


Abstract
The prostaglandin synthase cyclooxygenase-2 (COX-2) is produced by an immediate early response gene induced in most cells by a variety of stimuli. Several studies have shown that the immunosuppressant cyclosporin (CsA) interferes with prostanoid metabolism, but the mechanisms are unclear. Here we examine the effect of CsA on COX-2 mRNA induction in cultured rat vascular smooth muscle cells (VSMC) that natively express the nuclear factor of activated T-cells, a known mediator of CsA-sensitive transcription. CsA significantly suppresses strong COX-2 mRNA induction caused by the Ca2+-mobilizing mitogens UTP, angiotensin II, and platelet-derived growth factor-BB, and the synergistic induction caused by costimulation with ionomycin and a phorbol ester. Forskolin and interleukin-1β are substantially weaker COX-2 mRNA inducers, and CsA does not inhibit their effect. CsA strongly inhibits UTP-, angiotensin II-, and platelet-derived growth factor-BB-stimulated COX-2 gene transcription as measured by nuclear run-on or promoter-reporter studies, but has no effect on mRNA induction caused by post-transcriptional stabilization of a distal COX-2 mRNA 3′-untranslated region regulatory element. These data show that CsA selectively inhibits mitogen-induced COX-2 gene expression by a transcriptional mechanism that may involve the nuclear factor of activated T-cells.
Conversion of arachidonic acid to prostaglandin H2 by cyclooxygenase-2 (COX-2) is the key enzymatic step in prostaglandin synthesis. Prostaglandins are involved in several biological processes including inflammation, immune responses, cell growth, ovulation, and the regulation of vascular tone (Williams and DuBois, 1996). The two COX isoforms, COX-1 and COX-2, are encoded by separate genes (Fletcher et al., 1992; Hla and Neilson, 1992; Kraemer et al., 1992). Although their enzymatic function is similar, regulation of their cellular expression levels differs. COX-1 gene expression is largely constitutive whereas COX-2 gene expression is negligible under basal conditions but can increase dramatically in numerous cell types in response to a wide variety of stimuli (Smith and Dewitt, 1996).
The immunosuppressive drug cyclosporin A (CsA) has been reported to interfere with prostaglandin actions in several cell types, but the mechanisms are unresolved (Fan and Lewis, 1985; Stahl et al., 1989;Edwin et al., 1996; Iniguez et al., 1999). CsA forms a stable complex with cyclophilin, which binds to and inhibits the catalytic subunit of protein phosphatase 2B, otherwise known as calcineurin (Clipstone et al., 1994). Although calcineurin is thought to participate in a wide variety of cellular processes, relatively few specific substrates are known (Klee et al., 1998). The best understood calcineurin substrate is the nuclear factor of activated T-cells (NFAT), which is involved in controlling lymphocyte cytokine gene expression (for review, see Rao et al., 1997). Calcineurin dephosphorylates cytoplasmic NFAT, leading to its nuclear translocation, where it assembles on composite enhancers elements with transactivators stimulated by mitogen-activated protein kinase signaling. This mode of regulation explains a common characteristic of NFAT-regulated genes, which is synergistic induction by costimulation of Ca2+ and mitogen-activated protein kinase signaling.
Inhibition of NFAT-mediated transcriptional programs induced strongly by foreign antigens in lymphocytes is thought to be the principal therapeutic mechanism of action of CsA (Schreiber and Crabtree, 1992). NFAT targets in the immune system are well characterized, including interleukin (IL)-2, IL-3, IL-4, IL-5, tumor necrosis factor-α, interferon-γ, and granulocyte-macrophage colony-stimulating factor genes (Rao et al., 1997). Adverse therapeutic consequences of CsA treatment seen in almost all subjects include reversible hypertension and reduced renal function (Sander et al., 1996). Although NFAT was thought of initially as lymphoid specific, one or more of the four known NFAT genes is expressed in virtually all tissues (Hoey et al., 1995). Several recent reports have identified NFAT in a variety of cell types that include vascular smooth muscle cells (VSMC) (Ho et al., 1994; Cockerill et al., 1995; Boss et al., 1996; Abbott et al., 1998;Boss et al., 1998a,b). Using a heterologous NFAT-specific transcriptional reporter, recent studies have established that various phospholipase C-coupled receptors most likely serve as physiological NFAT regulators outside of the immune system (Boss et al., 1996; Boss et al., 1998a,b). The genes regulated by NFAT in VSMC and in most other nonlymphoid cell types are largely unknown, but their discovery may provide insights into the mechanisms of CsA-induced cardiovascular toxicity.
