


Abstract
Sequence analysis revealed a strong homology between the ligand-binding domain (LBD) of the human mineralocorticoid receptor (hMR) and glucocorticoid receptor (hGR). Nevertheless, steroids with bulky C11-substituents bind to hGR, unlike hMR. In this report, a mutant hMR, in which the residue Ala-773 facing the C11 steroid position was replaced by a glycine (A773G), was assayed for its capacity to bind steroids, to interact with receptor coactivators, and to stimulate transcription. The capacity of A773G to bind aldosterone and C11-substituted spirolactones was the same as that of the wild-type receptor. The agonist properties of aldosterone, as well as the antagonist feature of compounds bearing a 11β-allenyl group and a C17-ketone function, remain unchanged. In contrast, C11-substituted steroids with a 17γ-lactonic ring displayed antagonist properties with hMR and acted as potent agonists with A773G. An agonist-dependent hMR interaction with SRC-1 was observed for both the wild-type and the mutant receptors. The hMR activation process is discussed in the light of the hMR-LBD homology model based on the structural data of the human progesterone receptor LBD.
The mineralocorticoid receptor (MR) belongs to a large family of ligand-activated transcription factors that includes the other steroid receptors as well as thyroid, retinoid, and vitamin D receptors and also orphan receptors whose ligands have not yet been identified. All the members of this large family are characterized by a conserved DNA binding domain and a C-terminal ligand-binding domain (LBD) essential for chaperone protein interaction, receptor dimerization, and hormone-dependent transactivation (Arriza et al., 1987; Evans, 1988). Recently, the crystal structure of ligand-free and liganded LBDs has been solved for several nuclear receptors (NRs) (Bourguet et al., 1995;Renaud et al., 1995; Wagner et al., 1995; Wurtz et al., 1996; Brozowski et al., 1997). These crystal structures reveal a triple-layered antiparallel α-helical sandwich fold surrounding the ligand-binding cavity. The major difference between the ligand-free and the agonist-bound LBD is the folding back of the helix H12 toward the LBD core, allowing the binding of transcriptional coactivators (Nichols et al., 1998). Moreover, the helix H12 was demonstrated to be differently positioned after antagonist binding, preventing the coactivator-receptor interaction (Nichols et al., 1998). A three-dimensional model of the human MR (hMR)-LBD, based on the human retinoic acid receptor (hRARγ-LBD) crystal structure, has recently been proposed that allows the docking of various ligands within the ligand-binding cavity (Fagart et al., 1998). The identification of several amino acid residues involved in the interaction with agonists and antagonists has been made by mutagenesis. Gln-776 and Arg-817, two polar residues highly conserved within the steroid receptor family, anchor the C3-ketone function, common in mineralocorticoid agonist and antagonist ligands. At the opposite side of the ligand-binding cavity, the C20-ketone present in both agonist and antagonist ligands is interacting with Cys-942. The C21-hydroxyl moiety, which characterized the natural mineralocorticoid agonists, makes a hydrogen bond with Asn-770 (Fagart et al., 1998, Lupo et al., 1998). Although the interaction between Asn-770 and the 21-hydroxyl group seemed to be crucial for the stabilization of the active hMR conformation, other steroid substitutions could influence the agonist/antagonist activity. Sequence analysis reveals a strong homology between the hMR- and the human glucocorticoid receptor (hGR)-LBDs (56%). Recently, chimeras were made between hMR and hGR-LBD to characterize ligand-binding specificity (Rogerson et al., 1999). A region of the hMR-LBD that extends between amino acids 804 and 874 that is crucial for aldosterone binding specificity has been identified. Another study shows that the Gly-567 in the hGR-LBD is essential for glucocorticoid binding (Warriar et al., 1994). Indeed, the mutant G567A failed to bind either glucocorticoid agonists or antagonists, such as RU486. All steroid receptors that bind RU486 have a glycine residue at the corresponding position [Gly-708 in human aldosterone receptor and Gly-722 in human progestin receptor (hPR); Fig. 1]. A cysteine residue and an alanine residue at this position (Fig. 1) characterize PRs (and hMR) of chicken and wallaby, respectively, that are unable to bind RU486. Because glycine is devoid of any side chain, the presence of a cavity able to accommodate the 11β-dimethylaminophenyl substituent of RU486 is suggested. The presence of the alanine methyl group in hMR seemed to sterically hinder ligand binding. To analyze the role of a putative “hydrophobic hollow” located in the hMR ligand-binding pocket (LBP) facing the steroid C-11 position, we replaced alanine at the position 773 with a glycine residue and examined the steroid-binding capacity of the corresponding mutant hMR (A773G). Its ability to bind SRC-1, a coactivator known to interact with several members of the NR superfamily (Onate et al., 1995; Jenster et al., 1997; Shibata et al., 1997), was also tested and its transactivation activity was measured by cotransfection assays. The compounds tested were synthesized in our laboratory (Faraj et al., 1990; Claire et al., 1993); they differ at the C11- and/or C17-positions (Fig. 2). This study clearly shows that substitution of Ala-773 with a glycine residue does not affect the binding and activity of agonists, but it does modify the agonist/antagonist properties of antimineralocorticoids depending on the substituents at the C11- and C17-positions. The results are discussed in the light of a hMR-LBD homology model generated from the recently described hPR crystal structure (Williams and Sigler, 1998).

Sequence alignment of the hMR-LBD-Helix H3 region. The alignment includes MRs, GRs, PRs, and aldosterone receptors from numerous species. The amino acid corresponding to the position 773 of hMR is boxed. The position of helix H3 is shown down to the alignment. The numbering corresponds to the human MR, GR, PR, and AR, respectively. The figure was prepared using ALSCRIPT (Barton, 1993). an, Aotus nancymaae; bt, Bos taurus; cj,Callithrix jacchus; cp, Cavia porcellus; cu, Cnemidophorus uniparens; gg, Gallus gallus; hs, Homo sapiens; me, Macropus eugenii; mm, Mus musculus; oa, Ovis aries; oc, Oryctolagus cuniculus; om,Oncorhynchus mykiss; rn, Rattus norvegicus; sc, Serinus canaria; so,Saguinus oedipus; tb, Tupaia belangeri; xl, Xenopus laevis.

Structural formula of ligands.
Materials and Methods
Chemicals.
