


Abstract
Using cultured airway smooth muscle cells, we showed previously that the platelet-derived growth factor (PDGF) receptor uses the G-protein, Gi, to stimulate Grb-2-associated phosphoinositide 3-kinase (PI3K) activity. We also showed that this was an intermediate step in the activation of p42/p44 mitogen-activated protein kinase (p42/p44 MAPK) by PDGF. We now present two lines of evidence that provide further support for this model. First, we report that PDGF stimulates the Gi-mediated tyrosine phosphorylation of the Grb-2 adaptor protein, Gab1. This phosphorylation appears to be necessary for association of PI3K1a with the Gab1-Grb-2 complex. Second, PI3K appears to promote the subsequent association of dynamin II (which is involved in clathrin-mediated endocytic processing) with the complex. Furthermore, inhibitors of PI3K and clathrin-mediated endocytosis reduced the PDGF-dependent activation of p42/p44 MAPK, suggesting a role for PI3K in the endocytic signaling process leading to stimulation of p42/p44 MAPK. Together, these results begin to define a common signaling model for certain growth factor receptors (e.g., PDGF, insulin, insulin-like growth factor-1, and fibroblast growth factor) which use Gi to transmit signals to p42/p44 MAPK.
Mitogenic stimuli initiate cell proliferation via different classes of cell surface receptors that include both growth factor tyrosine kinase and G-protein-coupled receptors (GPCR). This involves the stimulation of the p42/p44 mitogen-activated protein kinase (p42/p44 MAPK) pathway. Both growth factors and GPCR agonists stimulate the tyrosine phosphorylation of Shc [Src homology 2 (SH2)-containing protein] and the sequential activation of Grb-2-mSos, Ras, c-Raf, MEK-1, and p42/p44 MAPK (Marshall, 1995). Gi-coupled receptor agonists also activate nonreceptor tyrosine kinases, such as c-Src tyrosine kinase, which function as intermediates between Giβγ and Ras-dependent p42/p44 MAPK activation (Van Biesen et al., 1995; Dikic et al., 1996; Wan et al., 1997). Furthermore, certain receptor tyrosine kinases appear to use classical GPCR signaling pathways to stimulate p42/p44 MAPK activation. For instance, Luttrell and colleagues have shown that pertussis toxin (which inactivates Gi) and an inhibitor of Giβγ-mediated signaling (C-terminal domain of β-adrenergic receptor kinase) blocked the activation of p42/p44 MAPK by insulin and insulin-like growth factor-1 (IGF-1) in rat fibroblasts (Luttrell et al., 1995). There is also evidence that fibroblast growth factor activation of p42/p44 MAPK involves Gi. In these studies, the C-terminal domain of β-adrenergic receptor kinase and pertussis toxin blocked the fibroblast growth factor stimulation of p42/p44 MAPK and promoted fibroblast differentiation (Fedorov et al., 1998).
Lefkowitz and colleagues have proposed that the GPCR-dependent signaling mechanism regulating activation of p42/p44 MAPK in response to IGF-1 and β-adrenergic agonists in fibroblasts involves an endocytic pathway mediated by β-arrestin I/II and dynamin II (Daaka et al., 1998; Ahn et al., 1999; Lin et al., 1999). Dynamin II is a Grb-2 adaptor protein that regulates the GTP-dependent pinching off of clathrin-coated endocytic vesicles, containing receptor signal complexes, from the plasma membrane (Takei et al., 1995). c-Src tyrosine kinases phosphorylate dynamin II in response to β-adrenergic agonists (Ahn et al., 1999), whereas lysophosphatidic acid-stimulated tyrosine phosphorylation of dynamin II is inhibited by pertussis toxin and wortmannin [phosphoinositide 3-kinase (PI3K) inhibitor] in Cos-7 cells (Kranenburg et al., 1997, 1999). Therefore, Gi/o and PI3K transmit signals to an unidentified tyrosine kinase, which can subsequently phosphorylate dynamin II.
In a recent study, we showed that platelet-derived growth factor (PDGF) uses both Gi-dependent and -independent routes to regulate p42/p44 MAPK in cultured airway smooth muscle (ASM) cells (Conway et al., 1999). We further characterized the Gi-dependent route and demonstrated the involvement of c-Src and a Grb-2-associated PI3K (Conway et al., 1999). In this article, we report that PDGF stimulates a Gi-mediated tyrosine phosphorylation of Gab1 [which is 30–47% homologous with the insulin receptor substrate-1 (IRS-1)]. Gab1 is a Grb-2 adaptor protein that has been implicated in IGF-1, epidermal growth factor, T-antigen, and gp130 cytokine receptor regulation of PI3K and p42/p44 MAPK (Holgado-Madruga et al., 1996; Takahashi-Tezuka et al., 1998; Von Willebrand et al., 1998; Lin et al., 1999). We have also demonstrated that the PDGF-stimulated tyrosine phosphorylation of Gab1 may be necessary for PI3K1a association with the Gab1-Grb-2 complex. Furthermore, the subsequently formed PI3K1a-Gab1-Grb-2 complex appears to have an important role in regulating dynamin II-mediated endocytic signaling to p42/p44 MAPK. These results begin to define a common signaling model for certain growth factor receptors that use Gi to transmit signals to p42/p44 MAPK.
Experimental Procedures
Materials.
All biochemicals, including collagenase, elastase, soya bean trypsin inhibitor, and PDGF-AB (specific for both PDGFα and β receptors) were from Sigma Chemical Co. (Dorset, UK). Pertussis toxin was from Calbiochem (Nottingham, UK). [γ-32P]ATP (3000 Ci/mol), MAPK Biotrak assay kits, and enhanced chemiluminescence reagents were from Amersham Pharmacia Biotech (Bucks, UK). Cell culture supplies were from Life Technologies (Paisley, UK). Anti-phospho-p42/p44 MAPK, p42 MAPK, PY20 horseradish peroxidase (HRP)-phosphotyrosine, and dynamin II antibodies were from Transduction Laboratories (Lexington, KY). Anti-Grb-2, p85 regulatory subunit of PI3K1a, Gab1 antibodies, and A431 and PC12 cell lysate were from Santa Cruz Biotechnology (Santa Cruz, CA). Reporter HRP-anti-mouse/rabbit antibodies were from the Scottish Antibody Production Unit (Carluke, Scotland). Male Dunkin-Hartly guinea pigs (200–400 g) were used for isolation of tracheal smooth muscle.
Cell Culture.