To determine whether there is a potential role for NFAT in VSMC COX-2 gene regulation, this study examined the effect of CsA on the ability of various agonists to induce COX-2 gene expression. We found that Ca2+-mobilizing mitogens acting through Gαq-coupled P2Y nucleotide and AT1 angiotensin receptors, or a growth factor receptor, are more efficacious inducers of the COX-2 mRNA then are non–Ca2+-mobilizing stimuli, including the cytokine IL-1β and the adenylyl cyclase activator forskolin. CsA selectively attenuates COX-2 mRNA induction caused by Ca2+-mobilizing receptors, most likely by a transcriptional mechanism. These and additional observations support the hypothesis that NFAT lies within activation pathways regulating VSMC COX-2 gene expression.
Experimental Procedures
Materials.
UTP, forskolin, angiotensin II, and luciferin were obtained from Sigma Chemical (St. Louis, MO). IL-1β and platelet-derived growth factor-BB (PDGF-BB) were purchased from Life Technologies Inc. (Grand Island, NY) and Calbiochem Inc. (San Diego, CA), respectively. CsA was a gift from Sandoz Pharmaceuticals. [α-32P]UTP was purchased from NEN Life Science Products (Boston, MA). Probes used in RNase protection assay (RPA) were synthesized using MAXIscript In Vitro Transcription Kit from Ambion, Inc. (Austin, TX), and RPA analysis was performed using the Ambion RPA II kit. Antibiotics, serum, and cell culture media were from Life Technologies. Phoenix retroviral producer cells (ATCC SD-3443) were obtained from the American Type Culture Collection (Rockville, MD). All retroviral reporter plasmids created for this study were maintained in Escherichia coli Top 10F′ from InVitrogen, Inc. (Carlsbad, CA) in 100 μg/ml ampicillin and 12.5 μg/ml tetracycline.
Cell Culture.
Rat thoracic aorta VSMC were studied between ∼25 to 70 population doublings after inception. They were grown until confluence in Dulbecco's modified Eagle's medium containing 3.7 mg/ml NaHCO3, 100 μg/ml streptomycin, 100 U/ml penicillin, and 10% calf serum in 5% CO2 at 37°C. After an additional 24-h incubation in serum-free media, confluent cells were treated with agonists. Phoenix Ampho retroviral producer cells were cultured as above except using 10% fetal bovine serum. Helper virus-free protocols for generating recombinant retroviruses and VSMC infection have been described in detail (Boss et al., 1998a).
Plasmids.
Retroviral luciferase reporter plasmids for measuring NFAT-, activator protein 1 (AP1)-, serum response element (SRE)-, and nuclear factor-κB (NFκB)-mediated transcription are described elsewhere (Abbott et al., 2000). pAR2, representing a full-length COX-2 promoter driving luciferase expression, was created by cloning 2.7 kilobase pairs (kb) of the COX-2 promoter region (Sirois et al., 1993) using the PCR primers G18 (5′AATTTACGCGTACTCCTGAAGCTCTCCG) and G19 (5′ATAGCGTCGACCCTGATAAAATTAGAACCAAA) into the SalI - MluI sites of pCAT/CUL, a retroviral vector wherein chloramphenicol acetyl transferase is also constitutively expressed from the 5′LTR promoter. A template for the 549-base COX-2 riboprobe (pAR1) was created by removing a 1.3 kb KpnI fragment from a pBluescriptSK+ vector containing the COX-2 cDNA (DuBois et al., 1994), and linearizing with BstBI. The template for the luciferase riboprobe (pKX37) was created by cloning a 540-baseEcoRI-XbaI fragment from pXF40LUC (Wang and Murphy, 1998) into pBluescriptKS+. pKX56 contains a 263-base HpaI-XhoI fragment encoding the most distal COX-2 3′-untranslated region cloned downstream of luciferase in pXF40LUC (Xu et al., 2000). The design and synthesis of the retroviral vectors that express trans-dominant NFAT inhibitor Zeo:eGFP:VIVIT (pTJ85) and the control retroviral vector expressing Zeo:eGFP (pTJ84) are described elsewhere (K. L. Abbott, J. R. Loss, A. M. Robida, and T. J. Murphy, submitted).