[1,2-3H]Aldosterone (40–60 Ci/mmol) was purchased from the Radiochemical Center (Amersham, Aylesbury, Buckinghamshire, UK). Aldosterone and progesterone were obtained from Sigma (St. Louis, MO), RU26752 from Roussel Uclaf Laboratories (Romainville, France) and spironolactone (SC9420) was from Searle Laboratories (Chicago, IL). 11β-Vinyl-3-oxo-19-nor-17α-pregna-4,9-diene-21,17-carbolactone (1), 11β-allenyl-3-oxo-19-nor-17α-pregna-4,9-diene-21,17-carbolactone (2), 11β-(3-hydroxypropyl)-3-oxo-19-nor-17α-pregna-4,9-diene-21,17-carbolactone (3), 11-ethylidene-3-oxo-19-nor-17α-pregna-4,9-diene-21,17-carbolactone (4), 11-(3-propenylidene)-3-oxo-19-nor-17α-pregna-4,9-diene-21,17-carbolactone (5), 4,9(10)-androstadiene-3,17-dione (6), and 11β-allenyl-3-oxo-19-nor-17α-pregna-4,9-diene-3,17-dione (7) were synthesized according to Faraj and Claire (Faraj et al., 1990; Claire et al., 1993). Structure and abbreviations of the steroids are given in Fig. 2.
Expression and Reporter Constructs.
The plasmid pchMR was constructed by cutting out, from the plasmid 3750 (1), aHindIII-HindIII fragment containing the entire coding sequence of the hMR gene and about 270 base pairs (bp) and 400 bp of the 5′- and 3′-untranslated regions. This fragment was then inserted into pcDNA3 (Invitrogen, NV leek, The Netherlands). The plasmid pchGR was constructed by cutting out, from the plasmid pRShGRα (Giguere et al., 1986), a KpnI-XhoI fragment, including the entire hGRα coding sequence and about 110 bp and 500 bp of the 5′- and 3′-untranslated regions and inserting it into pcDNA3.
Cell Culture and Transfections.
COS-7 cells were cultured in Dulbecco's minimal essential medium (Gibco-BRL, Cergy Pontoise, France) supplemented with 10% heat-inactivated fetal calf serum, 2 mM glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere with 5% CO2. Four hours before and throughout the transfection procedure, cells were maintained in a medium supplemented with 10% charcoal-stripped fetal calf serum. Cells were transfected using the phosphate calcium precipitation method according to the Promega system. The phosphate solution, prepared for a 6-well tray, contained 5 μg of one of the receptor expression vectors (wild-type or mutant pchMR or pchGR), 10 μg of pFC31Luc (which contains the Maloney murine tumor virus promoter driving the luciferase gene) and 5 μg of pSVβ containing the gene coding for the β-galactosidase enzyme. The steroids were added to the cells 12 h after transfection. After a 24-h incubation, cell extracts were assayed for luciferase (De Wet et al., 1987) and β-galactosidase activities (Herbomel et al., 1984). To standardize for transfection efficiency, the relative light units obtained in the luciferase assay were divided by the absorbance obtained in the β-galactosidase assay.
Site-Directed Mutagenesis.
The 3.6-kilobase pairHindIII fragment containing the entire coding sequence of the hMR was subcloned in the pAlter-1 vector. The mutation of Ala-773 into glycine was created by site-directed oligonucleotide mutagenesis using Altered Sites In Vitro Mutagenesis System (Promega, Charbonnières, France). Purified oligonucleotides were purchased from Genset (Paris, France). The primer used was 5′-CTCAACCGCTTAGGAGGCAAACAGATG-3′. Identification of the desired mutation was obtained by direct sequencing. Inserts encoding mutant sequences were subcloned in the expression vector pcDNA3 for in vitro expression of the mutant receptors in the rabbit reticulocyte lysate or subsequent transfections in COS-7 cells.
Coupled Cell-Free Transcription and Translation.
Plasmids (1 μg) containing cDNA coding for the wild-type or mutant hMRs were in vitro expressed using the T7-coupled rabbit reticulocyte lysate system purchased from Promega according to the manufacturer's instructions.
Competition Experiments.
After translation of the wild-type or mutant hMRs, the lysate was diluted (1:2) with ice-cold TEGWD buffer (20 mM Tris·HCl pH 7.4, 1 mM EDTA, 1 mM dithiothreitol, 20 mM sodium tungstate, and 10% glycerol) and incubated with 5 nM [3H]aldosterone and with or without unlabeled competitors (50 nM) for 2 h at 4°C. Bound and unbound steroids were separated by the charcoal-dextran method (Fagart et al., 1998).
Production of Glutathione S-Transferase Fusion Proteins.
The vectors pGEX2TK containing glutathioneS-transferase (GST) or GST fused with a SRC-1 amino acids sequence, SRC-1(570–780), were provided by M. G. Parker (Laboratory of Molecular Pharmacology, Imperial Cancer Research Foundation, London, UK). GST and GST fusion proteins were expressed as described by Kaelin et al. (1991). Overnight cultures ofEscherichia coli (DH5α) expressing the recombinant GST plasmids were diluted 1:10 in Luria-Bertani medium. When the absorbance at 600 nm reached 0.8, the induction was performed for 3 h with isopropyl β-d-thiogalactoside at a 0.1-mM final concentration. Bacteria were then collected by centrifugation, resuspended 1:10 in NETN (0.5% Nonidet P-40, 1 mM EDTA, 20 mM Tris·HCl, 100 mM NaCl, pH 8.0) containing proteases inhibitors, sonicated, and then centrifuged. Protein concentration was estimated by Bradford method and the bacterial proteins content was visualized by Coomassie blue staining after loading onto a SDS-polyacrylamide gel.
GST Pull-Down Assays.
An aliquot of the crude bacterial extract (1 ml) containing GST fusion proteins was incubated at 4°C with 25 μl glutathione-Sepharose beads, previously washed and resuspended [final concentration, 1:1 (v/v)] in NETN. The glutathione-Sepharose beads were then washed three times with NETN. The wild-type or mutant hMR (A773G) were transcribed and translated in vitro in rabbit reticulocyte lysate in the presence of35S-labeled methionine. The in vitro expressed receptors were incubated with 1 μM aldosterone, progesterone, or compound 2 for 10 min at 20°C and then incubated with the fusion proteins, loaded onto glutathione-Sepharose beads, for 1 h at 4°C (in a 100-μl total volume). Beads were then washed three times with NETN, resuspended in 20 μl of loading buffer, and analyzed by SDS-polyacrylamide gel electrophoresis. Signals were amplified with Entensify and gels were autoradiographed after −80°C overnight. Autoradiographs were scanned by image analysis (Optilab, Graftek, France) and absorbance results are expressed in arbitrary units.
Model Building.