The preparation of the primary cultures of guinea pig ASM cells has been described previously (Pyne and Pyne, 1993). Their identity was confirmed to be smooth muscle by the presence of α-actin using smooth muscle-specific mouse anti-α-actin monoclonal antibodies. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% (v/v) fetal calf serum and 10% (v/v) horse serum. Cells were routinely used at passage 3 to 4, where they exhibit a proliferative phenotype. These cells were maintained in Dulbecco's modified Eagle's medium supplemented with 0.1% (v/v) fetal calf serum and 0.1% (v/v) horse serum for 24 h before experimentation. Older cells (passage 5–6) become enlarged, exhibit a hypertrophic phenotype, and have a slow rate of proliferation.
p42/p44 MAPK Assays.
p42 MAPK phosphorylation was measured using the electrophoretic mobility shift assay according to Conway et al. (1999). Phosphorylated/activated p42 MAPK migrates on SDS polyacrylamide gel electrophoresis (SDS-PAGE) with a slower mobility compared with the dephosphorylated form. The phosphorylation of p42/p44 MAPK was also measured by Western blotting with anti-phospho-ERK-1/2 antibody. p42/p44 MAPK activity was measured in cell lysates using a specific p42/p44 MAPK peptide substrate (EGFR661–680 peptide synthesized to contain one phosphorylation site) as described by us previously (Conway et al., 1999). The treatment of cells with PD098059 (an inhibitor of MEK-1 activation) abolished PDGF-stimulated p42/p44 MAPK activity measured in both the kinase activity and shift assays (Pyne and Pyne, 1998).
Immunoprecipitation.
The medium was removed, and cells were lysed in ice-cold immunoprecipitation buffer A (1 ml, containing 20 mM Tris/HCl; 137 mM NaCl; 2.7 mM KCl; 1 mM MgCl2; 1 mM CaCl2; 1% (v/v) Nonidet P-40 (NP-40); 10% (v/v) glycerol; 1 mg/ml BSA; 0.5 mM sodium orthovanadate; 0.2 mM PMSF; and 10 μg/ml leupeptin, antipain, pepstatin, and aprotinin; pH 8) for 10 min at 4°C. The material was harvested, centrifuged at 22,000g for 5 min at 4°C, and 200 μl of cell lysate supernatant (equalized for protein, 0.5–1 mg/ml) was taken for immunoprecipitation with antibodies (5 μg of antibodies and 40 μl of 1 part immunoprecipitation buffer and 1 part protein A/protein G Sepharose CL4B). After agitation for 2 h at 4°C, the immune complex was collected by centrifugation at 22,000g for 15 s at 4°C. The immunoprecipitates were washed twice with buffer B containing 10 mM HEPES, pH 7, 100 mM NaCl, 0.2 mM PMSF, 10 μg/ml leupeptin, 20 μg/ml aprotinin, and 0.5% (v/v) NP-40 and once in buffer B without NP-40.
Immunoprecipitates were resuspended in 20 mM β-glycerophosphate, 5 mM Na4P2O7, 30 mM NaCl, and 1 mM dithiothreitol, pH 7.2 for PI3K activity assays or in boiling sample buffer (0.125 M Tris/HCl, pH 6.7, 0.5 mM Na4P2O7, 1.25 mM EDTA, 2.5% (v/v) glycerol, 0.5% (w/v) SDS, 25 mM dithiothreitol, 1% (w/v) bromophenol blue) for SDS-PAGE and Western blotting with antibodies. The specific immunoprecipitation of proteins was not evident when antibodies were omitted from the protocol (data not shown).
Blotting.
Immunoblotting was performed as described by us previously (Conway et al., 1999). After washing the blots, immunoreactive bands were visualized using the enhanced chemiluminescence detection kit.
PI3K Assay.
Resuspended anti-Grb-2 and phosphotyrosine immunoprecipitates (40 μl) were each combined with 20 μl of phosphatidylinositol (3 mg/ml) in incubation buffer containing 1% cholate. To each, 40 μl of [32P]ATP (3 μM Na2ATP, 7.5 mM MgCl2, 0.25 mCi/ml [32P]ATP, final specific activity was 0.083 μCi/pmol) was added. The reaction was performed at 37°C for 15 min and was terminated by adding 450 μl of chloroform/methanol (1:2 v/v). Organic and aqueous phases were resolved by adding 150 μl chloroform and 150 μl 1 M HCl. Samples were mixed and centrifuged (4200g for 10 min). This was repeated and the lower phase harvested and evaporated to dryness and [32P]phosphatidylinositol 3-phosphate resolved by thin-layer chromatography using chloroform/methanol/ammonia/H2O (210:300:45:75, v/v) in parallel with a nonradioactive standard. Radioactive bands were visualized by autoradiography, and samples corresponding to [32P]phosphatidylinositol 3-phosphate were scraped from the plate and subjected to Cerenkov counting.
