aligned α1 residues (Fig. 1). In χ65, residues corresponding to α1 Val-63, Arg-66, and Ser-68 were individually mutated to the corresponding γ2 amino acid residues (Ile-76, Ala-79, and Thr-81, respectively; Fig. 1). The three mutant subunits (χ65*) were each expressed with wild-type α1 and β2 subunits in HEK 293 cells, and the binding affinities for Ro15-1788 and Ro15-4513 were determined and compared with α1β2χ82 receptor values to identify mutations that increased affinity. The χ65 V76I mutation had no affect on Ro15-4513 or Ro15-1788 affinities (Table 1). The χ65 R79A and χ65 S81T mutations each increased the affinity for Ro15-4513 to α1β2χ82 receptor values. However, neither of these mutations increased Ro15-1788 binding affinity (Table 1). Overall, the results suggest that within the γ2 Asn-66-Trp-82 region, γ2 Ala-79 and γ2 Thr-81 preferentially influence Ro15-4513 binding. Because none of the α1-to-γ2 amino acid residue substitutions tested significantly increased Ro15-1788 affinity, it is likely that a combination of γ2 residues within the γ2 Asn-66-Trp-82 region is needed.
Analysis of Point Mutations at γ2 Ala-79 and Thr-81.
To confirm the importance of γ2 Ala-79 and γ2 Thr-81 for Ro15-4513 binding, a series of point mutations were made directly in the wild-type γ2 subunit. γ2 Ala-79 was mutated to arginine (γ2A79R), cysteine (γ2A79C), tyrosine (γ2A79Y), and glutamine (γ2A79Q), whereas γ2 Thr-81 was mutated to serine (γ2T81S), cysteine (γ2T81C), and alanine (γ2T81A). The mutant γ2 subunits were expressed with wild-type α1 and β2 subunits in HEK 293 cells, and the binding of flunitrazepam, Ro15-4513, and Ro15-1788 was measured and compared. Individual mutations altered the affinities of the three drugs by different amounts. For example, the γ2A79Y mutation had no significant effect on flunitrazepam binding but decreased Ro15-4513 and Ro15-1788 binding affinities by 52- and 21-fold, respectively, whereas the γ2 T81C mutation significantly decreased flunitrazepam, Ro15-4513, and Ro15-1788 binding affinities by 4-, 4-, and 2.5-fold, respectively (Fig. 5, Table2). In general, the mutations affected Ro15-1788 and Ro15-4513 binding more than flunitrazepam binding, and mutations at γ2 Ala-79 had much larger effects than mutations at γ2 Thr-81 (Fig. 6, Table 2). These results confirm and extend our chimeric data and indicate that γ2 Ala-79 and, to a lesser extent, γ2 Thr-81 are important for BZD binding. Moreover, the result suggests that γ2 Ala-79 preferentially influences imidazobenzodiazepine binding.

Affects of γ2 A79Y and γ2 T81C mutations on BZD binding. The myc-tagged γ2 (▪), γ2 A79Y (▴), and γ2 T81C (●) were expressed with wild-type α1 and β2 subunits in HEK 293 cells. Displacement of [3H]flunitrazepam binding by flunitrazepam (top left), Ro15-4513 (top right), and Ro15-1788 (bottom) is shown. Data are from single representative experiments; points are mean ± S.E. of triplicate determinations. Data were fit by nonlinear regression analysis. KIvalues are summarized in Table 2.
Binding affinities of BZD site ligands for myc-tagged γ2, γ2 A79 mutants, and γ2 T81 mutants

Analysis of point mutations at γ2 Ala-79 and γ2 Thr-81. The ratios of KI mutant toKI αβγmyc for flunitrazepam (FNZM, medium gray), Ro15-4513 (dark gray), and Ro15-1788 (light gray) are shown. KI values for each mutation are summarized in Table 2.
Discussion
We are interested in identifying the residues that form the BZD binding site. A binding site not only is formed by residues that directly contact agonists, antagonists, and/or inverse agonists but also includes other residues that maintain the structural integrity of the site and/or are involved in local conformational changes that occur when a ligand binds. In this study, the use of γ2/α1 intersubunit chimeras allows us to identify regions and residues unique to the γ-subunit that are required for the high-affinity binding of BZD ligands. In contrast to subunit subtype chimeras (e.g., γ3/γ2, γ1/γ2), which are useful for examining the subtle pharmacological differences between γ-subunit subtypes, γ2/α1 chimeric subunits can identify residues conserved between γ-subunit subtypes that are necessary for BZD binding.