The hMR-LBD homology model and the docking of the ligands were achieved according to the method described previously (Fagart et al., 1998). Briefly, the hMR-LBD homology model was generated by the Modeler package (version 4.0) (Sali and Blundel, 1993) using the hPR crystal structure as a template and is based on the hMR- and hPR-LBD sequence alignment. Ligands were docked manually in the pocket using the probe-accessible and van der Waals volumes as guides; these volumes were generated with VOIDOO (Kleywegt and Jones, 1994). The side chains in the vicinity of the ligand were positioned in favorable orientation using a rotamer library of the O package (Jones et al., 1991). The Charmm package (QUANTA/CHARMM; Molecular Simulation Inc. Burlington, MA) was used for all the calculations. The complexes were energy minimized in 2000 steps with a dielectric constant of 2, using the Powell procedure. During the minimization process, the hydrogen bonds were defined by upper harmonic distance restraints (60 kcal Å−2 force constant) and the overall structure of the LBD was maintained by harmonic position restraints (30 kcal Å−2 force constant) of the Cα atoms of residues defining the secondary structure elements.
Miscellaneous.
The protein concentration in the lysate was determined by the Bradford method, using BSA as standard (Bradford, 1976). The protein concentration of the rabbit reticulocyte lysate was about 50 mg/ml. Radioactivity was measured in a LKB liquid scintillation spectrometer after addition of 5 ml of OptiPhase “HiSafe” (counting efficiency ≈50%).
Results
Effect of Steroid Substitution on Steroid Binding to the Wild-Type and Mutant hMRs.
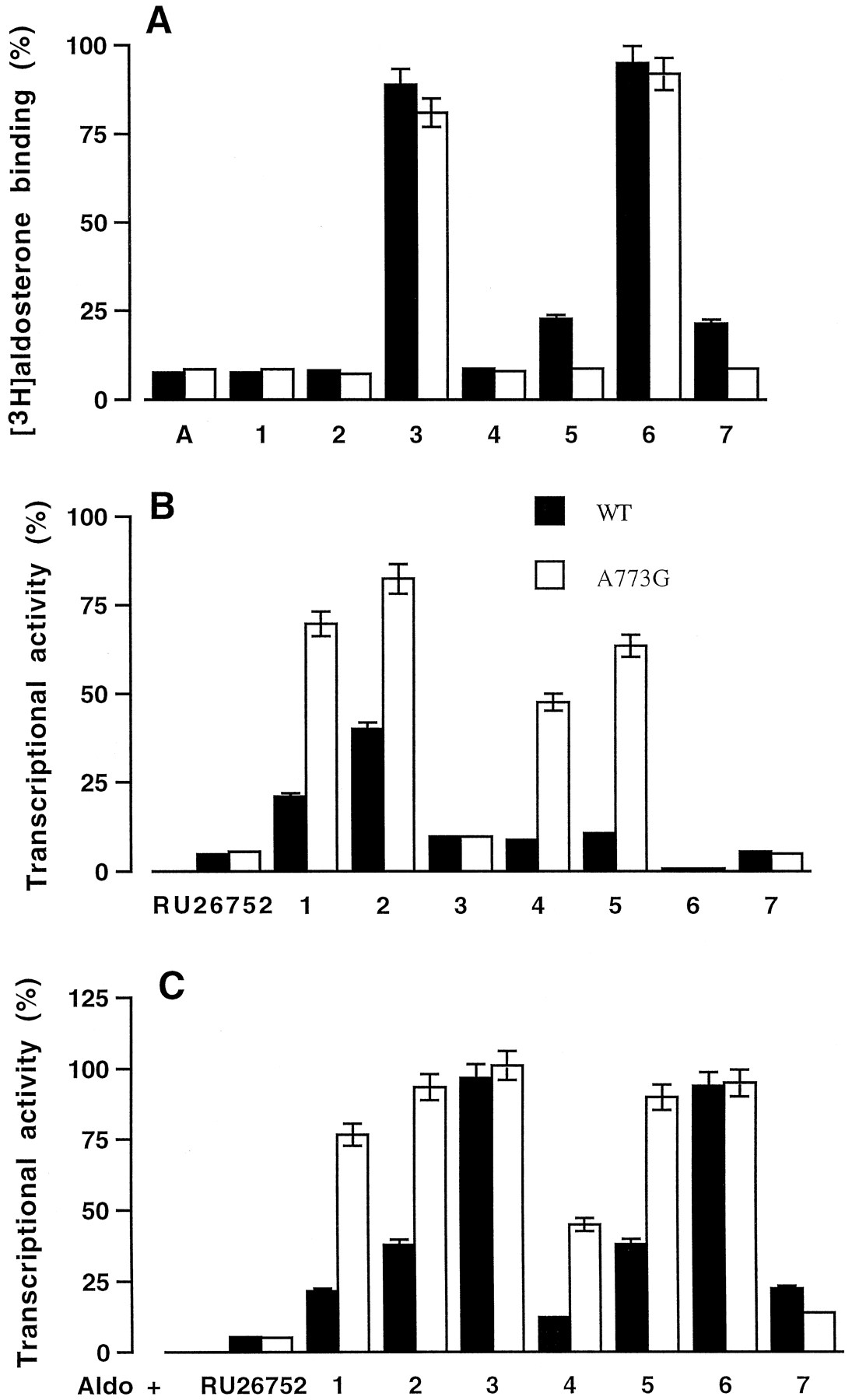
Wild-type and mutant hMRs were expressed in vitro and tested for their capacity to bind steroids substituted at the C11 and/or C17 position. As most of these compounds were available as unlabeled molecules, we measured their efficiency to inhibit [3H]aldosterone binding to the wild-type and mutant hMRs. [3H]Aldosterone binding to hMR was inhibited by 90% by aldosterone (Fig.3A). Except compound 3,characterized by a 11β-hydroxypropyl substituent, all the C11-substituted spirolactones (1, 2,4, and 5) were highly efficient at inhibiting [3H]aldosterone binding to hMR and A773G (Fig.3A). Compound 6, characterized by a 17-ketone-function and lacking of any substituent at the C11 position, was unable to inhibit [3H]aldosterone binding to hMR and A773G. In contrast, compound 7, with a C17-ketone function and a C11-allenyl substituent, was a potent competitor of aldosterone binding to the wild-type and mutant hMRs. These findings suggest that the C11- and C17-substituents are both determinant for the steroid binding to hMR and reveal that substitution of alanine 773 by a glycine residue does not modify the steroid-binding interaction.