Results
The Role of Gi in Regulating PDGF-Dependent Stimulation of p42/p44 MAPK.
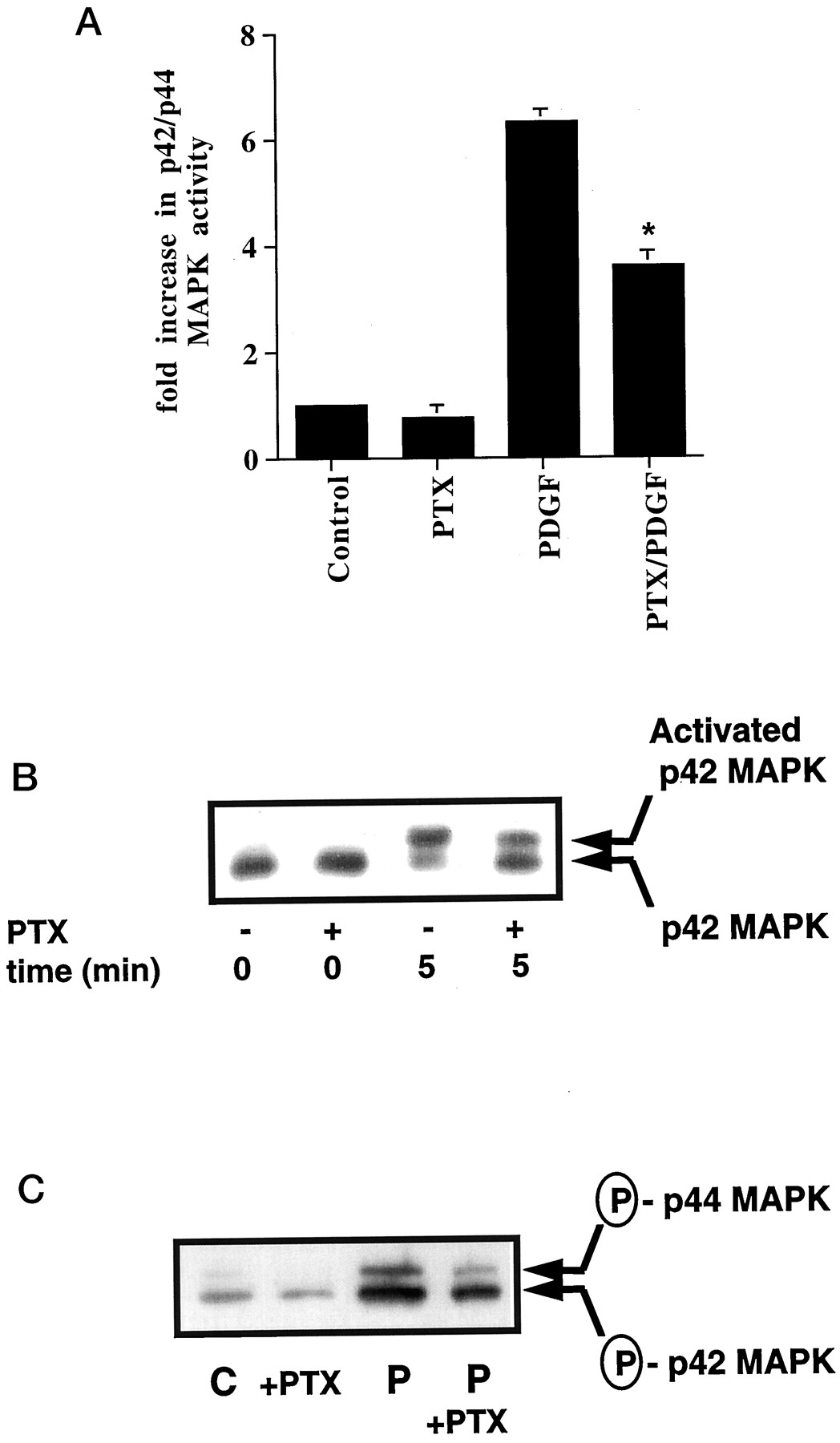
In previous studies, we showed that the pretreatment of ASM cells with 0.1 μg/ml pertussis toxin [which ADP ribosylates and inactivates all the Gi in the cell; Go is not expressed in ASM cells (Conway et al., 1999)] for 24 h reduced the PDGF-dependent activation of p42/p44 MAPK by approximately 50% (assessed using an in vitro kinase activity assay and by shift blot analysis) (Conway et al., 1999). These results are shown in Fig. 1, A and B. Similar results were obtained in cells stimulated with PDGF up to 30 min (Conway et al., 1999). In the current paper, we have also confirmed these results using a different approach, where samples were Western blotted with anti-phospho-p42/p44 MAPK-specific antibodies. Figure 1C shows that pertussis toxin induced a 50% reduction in the PDGF-stimulated phosphorylation of p42/p44 MAPK. Previous control experiments showed that pertussis toxin had no effect on PDGF receptor tyrosine phosphorylation and did not increase cyclic AMP levels (Conway et al., 1999). The latter was important because cyclic AMP inhibits PDGF-stimulated activation of p42/p44 MAPK in ASM cells (Pyne and Pyne, 1998).

The Gi-mediated regulation of p42/p44 MAPK in response to PDGF. ASM cells were treated with and without pertussis toxin (PTX; 0.1 μg/ml, 24 h) before stimulation with PDGF (P; 10 ng/ml, 5 min) as required. A, histogram showing effect of PTX on the PDGF-dependent activation of p42/p44 MAPK. Basal p42/p44 MAPK activity was 115 ± 23 pmol/min/mg of protein in control cells. Results are expressed as the fold increase over basal p42/p44 MAPK activity in control cells (means ± S.D. forn = 5–10 separate cell preparations; *P < .05 for PTX/PDGF versus PDGF alone, Student's t test). B, corresponding shift blot showing the phosphorylation of p42 MAPK. C, Western blot with anti-phospho-p42/p44 MAPK antibody. Reprobing the Western blot with anti-p42/p44 MAPK antibodies showed equal loading of these kinases in each sample (data not shown). B and C, representative results of an experiment performed on three separate cell preparations. C, control.
Role of Gab1 and PI3K1a.
Previously, we have shown that the PDGF receptor uses Gi to stimulate Grb-2-associated PI3K activity in ASM cells (Conway et al., 1999). Thus, PDGF stimulated PI3K activity in anti-Grb-2 immunoprecipitates was abolished in cells pretreated with pertussis toxin [fold increase in PI3K activity in anti-Grb-2 immunoprecipitates: control, 1 ± 0.2; pertussis toxin (0.1 μg/ml, 24 h), 0.5 ± 0.2; PDGF (10 ng/ml, 10 min), 5.4 ± 1; PDGF + pertussis toxin, 1.3 ± 0.2. Results are means ± S.D. for three separate cell preparations.]. An important role for PI3K was established using the PI3K inhibitor, wortmannin, which abolished PDGF-stimulated p42/p44 MAPK activation (Fig. 2, A and B). Therefore, in this study, we aimed to determine the identity of the Grb-2-associated PI3K and to investigate the Gi-dependent mechanism regulating their interaction.
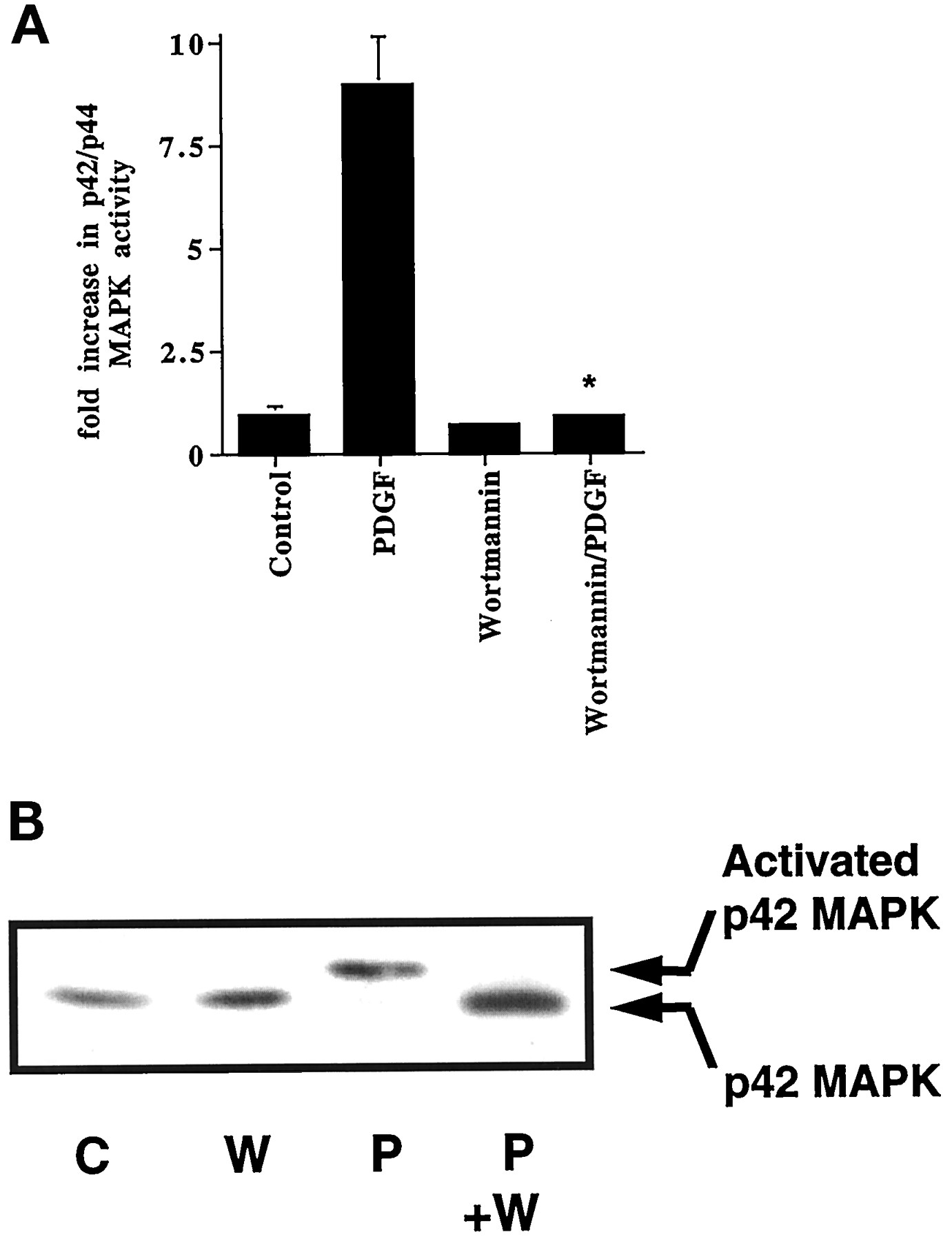
PDGF stimulation of p42/p44 MAPK is PI3K-dependent. ASM cells were treated with and without wortmannin (W; 50 nM, 15 min) before stimulation with PDGF (P; 10 ng/ml, 10 min) as required. A, histogram showing effect of wortmannin on the PDGF-dependent activation of p42/p44 MAPK. Basal p42/p44 MAPK activity was 115 ± 23 pmol/min/mg of protein in control cells. Results are expressed as the fold increase over basal p42/p44 MAPK activity in control cells (means ± S.D. for n = 5–10 separate cell preparations; *P < .005 for wortmannin/PDGF versus PDGF alone, Student's t test). B, corresponding shift blot showing phosphorylation of p42 MAPK. The blot is a representative result of an experiment performed on three separate cell preparations. C, control.
