


Abstract
Pertussis toxin (PTx), which inactivates Gi/o type G proteins, is widely used to investigate the involvement of Gi/o proteins in signal transduction. Activation of extracellular-regulated kinases 1 and 2 (ERK1/2) by G protein-coupled receptors has been described to occur either through a PTx-insensitive pathway involving activation of phospholipase C and protein kinase C (PKC), or through a PTx-sensitive pathway involving Giβγ-mediated activation of Src. Cholecystokinin (CCK) activates ERK1/2 by a PKC-dependent, and thus presumably PTx-insensitive, pathway. However, CCK has recently been shown to induce activation of Gi proteins in addition to Gq/11. In the present study, PTx partially inhibited CCK-induced ERK1/2 activation in pancreatic AR42J cells, although activation of phospholipase C was not reduced. PTx also inhibited ERK1/2 activation in response to the PKC activator 12-O-tetradecanoylphorbol-13-acetate (TPA) and epidermal growth factor (EGF) as well as activation of c-Raf-1 by EGF and CCK. In contrast, PTx, CCK, and EGF had only minor effects on A-Raf and B-Raf activity. Forskolin, a direct activator of adenylyl cyclase, inhibited CCK- and EGF-induced activation of c-Raf-1 and ERK1/2 in a manner similar to that of PTx. In PTx-treated cells, the cAMP content was increased and forskolin did not further inhibit CCK- and EGF-induced activation of c-Raf-1 or ERK1/2. In conclusion, the present study shows that PTx-sensitivity of receptor-induced ERK1/2 activation could be a consequence of disinhibition of the adenylyl cyclase signaling pathway, which in turn causes inhibition of c-Raf-1 activation rather than indicating involvement of a PTx-sensitive G protein in this signaling pathway.
Cellular responses such as proliferation or differentiation are regulated by a variety of external stimuli and involve the regulation of transcriptional events in eukaryotic cells through intracellular signaling pathways that activate kinases of the mitogen-activated protein kinase (MAPK) family [also termed extracellular signal-regulated kinases (ERKs)] (Treisman, 1996). MAPKs become activated in response to growth factors through signals triggered either by receptor tyrosine kinase or G protein-coupled receptors (GPCR) (Garrington and Johnson, 1999).
The mechanism of tyrosine kinase receptor-stimulated MAPK activation involves formation of complexes of the adaptor protein Grb2, the guanine nucleotide exchange protein Sos, and the adaptor-protein Shc, leading to activation of Ras, recruitment of Raf kinase to the membrane, which itself induces activation of MAPK kinase (MEK) and MAPK (Garrington and Johnson, 1999). Heterogeneity exists in the mechanisms whereby GPCRs activate MAPK. Depending on receptor and cell type, MAPK activation may be mediated by pertussis toxin (PTx)-sensitive and -insensitive G proteins and depends on activation of Src kinase, protein kinase C (PKC), phosphoinositide 3-kinase, and Ras (Luttrell et al., 1996; Dikic et al., 1996; Lopez-Ilasaca et al., 1997). The PTx-insensitive pathway involves Gq/11-mediated activation of phospholipase C followed by stimulation of protein kinase C (PKC). Receptors coupled to activation of PTx-sensitive G proteins, such as the lipoprotein A receptor, activate MAPK via Gβγ and Src kinase. Moreover, it has recently been demonstrated that Gi and Gq protein-coupled receptors (Daub et al., 1997) are capable of induce tyrosine phosphorylation of the epidermal growth factor (EGF) receptor, which seems to be an essential element of MAPK activation by these receptors. Thus, both intracellular tyrosine kinases as well as receptor tyrosine kinase receptors are involved in mitogenic signaling of GPCR.
Cholecystokinin (CCK) activates the two subtypes of the G protein-coupled CCK receptor, CCKA and CCKB, whereas gastrin is an agonist on the CCKB receptor only. CCK and gastrin stimulate MAPK through a PKC- and Src-dependent pathway (Dabrowski et al., 1996;Todisco et al., 1997; Daulhac et al., 1999). An interesting recent study reported that formation of heparin-binding epidermal growth factor (EGF)-like growth factor may be involved in gastrin-induced cell proliferation (Miyazaki et al., 1999). AR42J cells express both CCKA and CCKB receptors (Christophe, 1994). Both CCK receptor subtypes are able to induce MAPK activation (Dabrowski et al., 1997a). CCK receptor signaling involves activation of PTx-insensitive Gq/11-type G proteins, activation of phospholipase C-β1, and subsequent increases in the intracellular Ca2+ concentration (Zeng et al., 1996; Piiper et al., 1997a). In addition to PTx-insensitive Gq/11 proteins, CCK may also activate PTx-sensitive G proteins (Scemama et al., 1988; Schnefel et al., 1990;Yu et al., 1998; Pommier et al., 1999).