Luciferase Assay.
Drugs and agonists, or their vehicles, were added to cells cultured in 24-well plates from 100× concentrated stock solutions. The forskolin diluent is dimethyl sulfoxide, and that for CsA is a solution containing 60% EtOH and 2% Tween 80. Cells were pretreated with CsA or its vehicle 30 min before agonist addition. After incubation for 4 h at 37°C in 5% CO2, luciferase activity was measured as described previously (Takeuchi et al., 1993) using a Turner Designs model 20E Luminometer.
RPA.
The COX-2, luciferase, and cyclophilin riboprobes were synthesized using either T7 or T3 polymerase following Ambion Maxiscript Kit directions. The template for the cyclophilin probe, pTriCyp, was purchased from Ambion, Inc. pAR1 linearization with BstBI and pKX37 with XbaI serve as templates for 549 base-COX-2 and 600-base LUC riboprobes, respectively. RNA was isolated from cells treated in 35-mm dishes using Trizol (GibcoBRL) according to the manufacturer's directions. RPAs were performed using slight modifications of the Ambion RPA II kit protocols. In brief, aliquots (10–15 μg) of total RNA were placed in a 1.5-ml centrifuge tube and lyophilized under vacuum. The samples were then resuspended in 10 μl of hybridization buffer before adding 2 μl of a mixture of sequence gel-purified, [α-32P]UTP-labeled riboprobes (100,000 cpm per probe) also dissolved in hybridization solution. After heating to 95°C for 5 min, and hybridizing at 42°C for 12 to 18 h, the samples were digested for 30 min with a mixture of RNAases A and T1 (final concentrations, 2.3 and 90 U/ml, respectively) at 37°C. Protected fragments were then separated on a 5% polyacrylamide/urea denaturing gel. After exposure on storage phosphor screens for up to 24 h, images were acquired using a Molecular Dynamics PhosphorImager (Sunnyvale, CA). Hybridization signal quantification was performed using a volume integration protocol in the ImagineQuant program (ver. 3.3; Molecular Dynamics).
Nuclear Run-On Assay.
Cells grown to confluence on 150-mm plates were treated for 45 min with either vehicle, UTP, or angiotensin II in the absence or presence of CsA before harvesting nuclei and performing nuclear run-on using [α-32P]UTP as described previouosly (Nickenig and Murphy, 1994). Denatured plasmids (5 μg) containing cDNAs for COX-2 (pRDCOX-2), GAPDH (PIBI30; International Biotechnology, New Haven, CT), and negative control vector (pBluescriptSK+) were crosslinked to neutral nylon membranes (Hybond-N; Amersham Life Science) before hybridization with radiolabeled run-on RNA. After a 16-h hybridization, blots were washed in 50 ml of 2× SSC (300 mM sodium chloride, 30 mM sodium citrate, pH 7.4) for 15 min at room temperature, 2× SSC and 0.1% SDS for 15 min at 50°C, and then with 2× SSC containing 1 μg/ml of RNAase A for 5 min at room temperature. After exposure on storage phosphor screens for up to 24 h, images were acquired and quantified as above.
Data Analysis.
Statistical significance was determined using one or two-tailed paired t test using Prism v3.0 (GraphPad, Inc., San Diego, CA).
Results
VSMC COX-2 mRNA expression was measured using a quantitative duplex RPA in which COX-2 mRNA hybridization signals were normalized by the cyclophilin mRNA signal in each sample. Two characteristics of NFAT-mediated transcription are inhibition by CsA and synergistic activation by costimulation with calcium ionophores and phorbol esters (reviewed in Rao et al., 1997). As shown in Fig.1, peak COX-2 mRNA levels are induced on average 17- and 7-fold over basal maximally after treatment with either 100 nM ionomycin or 100 nM phorbol 12-myristate 13-acetate (PMA), respectively. The response to combined stimulation with ionomycin and PMA is both highly synergistic and inhibited markedly by CsA.