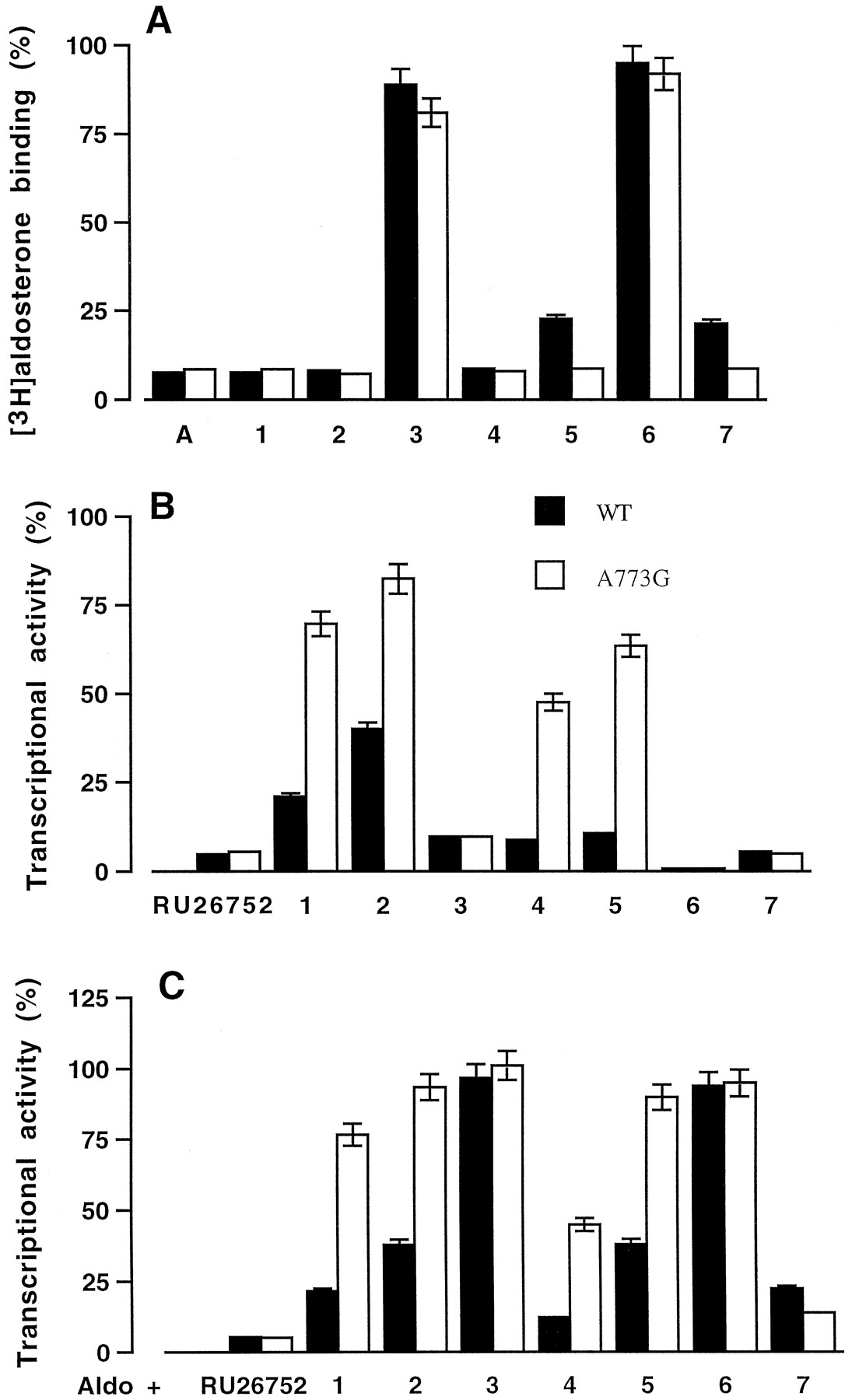
Steroid binding to the wild-type and mutant hMR. A, the wild-type and mutant hMR (A773G) were synthesized in vitro in rabbit reticulocyte lysate. The lysate was diluted 2-fold with TEGWD buffer and incubated with 5 nM [3H]aldosterone in the presence or absence of unlabeled competitors (50 nM) for 2 h at 4°C. Bound and unbound steroids (1–7) were separated by the charcoal-dextran method. Nonspecific binding was measured in a parallel translation experiment without any receptor expressing vector. Results are expressed as percent of specific [3H]aldosterone binding measured in the absence of competitor. Transactivation properties of wild-type and mutant hMRs in response to the C11 or C17 substituted derivatives. COS-7 cells were transfected with wild-type or mutant hMR expression vectors, pFC31luc as reporter plasmid, and a β-galactosidase internal reporter to correct for transfection efficiency. Before harvesting, cells were treated for 24 h with 10−6 M aldosterone, or RU26752 or compounds 1-7 (B), or with 10−9 M aldosterone plus 10−6 M of RU26752 or compounds1-7 (C). Transactivation was determined by luciferase activity, normalized to the internal β-galactosidase control, and expressed as percent of wild-type receptor activity measured in the presence 10−6 M aldosterone. Each point is the mean ± S.E.M. of three separate experiments. WT, wild-type.
Transactivation Properties of the Wild-Type and Mutant hMRs.
The ability of the wild-type and mutant hMRs to activate transcription in response to C11 and/or C17-substituted steroids was examined by cotransfection assays. A773G retained the ability of the wild-type receptor to stimulate transcription in response to aldosterone (100% activity, data not shown). In the presence of 10−6 M RU26752, a spirolactone devoid of any C11-substitution, compound 3, 6, or 7, the hMR activity was 5 to 10% that observed with aldosterone. This response was not modified by the replacement of alanine 773 by a glycine residue (Fig. 3B). In the presence of compound 1, 2,4, or 5, the hMR activity was 15 to 40% that observed with aldosterone, it increased to ∼50 to 85% on alanine-to-glycine substitution (Fig. 3B) indicating that these compounds act as agonists through A773G.
The antagonist activity of the C11- and/or C17-substituted compounds was tested by incubating the transfected cells with 10−9 M aldosterone in the absence (100% activity in Fig. 3C) or presence of 10−6 M of the steroids. The aldosterone induced activity of the wild-type and A773G remained unchanged in the presence of compound 3 or6. It decreased in the presence of RU26752 or compound7 to a value that accounts for 20% of the maximum aldosterone response. These results indicate that the antagonist properties of RU26752 and compound 7 are the same with the wild-type and the mutant hMRs. In the presence of compound1, 2, 4, or 5, the aldosterone-induced hMR activity of the wild-type hMR is 10 to 40% that observed with aldosterone alone and 50 to 90% in the case of A773G (Fig. 3C) indicating that the antagonist potency of compounds1, 2, 4, and 5 was lower with the mutant hMR than with the wild-type receptors. These results are in good agreement with the observation that these compounds are able to stimulate the transactivation function of the mutant A773G (Fig. 3A).