In the present study, we examined the possibility that PTx-sensitive G proteins are involved in CCK-induced activation of the MAPK pathway in pancreatic AR42J cells. We found that PTx indeed inhibited CCK-, phorbol ester-, and EGF-induced MAPK activation. However, this effect was caused by disinhibition of cAMP signaling cascade, which itself causes inhibition of c-Raf-1. Thus, our study demonstrates that sensitivity of a receptor pathway to PTx does not necessarily indicate an involvement of a PTx-sensitive G protein in this signaling pathway.
Experimental Procedures
Materials.
CCK octapeptide, human recombinant EGF, myelin basic protein, and affinity-purified, horseradish peroxidase-conjugated anti-mouse and anti-rabbit IgG were obtained from Sigma Chemical Co. (St. Louis, MO). Recombinant MEK1 and ERK2 were from Upstate Biotechnology Inc. (Lake Placid, NY). The antibody raised against dual-phosphorylated activated ERK1 and ERK2 and the MAPK assay kit were obtained from New England Biolabs (Beverly, MA). Antibodies recognizing ERK1, ERK2, c-Raf-1, A-Raf, and B-Raf were from Santa Cruz (Santa Cruz, CA). Protein G-Sepharose, the cAMP assay kit and enhanced chemiluminescence reagents were obtained from Amersham-Pharmacia Biotech (Piscataway, NJ). Dulbecco's modified Eagle's medium (DMEM), fetal calf serum, and penicillin/streptomycin were from Life Technologies, Inc. (Eggenstein, Germany). TPA, forskolin, U73122, and GF-109203X, all dissolved in dimethyl sulfoxide, were obtained from Calbiochem (La Jolla, CA). Fura 2-acetoxymethyl ester was from Molecular Probes (Eugene, OR). The inositol 1,4,5-trisphosphate (1,4,5-IP3) assay and [γ-32P]ATP (3000 Ci/mmol) were from DuPont NEN (Boston, MA).
Cell Culture.
AR42J (from the European Collection of Cell Cultures, Salisbury, UK) were cultured in DMEM containing 10% fetal calf serum and antibiotics. For the experiments, 70% confluent cells that had been serum-starved overnight were used. PTx treatment was carried out by incubating the cells with 200 ng/ml PTx for 12 h before the experiments.
Measurement of Intracellular 1,4,5-IP3 and cAMP.
The 1,4,5-IP3 production in AR42J cells was measured by displacement of bound [3H]1,4,5-IP3 using an 1,4,5-IP3-specific radioreceptor assay (Piiper et al., 1994). cAMP was measured using an enzyme immunoassay kit. AR42J cells cultured in 24-well plates were incubated for 24 h in serum-free DMEM in the presence or absence of PTx (200 ng/ml). Before the experiment, the medium was replaced by 200 μl of fresh DMEM followed by the addition of CCK, EGF, forskolin, or vehicle for 30 min. The intracellular cAMP content was measured according to the assay protocol provided by the manufacturer (Amersham-Pharmacia Biotech).
Determination of Intracellular Calcium Concentration ([Ca2+]i).
AR42J cells grown in 175 cm2 flasks were trypsinated, resuspended in DMEM without serum, and loaded with 2 μM Fura 2-acetoxymethyl ester for 30 min at 37°C. Fura 2-loaded cells were washed twice with DMEM and resuspended in 12 ml of fresh DMEM per flask. [Ca2+]i measurement was performed using a Hitachi dual-wavelength fluorescence spectrophotometer. Aliquots (2 ml) were incubated in a continuously stirred cuvette at 37°C, and the ratio of fluorescence at 510 nm after alternate excitation at 340 and 380 nm was used to calculate [Ca2+]i using the formula developed by Grynkiewicz et al. (1985).
Immunoblotting.
Immunoblotting was performed as recently described (Piiper et al., 1997a,b). Gel-resolved proteins were electrotransferred to nitrocellulose sheets. Protein transfer was visualized by Ponceau S stain. Antigen-antibody complexes were visualized using horseradish-peroxidase-conjugated antibodies and the enhanced chemiluminescence system. The specific bands on the autoradiograms were quantified by densitometry. Statistical analysis was performed using ANOVA with subsequent Bonferroni tests for multiple comparisons.
Phosphorylation of MAPK.