First, the p85 regulatory subunit of PI3K1a was detected in anti-Grb-2 immunoprecipitates by Western blotting with anti-p85 antibodies (Fig.3). Significantly, the amount of p85 PI3K1a associated with Grb-2 was increased in cells stimulated with PDGF and was prevented by pretreating cells with pertussis toxin (Fig.3). Equal amounts of Grb-2 were immunoprecipitated from these samples (see later). Therefore, changes in p85 PI3K1a association could be correlated with PI3K activity in anti-Grb-2 immunoprecipitates.

p85 PI3K1a interaction with Grb-2. ASM cells were treated with and without pertussis toxin (PTX; 0.1 μg/ml, 24 h) before stimulation with PDGF (P; 10 ng/ml, 10 min) as required. Western blot of anti-Grb-2 immunoprecipitates with anti-p85 PI3K1a antibodies, showing the effect of pertussis toxin on PDGF-stimulated p85 PI3K1a association with Grb-2. The p85 regulatory subunit of PI3K1a in A431 cell lysates was immunostained with antibodies as a positive control. This is a representative result of an experiment performed on three separate cell preparations. C, control; IP, immunoprecipitation; WB, Western blot.
Second, we evaluated the role of Gab1, which is 30 to 47% homologous with the insulin receptor substrate-1 (IRS-1). This protein was chosen for study because others have shown that it is a Grb-2 adaptor protein that binds and activates PI3K (Holgado-Madruga et al., 1996). Indeed, our experiments showed that Gab1 was constitutively associated with Grb-2 in ASM cells. This was based on the coimmunoprecipitation of Grb-2 with anti-Gab1 antibodies. Grb-2 (molecular mass of 24 kDa) was detected by Western blotting anti-Gab1 immunoprecipitates with an anti-Grb-2 antibody. The amount of Grb-2 coimmunoprecipitated from cell lysates by anti-Gab1 antibody was not affected by stimulating cells with PDGF or by pretreatment with pertussis toxin (Fig.4A). Similarly, the amount of Gab1 (molecular mass of 115 kDa) immunoprecipitated was unaffected (Fig.4B). Figure 4C shows the specificity of the anti-Grb-2 and Gab1 antibodies in the immunoprecipitation assays. First, Grb-2 immunoprecipitated with anti-Gab1 antibodies is aligned with comigrating Grb-2 in ASM cell lysates. Second, Gab1 immunoprecipitated with anti-Gab1 antibodies is aligned with comigrating Gab1 in ASM and A431 cell lysates. Identical results were obtained when anti-Grb-2 antibodies were used to coimmunoprecipitate the Grb-2-Gab1 complex (data not shown).

Gab1-Grb-2 interaction. ASM cells were treated with and without pertussis toxin (PTX; 0.1 μg/ml, 24 h) before stimulation with PDGF (P; 10 ng/ml, 10 min) as required. Western blots of anti-Gab1 immunoprecipitates with anti-Grb-2 antibodies (A) or anti-Gab1 antibodies (B). A431 cell lysates were run on the same SDS-PAGE and used as a positive control to detect Gab1 in B. C, anti- Gab1 immunoprecipitates Western blotted with anti-Grb-2 antibody (left) and anti-Gab1 and HRP-linked anti-phosphotyrosine antibodies (right) to show specificity of immunoprecipitation. Grb-2 is aligned with Grb-2 detected in ASM cell lysates, whereas Gab1 is aligned with Gab1 detected in ASM, PC12, and A431 cell lysates. Tyrosyl phosphorylated p85 PI3K in anti-Gab1 immunoprecipitates is also shown on the Western blot with HRP-linked anti-phosphotyrosine antibodies. Immunoprecipitations were from PDGF-treated cell samples. A through C, representative results of an experiment performed on three separate cell preparations. The line bar denotes the position of Gab1 and p85PI3K1a. C, control; IP, immunoprecipitation; P-Tyr, phosphotyrosine antibody; WB, Western blot.
PI3K1a was associated with Gab1 in anti-Gab1 immunoprecipitates. This association was stimulated by PDGF and blocked by pretreating cells with pertussis toxin [Fig. 5A; fold increase in PI3K association in anti-Gab1 immunoprecipitates: control, 1 ± 0.1; pertussis toxin (0.1 μg/ml, 24 h), 0.2 ± 0.1; PDGF (10 ng/ml, 10 min), 3.6 ± 0.3; PDGF + pertussis toxin, 0.9 ± 0.2. Results are means ± S.D. for three separate cell preparations.]. Equal amounts of Gab1 were immunoprecipitated from these samples (Fig. 4B). Therefore, our results show that PDGF stimulates the binding of PI3K1a to Gab1 in a pertussis toxin-sensitive manner, and this was correlated with p85 PI3K1a association with Grb-2 (Fig. 3). We also found that the p85 PI3K1a that binds to Gab1 is tyrosine phosphorylated (Fig. 4C).