CsA inhibits synergistic COX-2 mRNA induction by ionomycin and PMA. A, PhosphorImages of a representative RPA time-course study. VSMC were stimulated with 100 nM ionomycin, 100 nM PMA, or ionomycin plus PMA in the absence or presence of 1 μM CsA. RNA was collected at the indicated times thereafter. COX-2 and cyclophilin (Cyp) levels were measured simultaneously in each sample. B, COX-2 mRNA signals were normalized by those for cyclophilin and expressed as percentage of the maximal response within each experiment. Each point represents the mean ± S.E. from three independent experiments.
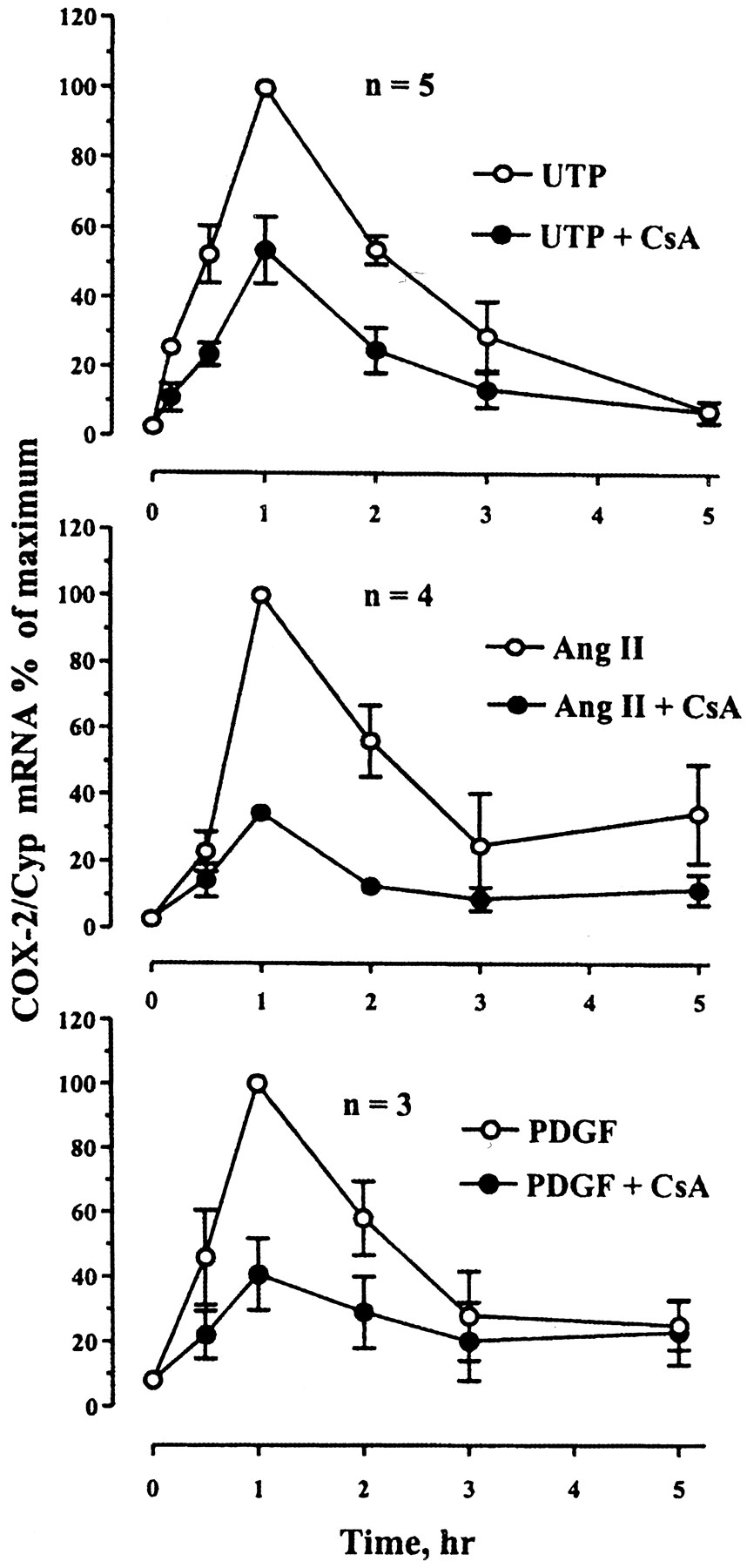
Gαq-coupled receptors and growth factors have been reported to induce NFAT-mediated transcription in smooth muscle cells, as assessed using an NFAT specific reporter (Boss et al., 1998a). We measured COX-2 mRNA responses in the absence or presence of 1 μM CsA during stimulation with the P2Y receptor agonist UTP (100 μM), the AT1 receptor agonist angiotensin II (100 nM), or with PDGF-BB (50 ng/ml) to stimulate a receptor tyrosine kinase. As shown in Fig. 2, UTP, angiotensin II, and PDGF-BB each rapidly increases COX-2 mRNA expression with peak responses at 1 h. As seen in Table1, UTP and angiotensin II tend to induce COX-2 mRNA to similar levels. CsA inhibits the responses to both Gαq-coupled receptor agonists but is slightly more efficacious at inhibiting the effect of angiotensin II (66 ± 2% inhibition) compared with UTP (46 ± 9% inhibition). COX-2 mRNA induction by PDGF-BB is less robust than is the response to Gαq agonists, but it is also inhibited by CsA (59 ± 11% inhibition).