AR42J cells were cultivated in 24-well plates and were treated with dexamethasone for 2 to 3 days. Cells were incubated with the desired reagents for various time periods in DMEM at 37°C, and incubation was terminated by washing with ice-cold PBS followed by lysis of the cells and Western blotting with an antibody that recognizes specifically the MAPK phosphorylated on Thr202 and Tyr204. To down-regulate PKC, cells were treated with TPA (100 nM) for 18 h. To confirm equal amounts of ERK1/2 in each lane, blots were either stripped and reprobed with anti-ERK1/2 or aliquots of the samples were analyzed by anti-ERK1/2 Western blotting.
MAPK Assay.
MAPK was immunoprecipitated from AR42J lysates with agarose-conjugated antibody. MAPK activity in the immunoprecipitates was measured by phosphorylation of recombinant Elk-1 and Western blotting with anti-phospho-Elk-1 antibody according to the protocol of the manufacturer.
Raf Kinase Assays.
Raf activity was measured by sequential activation of recombinant MEK, MAPK, and phosphorylation of myelin basic protein in the presence of [γ-32P]ATP. Individual Raf kinases were specifically immunoprecipitated from AR42J cell lysates with anti-c-Raf-1, A-Raf, or B-Raf antibodies; immune complexes were captured with protein A/G Sepharose, washed once with lysis buffer, and once with kinase buffer (20 mM 3-(N-morpholino)propanesulfonic acid, pH 7.2, 25 mM 2-glycerol phosphate, 5 mM EGTA, 1 mM Na3VO4, and 1 mM dithiothreitol). The immune complexes were incubated with inactive fusion proteins GST-MEK1 (0.4 μg), GST-ERK2 (1 μg), in a buffer containing 15 mM 3-(N-morpholino)propanesulfonic acid, pH 7.2, 20 mM 2-glycerol phosphate, 5 mM EGTA, 1 mM Na3VO4, 1 mM dithiothreitol, 150 μM ATP, and 25 μM MgCl2in a final volume of 50 μl for 30 min at 30°C with gentle agitation. Five microliters