By comparing the γ2/α1 crossover positions (Fig. 1) of a chimera that bound flunitrazepam with high affinity (χ65) with one that did not (χ56), we conclude that residues within the γ2 Met-57-Val-65 region are essential for high-affinity [3H]flunitrazepam binding (Fig.2, Table 1). The substitution of γ2 Met-57 in χ56 increases flunitrazepam binding affinity by more than 400-fold. When γ2 Met-57 and γ2 Tyr-58 are simultaneously substituted into χ56, flunitrazepam affinity increases by more than 1800-fold (Table 1). Thus, within the γ2 Met-57-Ile-62 region, γ2 Met-57 and Tyr-58 are key determinants for high-affinity flunitrazepam binding. To a lesser extent, the other unique γ2 residues in this region, γ2 Asn-60 and γ2 Ile-62, may also influence flunitrazepam binding. Only when all four γ2 residues are substituted into χ56 is high-affinity flunitrazepam binding completely restored to αβχ65 receptor levels (Table 1).
As in all mutagenesis experiments, it is difficult to establish whether these residues define a part of the BZD binding site pocket of the GABAA receptor or participate in nonlocal allosteric actions that affect BZD binding. Several lines of evidence, however, argue that the residues identified in this study, γ2 Met-57 and Tyr-58, are near the BZD binding site. The χ56 subunit is expressed in cell-surface GABAA receptors (Fig.3). Thus, the ability of γ2 Met-57 and γ2 Tyr-58 to restore high-affinity flunitrazepam binding to α1β2χ56 receptors is not due to an indirect affect of these residues on the assembly and/or expression of χ56 but more likely indicates that these residues are involved in flunitrazepam binding. Our conclusion that γ2 Met-57 and γ2 Tyr-58 are important for flunitrazepam binding is also supported by results using γ3/γ2 chimeras that suggest γ2 Met-57 influences zolpidem binding (Buhr and Sigel, 1997). Furthermore, a homologous region in the nicotinic acetylcholine receptor, γ Lys-34 and δ Ser-36, has been identified as being part of the acetylcholine binding site (Sine et al., 1995). Thus, we conclude γ2 Met-57 and Tyr-58 are involved in BZD binding. γ2 Met-57 is conserved in all γ2 subunits of various species, and γ2 Tyr-58 is conserved in all γ-subunit subtypes. We speculate that these residues interact with the aromatic groups of flunitrazepam by forming a hydrophobic subdomain in the BZD binding site.
The γ2 Met-57-Ile-62 region is the most amino-terminal region so far identified to contribute to flunitrazepam binding. If the GABA binding region of the α-subunit is homologous to the BZD binding region of the γ2 subunit, then our finding that the Met-57 region in the γ2 subunit is needed for BZD binding suggests that there may be another region in the α1 subunit, amino terminal to α1 Phe-64 (α1 Ile-44-Phe-49), that is involved in the formation of a GABA binding site. Alternatively, the γ2 Met-57 region may represent an area uniquely involved in the binding of BZDs.
Differences in the binding affinities of Ro15-4513 and Ro15-1788 for α1β2χ65 and α1β2χ82 receptors (Fig. 3, Table 1) indicate that an additional region of the γ2 subunit, γ2 Asn-66-Trp-82, influences BZD binding. By using two complementary approaches, mutagenesis of χ65 to identify mutations that increase BZD binding affinity and mutagenesis of wild-type γ2 to identify mutations that disrupt binding affinity, we conclude that γ2 Ala-79 and, to a lesser extent, γ2 Thr-81 are important for BZD binding. The inclusion of γ2 Ala-79 or Thr-81 in χ65 significantly increases Ro15-4513 binding affinity (Table 1). Consistent with these results, the substitution of γ2 Ala-79 with a variety of amino acid residues significantly decreases the affinities of flunitrazepam, Ro15-4513, and Ro15-1788 (Fig. 6, Table 2). Depending on the amino acid that is substituted, the mutation of γ2 Thr-81 also causes significant decreases in BZD binding affinities (Table 2). For this residue, the substitutions were fairly conservative (the side chains are all nearly the same size with similar hydrophilicities), which may explain the relatively small effects on affinity that were measured.