Dose-response curves were also generated by adding to the transfected cells 10−9 M aldosterone plus increasing concentrations (10−9 to 10−6 M) of progesterone, compound 2or 7 (antagonist effect) or by adding increasing concentrations (10−11 to 10−6 M) of aldosterone or compound 2(agonist effect). As shown in Fig. 4A, the aldosterone-induced activity of the wild-type hMR is inhibited by progesterone and compound 2 or 7 in a dose-dependent manner with IC50 values of ∼10−8, 5.10−8 M, 5.10−7 M, respectively. The ability of progesterone and compound 7 to inhibit the aldosterone-induced activity of A773G was the same as that observed for the wild-type receptor (data not shown). In contrast, compound2 stimulates the A773G activity in a dose-dependent manner with an ED50 value of ∼5.10−9 compared with 10−10 M for aldosterone (Fig. 4B). Compound5 exhibited the same characteristics (data not shown). Altogether these results show that the replacement of alanine 773 by a glycine residue in the hMR does not alter the agonist feature of aldosterone and the antagonist properties of progesterone and compound7. In contrast, it modifies the response to steroids bearing a hydrophobic chain at the C11-position and/or a γ-lactonic ring at the C17-position.
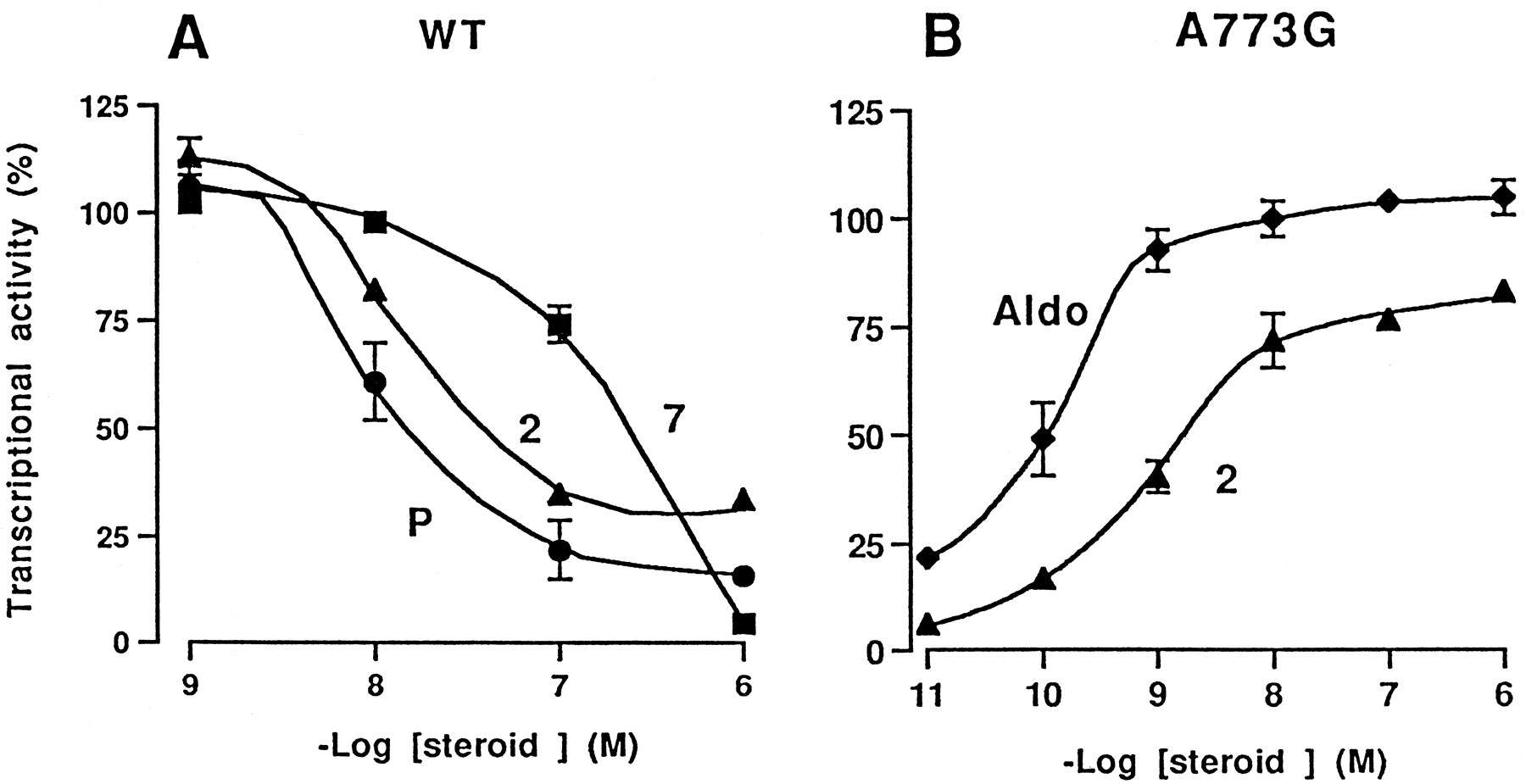
Transactivation properties of wild-type and mutant hMRs in response to increasing concentrations of steroids. COS-7 cells were transfected with wild-type hMR (A) or the A773G (B) expression vectors, pFC31luc as reporter plasmid, and a β-galactosidase internal reporter to correct for transfection efficiency. Before harvesting, cells were treated for 24 h with 10−9 M aldosterone plus 10−9 to 10−6 M of progesterone (●) or compound 2 (▴) or 7 (▪) (A) or with 10−11 to 10−6 M aldosterone (♦) or compound 2 (▴) (B). Transactivation was determined by luciferase activity, normalized to the internal β-galactosidase control, and expressed as percent of wild-type receptor (A) or A773G (B) activities measured in the presence of 10−6 M aldosterone. Each point is the mean ± S.E.M. of three separate experiments. WT, wild-type.
Because hGR is characterized by a glycine residue at position 567 (corresponding to alanine 773 in the hMR), we wonder whether compound2 was able to activate the hGR. COS-7 cells were transfected with a hGR expression vector and the plasmid pFC31luc and then incubated with 10−8 M dexamethasone, a potent glucocorticoid agonist, or compound 2 (agonist effect) or with 10−8 M dexamethasone in the presence of 10−6 M compound 2 (antagonist effect). At 10−6 M compound 2maximally increased the hGR activity and was unable to inhibit the dexamethasone-induced hGR activity, indicating that compound2 behaves as an agonist with the hGR (data not shown).
Interaction of Wild-Type and Mutant hMRs with SRC-1.