of the reaction was transferred to a cup containing 20 μg of myelin basic protein and 4 μCi of [γ-32P]ATP (final volume, 35 μl) in kinase buffer. The reaction was terminated by addition of 4×-concentrated Laemmli buffer. An aliquot of the reaction mixture was separated by SDS-polyacrylamide gel electrophoresis. Gels were stained, dried, and autoradiographed. The bands on the autoradiograms were quantified by densitometry.
Results
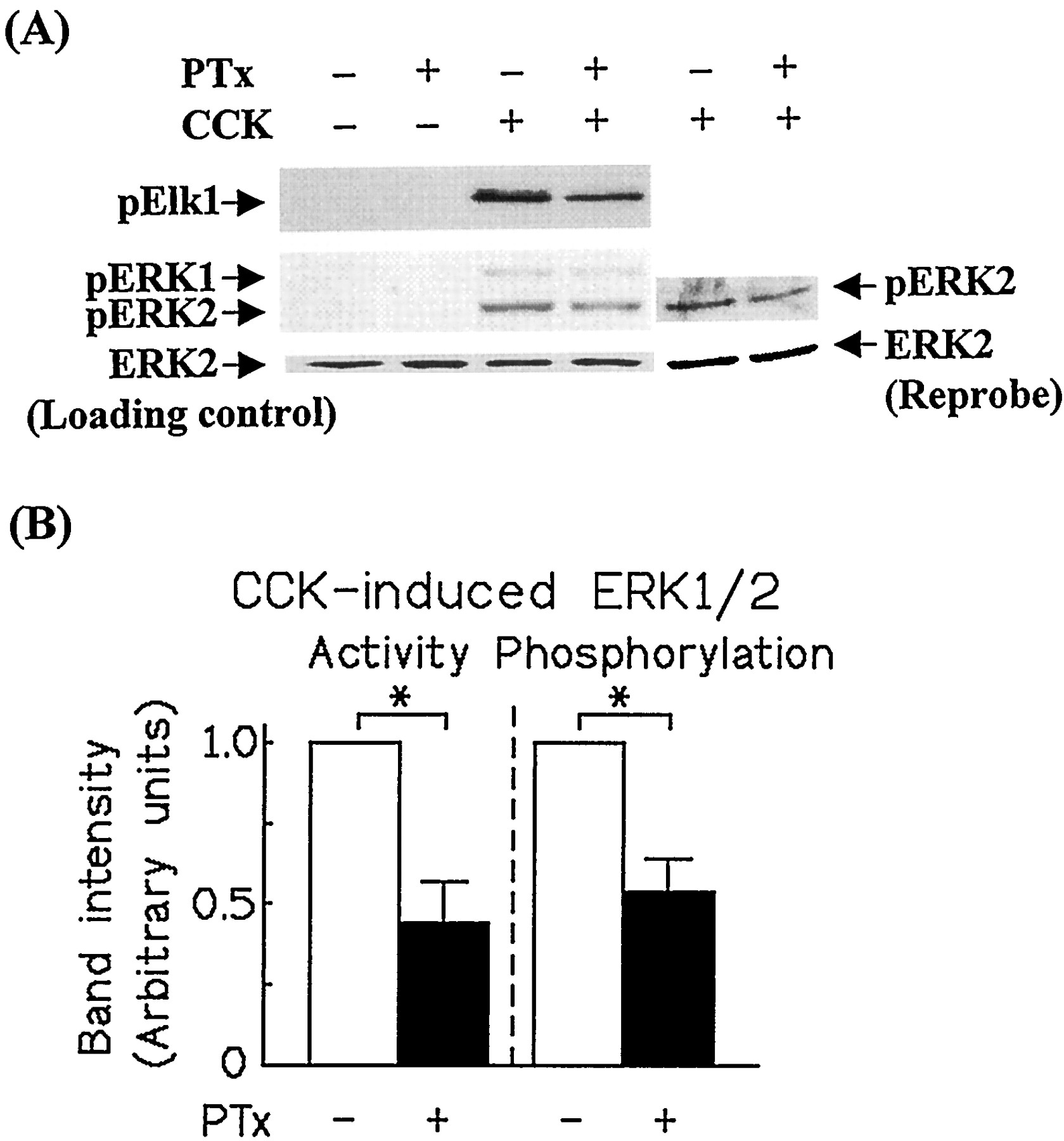
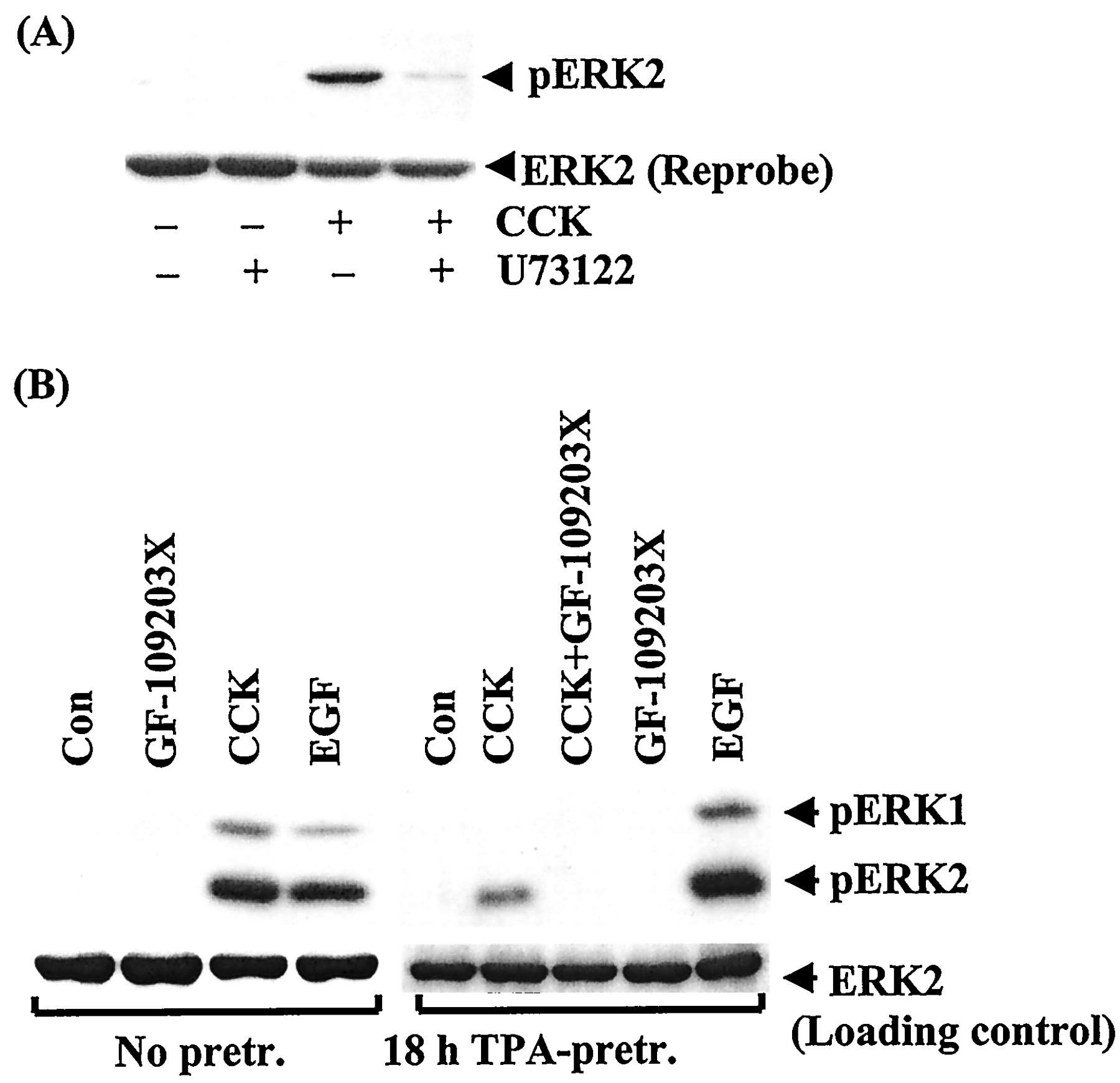
CCK (a CCKA and CCKBreceptor agonist) and gastrin (a CCKB receptor agonist) have been shown to induce PKC-dependent ERK1/2 activation (Dabrowski et al., 1996). Earlier studies reported the involvement of PTx-sensitive G proteins in CCK receptor signal transduction (Scemama et al., 1988; Schnefel et al., 1990; Yu et al., 1998; Pommier et al., 1999) and that βγ subunits from PTx toxin-sensitive G proteins can induce activation of ERK1/2 (Dikic et al., 1996; Luttrell et al., 1996). This prompted us to investigate the effect of PTx on CCK receptor-induced ERK1/2 activation in pancreatic AR42J cells, which express both CCKA and CCKBreceptors (Christophe, 1994). As illustrated in Fig.1, CCK (10 nM) caused strong activation of ERK1/2 that was inhibited by 50% in PTx-pretreated AR42J cells. A similar result was obtained when ERK1/2 activation was monitored by Western blotting using an antibody that specifically recognizes the dually phosphorylated, activated form of ERK1 and ERK2 (Fig. 1A, lower panel). Inhibition of phospholipase C by U73122 (10 μM) or of PKC by either GF109203X (3 μM) or down-regulation of PKC by overnight-incubation of the cells with TPA (100 nM) has been shown to inhibit gastrin-induced ERK1/2 activation (Daulhac et al., 1997;Todisco et al., 1997); this treatment also inhibited CCK-induced