The decreases in apparent BZD binding affinity after the mutation of γ2 Ala-79 and Thr-81 can be explained by one of two mechanisms. One possibility is that the mutations directly alter the BZD binding site and disrupt the free energy of ligand binding (i.e., the microscopic binding rate constants). Alternatively, the mutations may work indirectly, at a distance, to disrupt the structural integrity of the BZD binding site. Although it is experimentally difficult to distinguish between these two mechanisms (Colquhoun, 1998), several convergent lines of evidence argue that γ2 Ala-79 and Thr-81 are located near the BZD binding site. Using a simple allosteric receptor mechanism, it has been shown that unequal shifts in the binding sensitivities of different competitive ligands in response to a mutation indicate that the mutation directly disrupts the binding site (Zhang et al., 1994). As seen in Fig. 6, individual mutations of γ2 Ala-79 and Thr-81 alter the affinities of flunitrazepam, Ro15-4513, and Ro15-1788 by different amounts, suggesting that these residues are part of the BZD binding site. The identification of γ2 Ala-79 and Thr-81 as BZD binding site residues is concordant with their proximity to γ2 Phe-77, a previously identified BZD binding site residue (Buhr et al., 1997a; Wingrove et al., 1997). Covalent modification of γ2 A79C with sulfhydryl-specific reagents is slowed in the presence of BZD ligands, lending further support that γ2 Ala-79 is part of the BZD binding site (Teissére et al., 1999). γ2 Ala-79 and Thr-81 are in homologous aligned positions as α1 Arg-66 and Ser-68, which we have recently shown to be part of the GABA binding site (Boileau et al., 1999). Because the GABA and BZD binding sites appear to be conserved structures (Sigel and Buhr, 1997), it seems probable that γ2 Ala-79 and Thr-81 contribute to part of the BZD binding site.
γ2 Ala-79 and γ2 Thr-81 are conserved in the majority of GABAA receptor γ-subunit subtypes. In general, substitutions at γ2 Ala-79 affect the binding of Ro15-4513 and Ro15-1788 more than flunitrazepam binding. Tyrosine substitution of γ2 Ala-79 alters Ro15-4513 and Ro15-1788 binding but not flunitrazepam binding (Fig. 5). Interestingly, tyrosine substitution of γ2 Phe-77, an identified binding site residue located near γ2 Ala-79, affects flunitrazepam binding but not Ro15-1788 binding (Sigel et al., 1998). This region of the γ2 subunit most likely plays a role in BZD ligand discrimination.
The orientation of BZD ligands relative to these amino acid side chains and other identified binding site residues is not known. The stabilization of BZD binding most likely involves electrostatic, van der Waals, and hydrophobic interactions as well as hydrogen bonding between the different BZD component groups and the side chains of binding site amino acid residues. The aromatic binding site residues (e.g., α1 His-101, γ2 Phe-77) may be involved in π/π stacking interactions with the aromatic portions of BZD ligands. Recent evidence suggests that the pendant phenyl group of classic BZDs such as flunitrazepam may interact with α1 His-101 (McKernan et al., 1998), γ2 Phe-77 (Sigel et al., 1998), and/or γ2 Met-130 (Wingrove et al., 1997). Other residues, such as α1 Tyr-159, α1 Thr-206, and α1 Tyr-209, may be important for hydrogen bond interactions with the seven-member amino-lactam ring of BZDs. Ultimately, confirmation of these structural predictions is dependent on crystallization of the GABAA receptor.
Many of the γ/α-interface residues that have been identified as being important for BZD binding (γ2 Phe-77, α1 Tyr-159, α1 Thr-206, α1 Tyr-209) are homologous to the α/β-interface residues that are important for the binding of GABA (α1 Phe-64, β2 Tyr-157, β2 Thr-202, and β2 Tyr-205). In the aligned sequences of the subunits, these residues are identical. However, because the molecular structures of GABA and BZDs are quite distinct, it is likely that nonconserved residues are required to impart pharmacological specificity to these sites. Because γ2 Met-57 and γ2 Ala-79 are not conserved in the aligned positions of any α-subunit, we hypothesize that these particular amino acid side chains may be uniquely involved in BZD binding specificity.
In summary, our results are most simply explained by a model in which γ2 Met-57, Tyr-58, and Ala-79 line part of the BZD binding site. The identified residues are clustered in two distinct domains separated by about 20 residues in the linear γ2 sequence. Not all of these residues have to make contact with BZDs. Some of the residues may be important for maintaining the local physicochemical properties of the site or be involved in the local changes that occur at the binding site when BZDs bind. In the absence of a high-resolution crystal structure, identification of the amino acid residues involved in BZD binding is a first step toward building a detailed molecular model of the BZD binding site pocket.
Acknowledgments
We thank Joan Meister and Inge Siggelkow for expert technical help with the immunohistochemistry and Eric Dent for his expert help with the photomicroscopy.
Footnotes
-
Send reprint requests to: Cynthia Czajkowski, Ph.D., Department of Physiology, University of Wisconsin-Madison, 1300 University Ave., MSC Room 197A, Madison, WI 53706. E-mail:czajkowski{at}physiology.wisc.edu
-
This work was supported in part by grants to C.C. from the Alcoholic Beverage Association and NINDS-the National Institutes of Health. C.C. is a recipient of the Burroughs Wellcome Fund New Investigator Award in the Basic Pharmacological Sciences.
- Abbreviations:
- GABA
- γ-aminobutyric acid
- PCR
- polymerase chain reaction
- BZD
- benzodiazepine
- Received October 14, 1999.
- Accepted January 5, 2000.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}