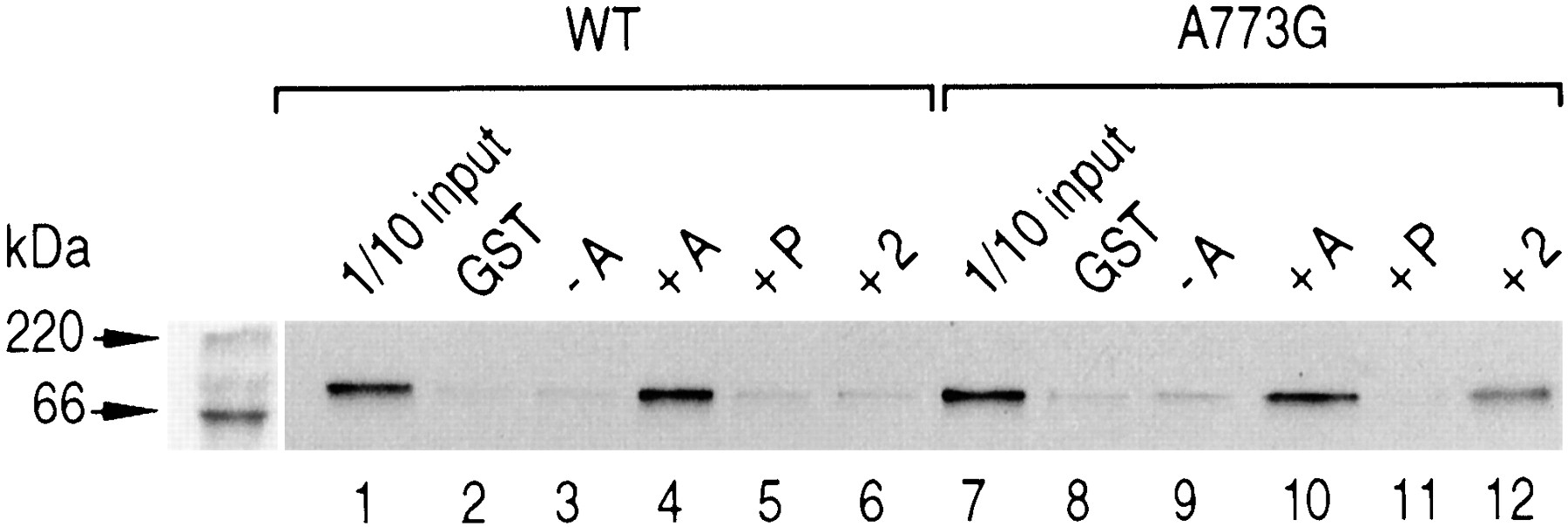
Numerous reports have emphasized the requirement of coactivator factors to promote the activity of NRs in response to their cognate ligand (Glass et al., 1997). The interaction of coactivators, namely SRC-1, with agonist-associated hMR has been shown by in vitro experiments (Hellal-Levy et al., 2000). It was therefore interesting to examine the binding of SRC-1 to the wild-type and mutant hMRs after binding of compound 2, which displays an antagonist activity with the wild-type receptor, whereas it acts as an agonist with the A773G. GST and GST-SRC-1 fusion proteins, recovered from bacterial extracts, were absorbed onto glutathione-Sepharose beads and incubated with in vitro synthesized 35S-labeled wild-type and mutant hMRs incubated or not with aldosterone, progesterone, or compound 2. In the absence of ligand or after incubation with progesterone, a mineralocorticoid antagonist, only weak or no interaction of SRC-1 with the wild-type or the mutant A773G was detected (Fig. 5, lanes 3,5,9 and 11). Incubation of the wild-type and mutant hMRs with aldosterone promoted an interaction with SRC-1 (Fig. 5, lanes 4 and 10). The wild-type hMR was unable to interact with SRC-1 after binding of compound2 (Fig. 5, lane 6), whereas A773G associated with compound2 was able to interact with SRC-1 (Fig. 5, lane 12). These results are in good agreement with the observations that compound2 acts as an antagonist with the wild-type hMR, whereas it displays agonist properties with A773G. Compound 5 exhibited the same characteristics (data not shown).

In vitro interaction of SRC-1 with wild-type and mutant hMR. [35S]Methionine-labeled wild-type or mutant hMR was incubated without or with 1 μM aldosterone (A), progesterone (P) or compound 2 for 10 min at 20°C and then incubated with Sepharose glutathione beads preincubated with GST fusion proteins with SRC-1. The beads were washed and boiled in Laemmli buffer, and the samples were analyzed by SDS-polyacrylamide gel electrophoresis followed by autoradiography. As control, incubation of [35S]methionine-labeled wild-type or mutant hMR with GST alone (lanes 2 and 8) and 1/10 of the receptors input used in the assay (lanes 1 and 7) are shown.
Ligand Docking.
We have reported a three-dimensional model of the hMR-LBD based on the hRARγ-LBD crystal structure (Fagart et al., 1998). The human estrogen receptor- and hPR-LBD crystal structures have recently been determined to show minor differences compared with the hRARγ structure. The N-terminal part of helix H3 is shifted toward the core of the LBD (∼2 Å) pushing away helix H6 and the loop connecting H6 to H7 out of the core (∼6 Å). Because the sequence homology between hMR- and hRARγ-LBDs is low (<20%) compared with that between hMR and hPR (56%), we constructed a three-dimensional homology model of the hMR-LBD using the crystal structure of the hPR-LBD as a template and docked aldosterone within the LBP. The hydrogen bond network described previously is still present in this refined model. The C3-ketone function is anchored by Gln-776 and Arg-817, and Asn-770 forms a hydrogen bond with the C21-hydroxyl group of aldosterone (Fig. 6A). Interestingly, in the refined hMR, the Asn-770 carbonyl moiety is at a favorable distance and orientation to connect the C18-hydroxyl group (2.8 Å), and Ala-773 is in a close contact with the oxygen of the hemiketal group (3.4 Å) of aldosterone.