ERK1/2 phosphorylation (Fig. 2A). In contrast, down-modulation of PKC by TPA (Fig. 2B) or GF109203X (data not shown) had no effect on the response to EGF, indicating that CCK/gastrin-induced ERK1/2 activation is mediated by sequential activation of phospholipase C and PKC. To examine whether the inhibitory effect of PTx on CCK receptor-induced ERK1/2 activation is caused by reduced activation of phospholipase C, we examined the effect of PTx on CCK-induced 1,4,5-IP3 production and increase in [Ca2+]i. As shown in Table 1, PTx did not inhibit CCK-induced 1,4,5-IP3 production or increase in [Ca2+]i. In fact, CCK-induced increase in 1,4,5-IP3 and [Ca2+]i tended to be higher in PTx-treated cells compared with untreated cells. Thus, the inhibitory effect of PTx on CCK receptor-mediated ERK1/2 activation was not caused by inhibition of phospholipase C activation.

PTx inhibits CCK-induced MAPK phosphorylation and activation in AR42J cells. A, PTx-treated and untreated control cells grown in six-well plates were exposed to CCK (10 nM) for 3 min at 37°C in serum-free DMEM. After cell lysis, MAPK was immunoprecipitated, and its activity was assayed with recombinant Elk-1 as a substrate by anti-phospho-Elk-1 Western blotting (A, top). Bottom, detection of MAPK phosphorylation by Western blotting of cellular lysates with an antibody that specifically recognizes the dually phosphorylated activated form of ERK1 and ERK2. For loading control, the blot was stripped and reprobed with anti-ERK1/2 or the same samples were analyzed by anti-ERK1/2 on a separate blot. B, densitometric quantitation of bands corresponding to MAPK activity and phosphorylation. The data shown are means ± S.E. of five independent experiments. Asterisks indicate a significant effect of PTx treatment compared with the untreated control cells (P < .05).