PDGF-stimulated PI3K1a-Gab1 complex formation. ASM cells were treated with and without pertussis toxin (PTX; 0.1 μg/ml, 24 h) before stimulation with PDGF (P; 10 ng/ml, 10 min) as required. A, Western blot of anti-Gab1 immunoprecipitates with anti-p85 PI3K1a antibodies showing the effect of pertussis toxin on PDGF-stimulated p85 PI3K1a association with Gab1. A431 cell lysates were run on the same SDS-PAGE and used as a positive control to detect p85 PI3K1a. B, anti-Gab1 immunoprecipitates and ASM, PC12, and A431 cell lysates Western blotted with anti-p85 PI3K1a antibody to show specificity of immunoprecipitation. Immunoprecipitations were from PDGF-treated cell samples. A and B, representative results of an experiment performed on three separate cell preparations. C, control; IP, immunoprecipitation; WB, Western blot.
PI3K activity present in anti-phosphotyrosine immunoprecipitates was stimulated by 30-fold in cells treated with PDGF and was not blocked by pertussis toxin [fold increase in PI3K activity in anti-phosphotyrosine immunoprecipitates: control, 1 ± 0.1; pertussis toxin (0.1 μg/ml, 24 h), 1.4 ± 0.3; PDGF (10 ng/ml, 10 min), 30 ± 1.2; PDGF + pertussis toxin, 31 ± 2. Results are means ± S.D. for three separate cell preparations.] (Conway et al., 1999). Thus, the binding of PI3K1a to Gab1 is pertussis toxin-sensitive, whereas its PDGF-stimulated tyrosyl phosphorylation is insensitive to this toxin.
Figure 5B shows the specificity of p85 PI3K1a immunoprecipitation with anti-Gab1 antibodies. p85 PI3K1a in anti-Gab1 immunoprecipitates is aligned with comigrating kinase in ASM, PC12, and A431 cell lysates.
Tyrosine Phosphorylation of Gab1.
Others have shown that growth factors stimulate the tyrosine phosphorylation of Gab1 to promote the binding of PI3K (Holgado-Madruga et al., 1996;Takahashi-Tezuka et al., 1998; Von Willebrand et al., 1998). Indeed, PDGF stimulated the tyrosine phosphorylation of Gab1 in ASM cells. Thus, a 115-kDa tyrosine phosphorylated protein, corresponding to Gab1 and comigrating with Gab1 in an A431 lysate standard, was immunoprecipitated from PDGF-stimulated, but not control, ASM cells using anti-Gab1 antibodies (Fig. 6). Furthermore, pertussis toxin pretreatment abolished the PDGF-stimulated tyrosine phosphorylation of Gab1. These represent changes in the phosphorylation state of Gab1 because equal amounts of this protein were immunoprecipitated with anti-Gab1 antibodies (Fig. 4B). Our findings, therefore, link the Gi dependence of Gab1 tyrosine phosphorylation with the binding of PI3K1a to the Gab1-Grb-2 complex. Figure 4C shows anti-Gab1 immunoprecipitates probed with HRP-linked anti-phosphotyrosine antibody to define the specificity of the immunoprecipitation and to demonstrate the presence of tyrosyl phosphorylated Gab1 and p85 PI3K1a. Interestingly, in older cells that exhibited a hypertrophic phenotype, PDGF stimulated a more robust tyrosine phosphorylation of Gab1 that was pertussis toxin-insensitive (data not shown).
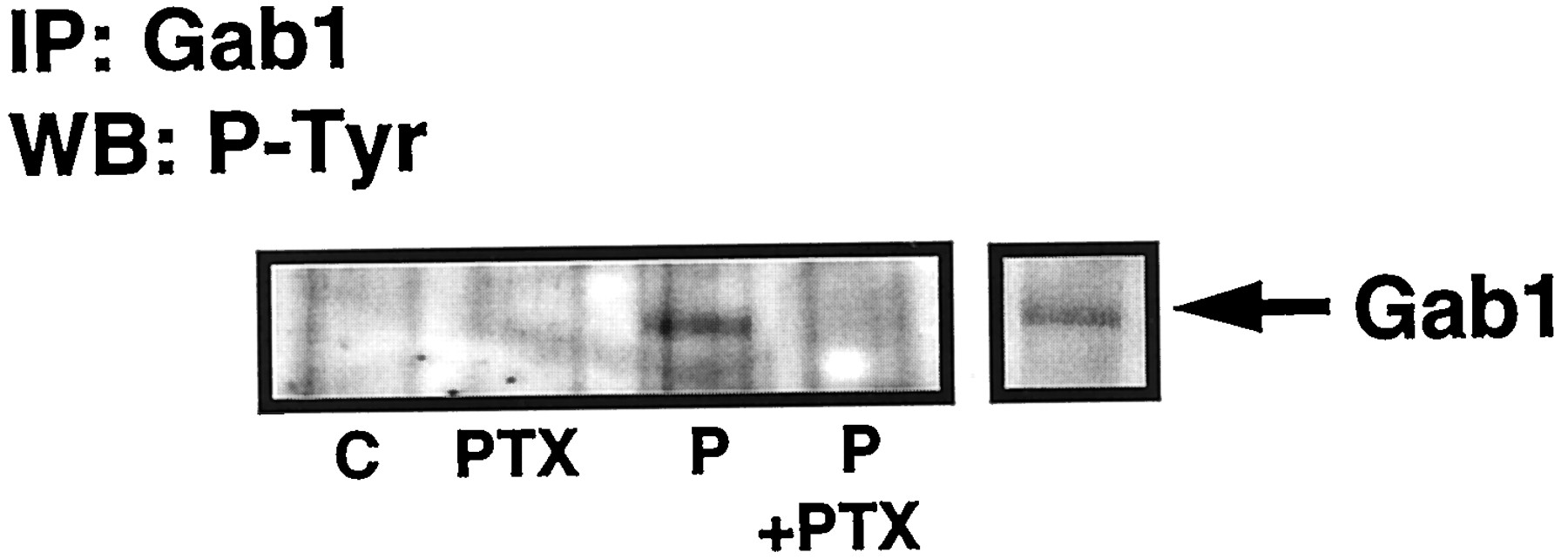
Gab1 tyrosine phosphorylation. ASM cells were treated with and without pertussis toxin (PTX; 0.1 μg/ml, 24 h) before stimulation with PDGF (P; 10 ng/ml, 10 min) as required. Western blot of anti-Gab1 immunoprecipitates with HRP-linked anti-phosphotyrosine antibodies showing effect of pertussis toxin on PDGF-stimulated tyrosine phosphorylation of p115 Gab1. A431 cell lysates were run on the same SDS-PAGE and used as a positive control to detect Gab1. This is a representative result of an experiment performed on three separate cell preparations. C, control; IP, immunoprecipitation; WB, Western blot.