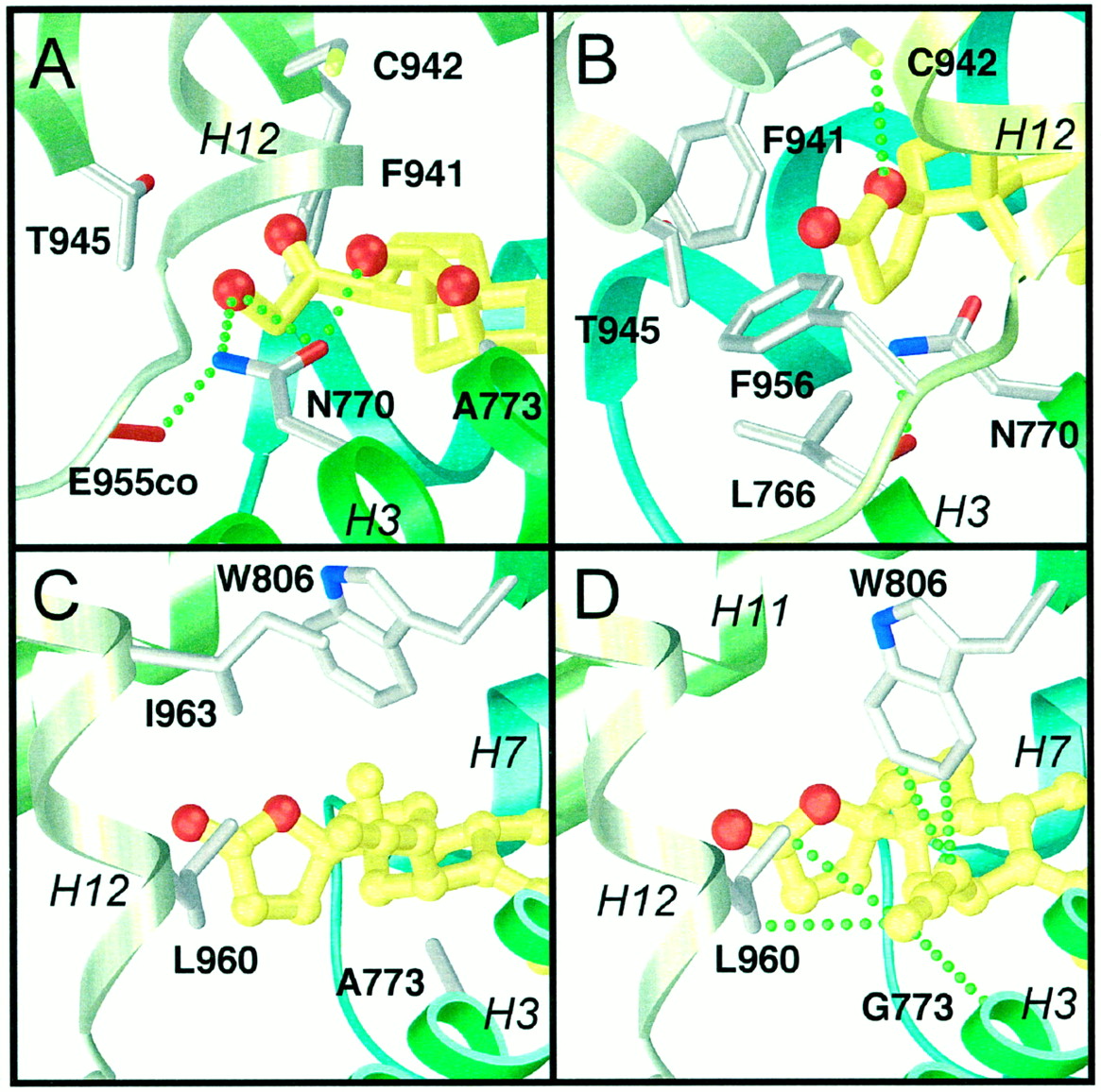
Anchoring of the ligand D-ring in the hMR-LBP. The ligand is colored in yellow, with its oxygen atoms in red. The hydrogen bonding network anchoring the D-ring as discussed in the text is depicted as green dashed lines. A, close-up view of the aldosterone D-ring anchoring with Asn-770. B, the contacts of RU26752 γ-lactonic ring with Leu-766, Phe-941, Thr-945, Phe-956, and Cys-942. The figure was prepared with SETOR (Evans, 1993). Anchoring of the compound2 in the hMR-LBP. C, close-up view showing the constraints between the compound 2 11β-allenyl side chain and Leu-960, and between Trp-806 and Ile-963 in the wild-type hMR. D, close-up view of the stabilizing van der Waals contacts, depicted as green dashed lines, between the compound 2 11β-allenyl side chain and Gly-773, Trp-806, and Leu-960 in the A773G mutant hMR. The figure was prepared with SETOR (Evans, 1993).
To better understand the effect of C11- and C17-substitution on the steroid agonist versus antagonist properties, we docked the antagonist RU26752 and compounds 1 to 7 in the hMR-LBP model. In this model, the RU26752 lactonic ring is located in a region delimited by helices H3, H11, and H12 and forms van der Waals contacts with Leu-766, Phe-941, Thr-945, and Phe-956 (Fig. 6B). The 17β-oxygen atom makes a weak hydrogen bond with Cys-942. Compounds 6and 7, lacking the lactonic ring, are unable to establish the van der Waals and polar interactions described above. Because of steric hindrances between the alanine 773 and the steroid C11-substituents, compounds 1 to 5 cannot adopt an orientation similar to RU26752. A rotation of the ligand around the C3 to C17 axis and a displacement of the Trp-806 side chain are both required for ligand accomodation. In such a configuration, constraints occur between the C11-substituent and Leu-960 in one part and between Trp-806 and Ile-963, preventing the positioning of helix H12 in the active orientation (compound 2, Fig. 6C). Replacement of the Ala-773 with a glycine residue within the hMR generates a tight hydrophobic hollow delimited by Gly-773 (helix H3), Trp-806 (helix H5), and Leu-960 (helix H12) (Fig. 6D). The polar and bulky hydroxypropyl group of compound 3 cannot be accommodated in this tight cavity. In contrast, the C11-substituents of compounds 1,2, 4, 5, and 7 fit well, this is illustrated in Fig. 6D for compound 2. The allenyl side chain of this compound forms van der Waals contacts with the Gly-773 backbone, Trp-806 and Leu-960. Moreover, it is likely that π interactions could stabilize the interaction between the substituent double bonds and the Trp-806 indole ring. The terminal carbon atom of the C11-substituent of compounds 2 and5 are at a short distance from the carbon atom of the glycine 773 carbonyl group (3.2 Å). The electrostatic fitted charges of these two compounds, characterized by an allenyl or a propenylidenyl group at the C11-position were determined using the Spartan ab initio program with a 6–31G* basis set. It reveals that the terminal carbon of both compounds is characterized by a partial negative charge (−0.50 and −0.46 e, respectively). These data suggest that a dipole-dipole stabilizing interaction could exist at this level between the glycine carbonyl and the C11-substituent terminal C-H groups.
Discussion
This study was carried out to better understand how the contacts between MR and agonist and antagonist ligands modulate the receptor activity. We measured the ability of synthetic steroids with various substituents at the C11- and/or C17-position to bind and activate the wild-type hMR and a mutant hMR in which the alanine residue at position 773, facing the 11β-steroid substituent, was substituted with a glycine residue (A773G). The determinant for mineralocorticoid agonism and antagonism was analyzed by using a three-dimensional model of the human MR constructed by taking as a template the recently published structural data of the hPR (Williams and Sigler, 1998), whose LBD displays a 57% homology with that of the hMR.