U73122, GF-109203X, and down-regulation of PKC inhibit CCK-induced MAPK phosphorylation. AR42J cells grown in six-well plates were exposed to CCK (10 nM) or EGF (20 nM) for 3 min in the presence of either U73122 (10 μM) or solvent (top). Bottom, cells were preincubated with or without TPA (100 nM) for 18 h before stimulation with CCK (10 nM) or EGF in the presence or absence of GF-109203X (4 μM). MAPK phosphorylation was detected by Western blotting with an antiphospho-MAPK specific antibody. For loading control, the blot was stripped and reprobed with anti-ERK1/2 (A), or the same samples were analyzed by anti-ERK1/2 on a separate blot (B). The blots shown are representative of three others.
Effect of PTx on CCK-induced 1,4,5-IP3 and [Ca2+]i in AR42J cells
To further investigate the inhibitory effect of PTx on CCK-induced ERK1/2 activation, we determined the effect of PTx on activation of ERK1/2 by TPA, an activator of PKC, which links the CCK receptor to MAPK activation distal from the receptor-coupling G proteins and should activate ERK1/2 by a PTx-insensitive mechanism. To our surprise, PTx-pretreatment inhibited TPA-induced activation and phosphorylation of ERK1/2 by approximately 50%, a degree similar to that of CCK-induced ERK1/2 activation (Fig. 3). This suggests that the PTx-sensitive target inhibits ERK1/2 activation at a site distal from activation of PKC. Next, we tested the effect of PTx on ERK1/2 activation by EGF, which is supposed to activate ERK1/2 by a G protein-independent mechanism. However, PTx also inhibited EGF-induced ERK1/2 phosphorylation and activation by approximately 50% (Fig. 3). PTx had no significant effect on EGF-induced protein tyrosine phosphorylation, EGF receptor autophosphorylation, or EGF-induced tyrosine phosphorylation of Shc and phospholipase C-γ1 (data not shown).

PTx inhibits TPA- and EGF-induced MAPK phosphorylation in AR42J cells. PTx-treated and untreated control AR42J cells grown in six-well plates were exposed to TPA (10 nM) or EGF (20 nM) for 3 min at 37°C in serum-free DMEM. Then, MAPK phosphorylation was determined. For loading control, the blot was stripped and reprobed with an anti-ERK1/2. Bottom, densitometric quantitation of the bands represent means ± S.E. of three independent experiments. Asterisks indicate a significant effect of PTx treatment compared with the untreated control cells (P < .05).
Because agents that increase the intracellular cAMP concentration block growth factor-stimulated ERK1/2 activation in a number of cell types (Burgering et al., 1993; Sevetson et al., 1993), we examined the possibility that inactivation of Gi/o type G proteins by PTx may increase the intracellular cAMP level, which could mediate inhibition of CCK- and EGF-induced ERK1/2 activation in AR42J cells. As illustrated in Fig. 4, PTx caused a 3-fold increase in the basal cAMP concentration. CCK and EGF did not significantly alter the elevated cAMP level in PTx-treated cells.

Effect of PTx on the intracellular cAMP concentration. AR42J cells were grown in 24-well plates in the presence or absence of PTx for 18 h. Then, cells were incubated with CCK, EGF, or forskolin for 30 min at 37°C, followed by determination of cellular cAMP content in each well. Open bars, control; hatched bars, PTx-treated cells. The data shown are means ± S.E. of three independent experiments. Asterisks indicate a significant difference (*P < .05; **P < .01).
Additional evidence for the assumption that an increased cAMP level mediates the effect of PTx on ERK1/2 activation came from experiments in which we studied the effect of forskolin, a direct activator of adenylyl cylase, on CCK- and EGF-induced ERK1/2 activation in PTx-treated and untreated control cells. Figure5 shows that forskolin mimicked the inhibitory effect of PTx on CCK-induced ERK1/2 activation. PTx did not further inhibit CCK-induced ERK1/2 activation in the presence of forskolin, indicating that forskolin and PTx inhibit CCK-induced ERK1/2 activation by similar mechanisms.

Forskolin mimics the inhibitory effect of PTx on CCK-induced MAPK activation. Top, PTx-treated and untreated cells grown in six-well plates were exposed to CCK (10 nM) in the presence or absence of forskolin (10 μM) for 3 min at 37°C in serum-free DMEM. Then, MAPK activity was determined. Bottom, densitometric quantitation of bands. The values shown are means ± S.E. of three independent experiments. Asterisks indicate a significant effect of PTx compared with the untreated condition (P < .05).