The Role of Dynamin II.
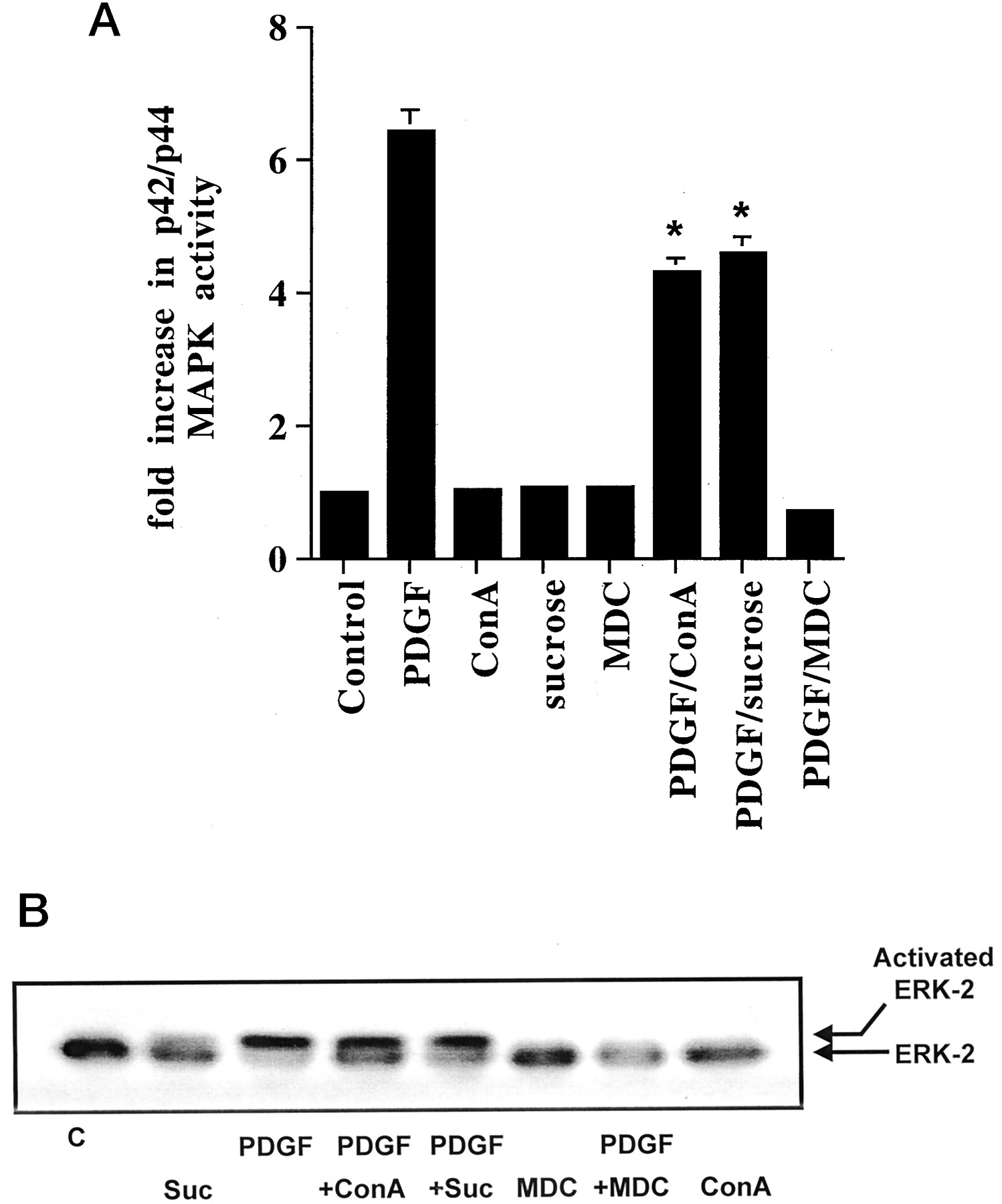
The role of PI3K in regulating p42/p44 MAPK activation was further investigated. We were interested in the possibility that PI3K might regulate dynamin II, a Grb-2 adaptor protein that has been implicated in clathrin-mediated endocytic signaling to p42/p44 MAPK (Daaka et al., 1998; Ahn et al., 1999; Lin et al., 1999). In previous studies, inhibitors of clathrin-mediated endocytosis, such as concanavalin A (Con A), which prevents growth factor receptor clustering; hypertonic sucrose, which blocks clathrin association; and monodansylcadaverine (MDC), have been shown to ablate IGF-1-stimulated p42/p44 MAPK activation in fibroblasts (Daaka et al., 1998). In this study, we show that these inhibitors also reduce the stimulation of p42/p44 MAPK by PDGF (Fig.7, A and B). Con A and hypertonic sucrose reduced the PDGF-dependent activation of p42/p44 MAPK [by 38.6 ± 5.5% (n = 5) and 33.6 ± 10% (n= 3), respectively], whereas MDC abolished the response (n = 3). The reason for the stronger effect of MDC is not known, although it may be a more effective endocytic inhibitor than Con A/sucrose, or it may inhibit additional intermediates in the p42/p44 MAPK cascade.