The results reported in the present study reveal that the nature of the C17 substituent of the D ring is a determinant for steroid binding to the hMR. Progesterone, with a C17 carboxymethyl group, binds to the hMR with high affinity, as does aldosterone, the natural mineralocorticoid agonist characterized by a C17-hydroxymethyl ketone moiety (Fagart et al., 1998). Similarly, RU26752, a synthetic steroid of spirolactone group, harboring a 17γ-lactone ring, also binds the hMR with high affinity. In contrast, the synthetic compound 6, with a 17-ketone, and testosterone, with a 17-hydroxyl group, are devoid of any affinity for the hMR. This is also the case for two glucocorticoid ligands RU26988 and RU28362 (Gomez-Sanchez and Gomez-Sanchez, 1983;Rafestin-Oblin et al., 1986) bearing a 17β-hydroxyl moiety and a 17α-propynyl group and also for 17O-methyl canrenoic acid, which is the opened form of the 17γ-lactonic moiety (Funder et al., 1974; Peterfalvi et al., 1980). The hMR-LBD homology model reveals that the hydroxymethyl ketone of aldosterone forms numerous contacts with the hMR through Asn-770, Phe-941, Cys-942, Thr-945, and Phe-956. Progesterone and RU26752 contact Cys-942 through the C20-ketone group and the 17β-oxygen of the lactonic ring, respectively. Additional stabilizing hydrophobic contact with the receptor through the lactonic ring is observed for RU26752. Conversely, compound 6, with a small 17-ketone function, is unable to establish such contacts. Interestingly, the substitution of compound6 at the C11 position by an allenyl group (compound7) restores the binding to the hMR. The allenyl group forms favorable hydrophobic contacts with the hMR-LBP, as revealed by the model. Thus, the contacts between the C3-ketone function and Gln-776 and Arg-817 are not sufficient for steroid binding to MR and additional contacts through the C11 or C17 substituents are required.
Recent crystal structure analyses of several NRs revealed that the major difference between the ligand-free and the agonist-associated receptor states is the positioning of the helix H12 that harbors the ligand activated transactivation function (AF-2) (Bourguet et al., 1995; Renaud et al., 1995; Wagner et al., 1995; Wurtz et al., 1996;Brozowski et al., 1997; Fagart et al., 1998; Nichols et al., 1998). In the unliganded state, the helix H12 points away from the receptor, whereas in the agonist-associated state, it is folded back toward the core of the LBD. The repositioning of H12, together with additional structural changes, such as the bending of H3, brings it into a distinct receptor environment, thus creating an interface suitable for NR coactivator binding (Glass et al., 1997). The active receptor conformation is insured by specific steroid-receptor contacts. The C21-hydroxyl group of aldosterone forms strong hydrogen bond with Asn-770 (H3) (Fagart et al., 1998). Moreover, this residue anchors the oxygen atom of the Glu-955 main-chain, allowing the folding back of H12 in its active position. Compounds 1 and 2characterized by a 17γ-lactone ring and a C11 hydrophobic substituent but lacking the 21OH group display a partial agonist activity when acting through the wild-type hMR and are almost full agonists with A773G. The docking of compound 2 within the A773G model (Fig. 6D) suggested that the C11-allenyl group can be accommodated in the tight hollow delimited by the helices H3, H5, and H12, a position that stabilizes H12 in its active conformation by strong van der Waals contacts. In the model of the wild-type hMR, the presence of the Ala-773 side-chain displaces the C11-substituent, thus preventing the nearby H12 from adopting its optimal position. The partial agonist activity of compounds 1 and 2 with the wild-type hMR compared with the low agonist activity of compounds 4and 5 might be related to the nature of the C11 substituent. In compounds 1 and 2, the C11 carbon has an sp3 hybridization. The substituent is thus bent and can turn around the C11—C11′ bond to be accommodated. On the other hand, compounds4 and 5 have a sp2 hybridization and the conjugated substituents have no flexibility with an equatorial orientation.
Numerous studies have reported that the interaction of coactivators with NRs is observed when receptors are transcriptionally active (Glass et al., 1997). Both the hMR and A773G are unable to interact with SRC-1 after binding the antagonist progesterone, whereas the interaction of both receptors is detected after incubation with aldosterone. Compound2, which is unable to stimulate maximally the hMR transactivation, does not allow the SRC-1 recruitment, whereas it activates the mutant hMR and facilitates the interaction with the coactivator, confirming the agonist-dependent interaction of the receptor with the coactivators.
It has been proposed that antagonism in the hMR on binding of RU26752 and progesterone was caused by a loss of contact between antagonist ligands and the H12 region (Fagart et al., 1998). Similarly, the antagonist properties of compound 7, which are observed with both the wild-type and the mutant A773G, are caused by a loss of anchoring of the D ring. Docking of the 11β-substituted spirolactones (compounds 1, 2, 4, and 5) in the hMR-LBD model suggests that their antagonist properties may be explained by a mechanism distinct from that observed with RU26752 and progesterone. Constraints between the 11β-steroid substituent and Trp-806 in helix H5 lead to the expulsion of the hMR helix H12 from its active position. Such a mechanism could be compared with the antagonist effect of raloxifene and tamoxifen, where the bulky substituent prevents the positioning of estrogen receptor helix H12 in its active orientation (Brozowski et al., 1997).
Altogether, these results suggest that the C11 and C17 substituents contribute differently to the mineralocorticoid agonist/antagonist properties and point out two possible mechanisms for aldosterone antagonism. The possibility of additional hydrophobic contacts between the steroid and the hMR changing the agonist/antagonist feature of the steroid without altering ligand affinity might be extended to other steroid positions, such as the C7 position, allowing a new way toward synergic influence of both C11- and C7α-substitutions on the antimineralocorticoid specificity.
Acknowledgments
We are grateful to V. Cavaillès, H. Richard-Foy, and F. Gouilleux for the generous gifts of the plasmids.
Footnotes
- Received March 9, 2000.
- Accepted June 23, 2000.
-
Send reprint requests to: Gilles Auzou, INSERM U439, 70 rue de Navacelles, 34090 Montpellier, France. E-mail:auzou{at}montp.inserm.fr
-
↵1 Considered jointly as first authors.
-
This work was supported by INSERM (APEX 9834, MERO). This work was presented as a poster to the Forefront meeting of the International Society of Nephrology: “News in Aldosterone Action” Château de Montvillargenne, Paris, France, August 1999.
Abbreviations
- MR
- mineralocorticoid receptor
- LBD
- ligand binding domain
- NR
- nuclear receptor
- hMR
- human mineralocorticoid receptor
- hRARγ
- human retinoic acid receptor
- GR
- glucocorticoid receptor
- PR
- progestin receptor
- LBP
- ligand-binding pocket
- bp
- base pairs
- GST
- glutathione S-transferase
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}