An increased cAMP concentration has been reported to inhibit activation of c-Raf-1, suggesting that cAMP-mediated inhibition are caused by inhibition of c-Raf-1 (e.g., Burgering et al., 1993; Sevetson et al., 1993). Western blotting analysis showed that AR42J cells express all three different Raf proteins (c-Raf-1, Raf-A, and Raf-B) (Fig.6, upper part), all of which can be activated by growth factor stimulation and activate MEK, which then activates ERK1/2 (Kyriakis et al., 1992; Jaiswal et al., 1994;Pritchard et al., 1995). Therefore, we determined activation of each individual Raf kinase by immunoprecipitating the Raf kinases from PTx-treated and untreated cells that had been exposed to CCK, EGF, or vehicle, and measured Raf kinase activity by an immune complex kinase assay. As illustrated in Fig. 6, EGF and CCK strongly activated c-Raf-1. In contrast, EGF and CCK induced only low A-Raf and B-Raf activation, if any, indicating that c-Raf-1 is the major mediator of both CCK- and EGF-induced MAPK activation in AR42J cells. Activation of c-Raf-1, but not of A-Raf or B-Raf, was significantly inhibited in PTx-pretrated cells (Fig. 6). Similarly, forskolin inhibited CCK- and EGF-induced activation of c-Raf-1, whereas its effect on A-Raf activity was not significant (Fig. 6). The lack of effect of cAMP-dependent protein kinase A on B-Raf activation has been reported recently (Sutor et al., 1999). Thus, the inhibitory effect of PTx and forskolin on CCK- and EGF-induced ERK1/2 activation strongly correlated with inhibition of c-Raf-1 activation, indicating that PTx inhibits CCK- and EGF-induced ERK1/2 activation by interfering with activation of c-Raf-1.

Effect of CCK, EGF, PTx, and forskolin on activity of c-Raf-1, A-Raf, and B-Raf. PTx-treated and untreated cells grown in six-well plates were exposed to CCK (10 nM), EGF (20 nM), and/or forskolin (10 μM) for 3 min at 37°C in serum-free DMEM, followed by cell lysis. A, top, cell lysates were examined for presence of Raf isoenzymes by Western blotting. Bottom, activity of c-Raf-1, A-Raf, and B-Raf was determined by an immune complex kinase assay using myelin basic protein as a substrate. B, densitometric quantification of bands. The values shown are means ± S.E. of three to five independent experiments. Asterisks indicate a significant effect of PTx treatment compared with the untreated condition (P < .05).
Discussion
PTx-catalyzed ADP-ribosylation and thereby inactivation of Gi/o-type G proteins is widely used as specific tool to investigate involvement of a Gi/o-type G protein in intracellular signaling pathways. Numerous studies have reported inhibition of PTx on ERK1/2 activation by receptors that have not been demonstrated to couple to PTx-sensitive G proteins or that couple to several different G proteins such as Gqand Gi. For instance, in cardiac myocytes, PTx, cAMP analogs, and inhibition of PKC decreases the ability of TPA and endothelin-1 to activate ERK1/2 (Chiloeches et al., 1999). Moreover, stimulation of c-Raf-1 and A-Raf by endothelin-1 is decreased by PTx (Bogoyevitch et al., 1995). Similarly, in adrenal glomerulosa cells, stimulation of the angiotensin II receptor, a Gq-coupled receptor, leads to ERK1/2 activation that is partially inhibited by PTx (Smith et al., 1999). In hepatocytes, PTx inhibits angiotensin II-, vasopressin-, norepinephrine-, prostaglandin F2α-, as well as EGF-induced MAPK activation (Melien et al., 1998), and ERK1/2 activation is also inhibited by cAMP (Gines et al., 1996). In most studies, the mode of action of PTx remained unclear. In the present study, we demonstrate that PTx can inhibit ERK1/2 activation by disinhibition of adenylyl cyclase. The subsequent increase in the cAMP level mediates inhibition of Raf, most likely c-Raf-1, which itself activates ERK1/2 via MEK. These conclusions are based on the following findings: 1) PTx partially inhibited CCK- and EGF-induced activation of ERK1/2 and c-Raf-1; 2) these inhibitory effects of PTx were mimicked by activation of adenylyl cyclase; 3) PTx did not further inhibit CCK- and EGF-induced activation of c-Raf-1 or ERK1/2 in the presence of forskolin; and 4) CCK, EGF, and PTx had only minor effects on activity of A-Raf and B-Raf; similar to native pancreatic acini (Willems et al., 1987), PTx increased the cellular cAMP content. Thus, our data show that PTx-sensitivity of receptor agonist-induced ERK1/2 activation does not necessarily indicate that a PTx-sensitive G protein is involved in this receptor pathway.