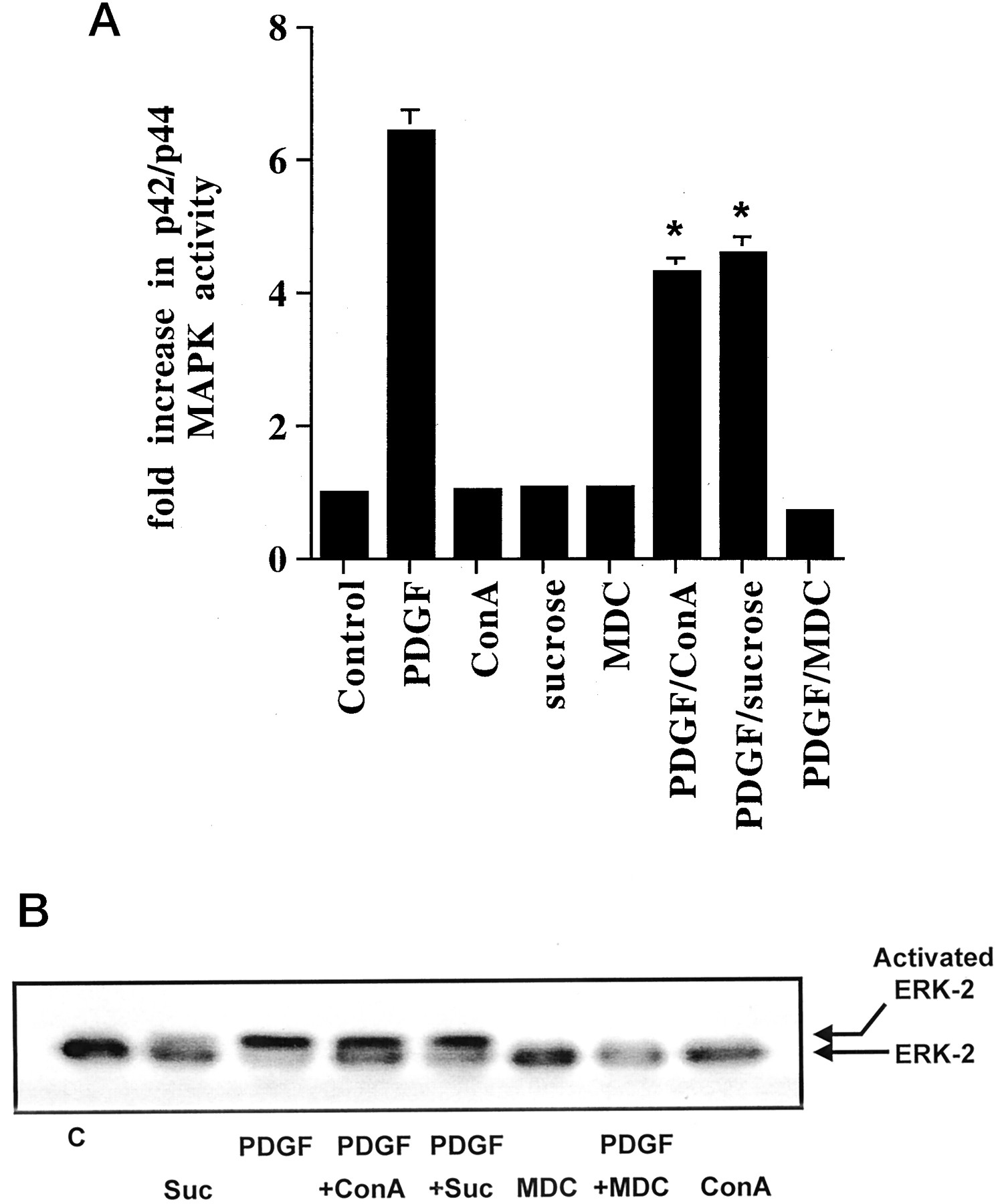
Inhibitors of clathrin-mediated endocytosis block p42/p44 MAPK activation. ASM cells were treated with and without Con A (0.25 mg/ml), sucrose (0.4 M), and MDC (500 μM) for 15 min before stimulation with PDGF (10 ng/ml, 10 min) as required. A, histogram showing the effect of Con A, sucrose, and MDC on PDGF-stimulated p42/p44 MAPK activity. Basal p42/p44 MAPK activity was 100 ± 13 pmol/min/mg of protein in control cells. Results are expressed as the fold increase over basal p42/p44 MAPK activity in control cells (means ± S.D. for n = 3–5 separate cell preparations; *P < .05 versus PDGF alone, Student's t test). B, corresponding shift blot showing phosphorylation of p42 MAPK. The immunoblot is a representative result of an experiment performed on three separate cell preparations. C, control; Suc, sucrose.
Our experiments showed that PDGF stimulated the association of dynamin II with Grb-2 in a pertussis toxin- and wortmannin-sensitive manner. Dynamin II (molecular mass of 100 kDa) was detected by Western blotting anti-Grb-2 immunoprecipitates with anti-dynamin II antibodies (Fig.8A). In cells stimulated with PDGF, the amount of the dynamin II immunoprecipitated with Grb-2 antibodies was increased (Fig. 8A, inset) under conditions where identical amounts of Grb-2 were coimmunoprecipitated (Fig. 8B). The pretreatment of cells with either pertussis toxin or wortmannin reduced the amount of dynamin II in Grb-2 immunoprecipitates (Fig. 8A). Wortmannin did not completely reduce formation of this complex, suggesting that a small proportion is regulated in a PI3K-independent manner. However, this small pool of PI3K-independent Grb-2-dynamin II does not appear to support stimulation of p42/p44 MAPK because the latter was completely abolished by wortmannin (Figs. 2, A and B). We did not detect the tyrosine phosphorylation of dynamin II in response to PDGF (data not shown).

PDGF stimulated dynamin II-Grb-2 association. ASM cells were treated with and without pertussis toxin (0.1 μg/ml, 24 h, upper panel) or wortmannin (W; 50 nM, 15 min, lower panel) before stimulation with PDGF (P; 10 ng/ml, 10 min) as required. A, Western blot of anti-Grb-2 immunoprecipitates with anti-dynamin II antibodies showing effect of pertussis toxin and wortmannin on PDGF-stimulated Grb-2-dynamin II complex formation. These are representative results of an experiment performed on three separate cell preparations. Inset is a histogram showing the fold increase in dynamin II-Grb-2 complex formation over controls as determined by densitometry (n = 3 separate cell preparations; results expressed as means ± S.D, *P < .05 for PDGF versus control, pertussis toxin + PDGF and wortmannin + PDGF, Student's t test). B, corresponding Western blot with anti-Grb-2 antibodies showing equivalent immunoprecipitation of Grb-2. C, anti-Grb-2 immunoprecipitates and ASM, PC12, and A431 cell lysates Western blotted with anti-dynamin II antibodies to show specificity of immunoprecipitation. Also shown is an alignment with Gab1 in anti-Gab1 immunoprecipitates to demonstrate the difference in electrophoretic mobility with dynamin II. Immunoprecipitations were from PDGF-treated cell samples. A through C, representative results of an experiment performed on three separate cell preparations. C, control; IP, immunoprecipitation; WB, Western blot.
Figure 8C shows the specificity of dynamin II immunoprecipitation with anti-Grb-2 antibodies. Dynamin II in anti-Grb-2 immunoprecipitates is aligned with comigrating dynamin II in ASM, PC12, and A431 cell lysates. These blots were compared with Gab1 in anti-Gab1 immunoprecipitates to show the difference in the electrophoretic mobility with dynamin II.
Discussion
The findings in this article show that the PDGF receptor uses Gi to stimulate activation of p42/p44 MAPK. This accounts for approximately 50% of the activation and involves intermediate c-Src and Grb-2-associated PI3K, the stimulation of which is blocked by pertussis toxin. Indeed, other GPCR agonists that use Gi stimulate this common pathway in ASM cells (Rakhit et al., 1999). For example, sphingosine 1-phosphate binds to the heterotrimeric Gi-coupled receptor, Edg1, to stimulate c-Src, Grb-2-associated PI3K, and p42/p44 MAPK, which are completely abolished by pertussis toxin. The mechanism used by the PDGF receptor to couple to Gi is not known, although other growth factor receptors have also been shown to interact with G-proteins (Rothenberg and Kahn, 1988; Luttrell et al., 1990). Recently, the IGF-1 receptor has been reported to activate Gi and causes release of βγ subunits (Hallak et al., 2000), which initiate activation of the p42/p44 MAPK pathway (Luttrell et al., 1995).