CCK, which activates both CCKA and CCKB receptors, and gastrin, a CCKB receptor agonist, stimulate ERK1/2 activation through a PKC-dependent pathway and induce tyrosine phosphorylation of Shc and its complex formation with Grb2 (Dabrowski et al., 1996; Daulhac et al., 1999). CCK receptor signaling is initiated by binding to a heptahelical receptor and activation of PLC-β1 via stimulation of PTx-insensitive Gq/11-type G proteins (Zeng et al., 1996; Piiper et al., 1997a). In addition to PTx-insensitive Gq/11 proteins, CCK may also activate PTx-sensitive G proteins in pancreatic acini (Schnefel et al., 1990) as well as in AR42J cells (Scemama et al., 1988) and cat gallbladder muscle (Yu et al., 1998). The CCKB receptor has recently been shown to couple to both PTx-sensitive and -insensitive G proteins (Pommier et al., 1999). Thus, it might be possible that PTx-sensitive G proteins participate in CCK/gastrin-induced stimulation of ERK1/2 in AR42J cells expressing both CCKA and CCKB receptors, both of which can mediate ERK1/2 activation (Dabrowski et al., 1997a). Our data indicate that PTx-sensitive G proteins are not involved in CCK and EGF receptor-induced ERK1/2 activation because PTx had no further inhibitory effect on CCK- and EGF-induced ERK1/2 activation in the presence of forskolin.
Surprisingly, PTx slightly augmented rather than inhibited the ability of CCK to increase the 1,4,5-IP3 level and the intracellular concentration of calcium. Similar to its effect on ERK1/2 activation, this effect is likely to be a consequence of PTx-induced increase in the intracellular cAMP concentration, because elevation of the intracellular cAMP level leads to an increase in the intracellular concentrations of 1,4,5-IP3 and free calcium in several cell types (Chew, 1986; Yamaguchi et al., 1987; Dehaye et al., 1993), including AR42J (Bold et al., 1995), indicating that stimulation of protein kinase A can activate phospholipase C-β to generate a calcium signal. A recent study suggested that sequential activation of Gs and Gi, mediated by protein kinase A, may mediate this response (Luo et al., 1999).
Raf proteins are a family of serine/threonine kinases consisting of A-Raf, B-Raf, and c-Raf-1, and are activated by growth factor receptor tyrosine kinases as well as by GPCRs (Treisman, 1996). In the present study, we found that, similar to native pancreatic acini (Dabrowski et al., 1997b), AR42J cells express all three Raf isoforms. Whereas CCK seems to activate all three Raf isoforms to a similar degree in native pancreatic acini (Dabrowski et al., 1997b), in AR42J cells, c-Raf-1 was strongly activated by CCK and EGF, whereas A-Raf and B-Raf were only slightly activated. Thus, although all three isoforms of Raf are able to activate MEK (Kyriakis et al., 1992; Jaiswal et al., 1994; Pritchard et al., 1995), in AR42J cells, CCK and EGF seem to activate ERK1/2 mainly through activation of c-Raf-1. This assumption is supported by the finding that the extent of inhibition of CCK- and EGF-induced ERK1/2 activation by PTx and forskolin correlates well with inhibition of c-Raf-1.
In conclusion, the present study, using pancreatic AR42J cells, shows that PTx-sensitivity of receptor-induced ERK1/2 activation could be a consequence of disinhibition of the adenylyl cyclase signaling pathway, which in turn causes inhibition of c-Raf-1 activation rather than indicating involvement of a PTx-sensitive G protein in this signaling pathway. Thus, our study demonstrates that sensitivity of a receptor pathway to PTx does not necessarily indicate an involvement of a PTx-sensitive G protein in this signaling pathway.
Footnotes
- Received February 29, 2000.
- Accepted May 25, 2000.
-
Send reprint requests to: Albrecht Piiper, M.D., Ph.D., or Stefan Zeuzem, M.D., University of Frankfurt, Department of Medicine II, Theodor-Stern-Kai 7, D-60590 Frankfurt A.M., Germany. E-mail:piiper{at}em.uni-frankfurt.de orzeuzem{at}em.uni-frankfurt.de
-
This work was partly supported by grants from the Deutsche Forschungsgemeinschaft (Ze 237/4–5) and Nachlaβ Held/Hecker.
Abbreviations
- MAPK
- mitogen-activated protein kinase
- ERK
- extracellular signal-regulated kinase
- GPCR
- G protein-coupled receptor
- MEK
- mitogen-activated protein kinase kinase
- PTx
- pertussis toxin
- PKC
- protein kinase C
- CCK
- cholecystokinin
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- EGF
- epidermal growth factor
- DMEM
- Dulbecco's modified Eagle's medium
- IP3
- inositol trisphosphate
- [Ca2+]i
- intracellular calcium concentration
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}