We have also reported here that the stimulation of Grb-2-PI3K complex formation by PDGF involves the Grb-2 adaptor protein, Gab1. This was based on several lines of evidence. First, we show that PDGF uses a Gi-mediated mechanism to stimulate the tyrosine phosphorylation of Gab1 because this was abolished by pertussis toxin. The tyrosine phosphorylation of this protein appears to promote binding of p85 PI3K1a to the Gab1-Grb-2 complex because this was also abolished by pertussis toxin. This is further supported by studies that have shown that tyrosine phosphorylation site(s) in Gab1 can bind p85 PI3K via an SH2 domain within the kinase (Holgado-Madruga et al., 1996). In ASM cells, Gab1 appears to be constitutively bound to Grb-2. Moreover, PI3K association with the Gab1-Grb-2 complex appears to be linked to p42/p44 MAPK activation based on studies with pertussis toxin and wortmannin, which reduce PDGF-stimulated p42/p44 MAPK activation.
The p85 regulatory subunit of PI3K1a that binds Gab1 is also tyrosine phosphorylated. Although the binding of this phosphorylated protein to the Gab1-Grb-2 complex is pertussis toxin-sensitive, itstyrosine phosphorylation appears to be insensitive to this toxin. This is based on data that showed that the PDGF-stimulated tyrosine phosphorylation of PI3K in anti-phosphotyrosine immunoprecipitates was not affected by pretreating ASM cells with pertussis toxin (this study and Conway et al., 1999).
A small amount of PI3K1a is bound to Gab-1-Grb-2 in unstimulated cells. This may be due to direct binding of PI3K to Grb-2 via an SH3 domain in this protein, as has been shown in other studies (Holgado-Madruga et al., 1996), or it might represent a very small pool of preactivated PI3K1a-Gab1-Grb-2 complex.
In summary, there appear to be two pathways that are convergent on the Grb-2 adaptor protein, Gab1. One is Gi-dependent and involves the tyrosine phosphorylation of Gab1 and subsequent association of PI3K1a, whereas the other route is Gi-independent, involving the tyrosine phosphorylation of PI3K1a (see Scheme FS1). The purpose of this convergence may be to localize tyrosine phosphorylated and activated PI3K with the Gab1-Grb-2 complex to promote subsequent binding of dynamin II. This is supported by the finding that PDGF stimulates the binding of dynamin II to the PI3K-Gab1-Grb-2 complex in a pertussis toxin- and wortmannin-sensitive manner. Others have shown that dynamin II binds to an SH3 domain in Grb-2. Interestingly, others have also shown that dynamin II GTPase activity is stimulated by phosphoinositides and Grb-2 (Barylko et al., 1998). The increased GTPase accelerates the rate of pinching off of endocytic vesicles containing active receptor signaling complexes, the final step at the plasma membrane required for the internalization and redistribution of active receptor signaling complexes with cytoplasmic p42/p44 MAPK. This is consistent with our results using inhibitors of clathrin-mediated endocytosis that reduced the PDGF-dependent activation of p42/p44 MAPK. Therefore, the association of PI3K with the Gab1-Grb-2 complex may be a catalytic step that triggers endocytosis of the receptor signal complex required to activate p42/p44 MAPK.

Proposed model for the PDGF receptor-dependent regulation of p42/p44 MAPK. First, the PDGF receptor stimulates a Gi-mediated tyrosine phosphorylation of the Grb-2 adaptor protein, Gab1, which results in the association of PI3K1a with the Gab1-Grb-2 complex. Second, the PI3K1a is tyrosine phosphorylated in a Gi-independent manner. Furthermore, the PI3K in the Gab1-Grb-2 complex may promote the subsequent association of dynamin II. This may have a critical role in regulating clathrin-mediated endocytic processing, leading to stimulation of p42/p44 MAPK. There may also be an additional Gi-independent route leading to p42/p44 MAPK activation that bypasses the dynamin II step and that is also inhibited by wortmannin.
The role of PI3K and dynamin II in regulating endocytic signaling was further supported by studies from Malbon and colleagues (Karoor et al., 1998). These authors showed that insulin stimulates the phosphorylation of the β-adrenergic receptor on Tyr-350 and promotes the binding of the receptor to Grb-2 via an SH2 domain. The Grb-2-β-adrenergic receptor complex is then targeted to clathrin-coated pits for sequestration. These authors also showed that insulin stimulates the formation of a complex between PI3K, Grb-2, and dynamin II. This complex has a critical role in regulating β-adrenergic receptor sequestration, based on experiments with the PI3K inhibitor, wortmannin, which was shown to prevent dynamin II binding to Grb-2 and to block insulin-stimulated β-adrenergic receptor sequestration.
The GPCR-mediated pathway described here for PDGF represents only part of the mechanism regulating p42/p44 MAPK because complete inactivation of Gi by pertussis toxin reduces, but does not abolish, stimulation of this kinase. This suggests the presence of an additional Gi-independent route(s) (that does not involve Gab1) regulating p42/p44 MAPK. Interestingly, this additional Gi-independent route is inhibited by wortmannin, suggesting that PI3K may also be involved in a pathway that bypasses dynamin II (see Scheme FS1). One possibility is that PI3K might regulate atypical protein kinase C isoforms, which are implicated in the activation of p42/p44 MAPK by PDGF in ASM cells (Pyne and Pyne, 1998).
In conclusion, the results presented here provide important information on the integration of GPCR and receptor tyrosine kinase signals to produce efficient stimulation of the p42/p44 MAPK cascade. These studies are the first to show that PDGF stimulates the tyrosine phosphorylation of Gab1 and to implicate this protein in the GPCR-mediated activation of p42/p44 MAPK by PDGF. The goal of future studies will be to identify the GPCR-regulated tyrosine kinase (e.g., c-Src tyrosine kinase possibly) responsible for the phosphorylation of Gab1 and to define the role of PI3K in endocytic signaling to p42/p44 MAPK.
Footnotes
- Received December 30, 1999.
- Accepted May 8, 2000.
-
Send reprint requests to: Dr. Nigel Pyne and Dr. Susan Pyne, Department of Physiology and Pharmacology, University of Strathclyde, Strathclyde Institute for Biomedical Sciences, 27 Taylor St., Glasgow G3 0NR, UK. E-mail: n.j.pyne{at}strath.ac.uk andsusan.pyne{at}strath.ac.uk
-
This study was supported by the Wellcome Trust and the Medical Research Council. S.P. is a Wellcome Trust Senior Biomedical Research Fellow.
Abbreviations
- GPCR
- G-protein-coupled receptor
- SH
- Src homology
- PDGF
- platelet-derived growth factor
- ASM
- airway smooth muscle
- MAPK
- mitogen-activated protein kinase
- PI3K
- phosphoinositide 3-kinase
- IGF
- insulin-like growth factor
- HRP
- horseradish peroxidase
- PAGE
- polyacrylamide gel electrophoresis
- NP-40
- Nonidet P-40
- Con A
- concanavalin A
- MDC
- monodansylcadaverine
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}