CsA inhibits COX-2 mRNA induction by phospholipase C-coupled receptors. The cells were stimulated with a bolus of 100 μM UTP, or 100 nM angiotensin II (Ang II), or 50 ng/ml PDGF-BB, in the absence or presence of 1 μM CsA, before collecting RNA at the indicated times. Each point represents the mean ± S.E. of three to five independent experiments, expressed as maximal response to each agonist within each experiment.
Peak COX-2 mRNA responses expressed as fold induction over basal and inhibition by CsA
COX-2 mRNA levels were also measured after stimulation with either forskolin (10 μM) to activate adenylyl cyclase or with IL-1β (10 ng/ml) as a reference inflammatory cytokine, in the absence or presence of CsA. The maximal induction by forskolin occurs at 1 h, with recovery to basal expression levels within 3 h (Fig.3). The response to IL-1β is more gradual then any other agonist tested, with the highest level of induction occurring at the latest time-point assayed (Fig. 3). CsA has no effect on COX-2 mRNA induction by these two agonists, and instead selectively inhibits responses to those that stimulate phospholipase C activity. Notably, the maximal induction of COX-2 mRNA by Ca2+-mobilizing agonists is greater than effects caused by forskolin or IL-1β (Table 1).

CsA does not inhibit COX-2 mRNA induction by IL-1β and forskolin. Cells were stimulated with a bolus of either 10 μM forskolin or 10 ng/ml IL-1β in the absence or presence of 1 μM CsA. RNA was collected at the indicated times thereafter. Each point is the mean ± S.E. from three independent experiments, expressed as percentage of the maximal response to each agonist.
To gain insight into whether CsA inhibits NFAT-mediated transcription selectively in this preparation, we examined luciferase activity in four different cell lines created to report the function of the activator protein-1 (AP-1; e.g., c-fos, c-jun), SRE, NFκB, and NFAT using retroviral luciferase reporters. As expected, CsA treatment significantly reduces UTP-, angiotensin II-, and PDGF-BB-stimulated NFAT-mediated luciferase reporter activity, which is not stimulated by forskolin or IL-1β treatment (Fig. 4). In contrast, CsA treatment has no effect on the differential ability these various agonists display in stimulating SRE, AP1, or NFκB reporter activity (Fig. 4). Thus, from this limited survey, CsA selectively inhibits NFAT-mediated transcription in this preparation.

CsA selectively inhibits NFAT-mediated luciferase expression. VSMC lines stably transgenic for retroviral-based NFAT-, NFκB-, AP1- and SRE-specific luciferase transcriptional reporters were treated with either 100 μM UTP, 100 nM angiotensin II (Ang II), 50 ng/ml PDGF-BB, 10 μM forskolin, or 10 ng/ml IL-1β in the absence or presence of 1 μM CsA. Each bar represents the mean ± S.E. from three independent experiments, each with duplicate determinations. The data are normalized to the maximal response to UTP within each cell line, except for NFκB, for which responses were standardized to the effects of IL-1β. B, basal; U, UTP; A, AngII; P, PDGF; F, forskolin; I, IL-1β; ■, −CsA; ▪, +CsA. ∗, statistically significant difference paired ttest (P < .05).
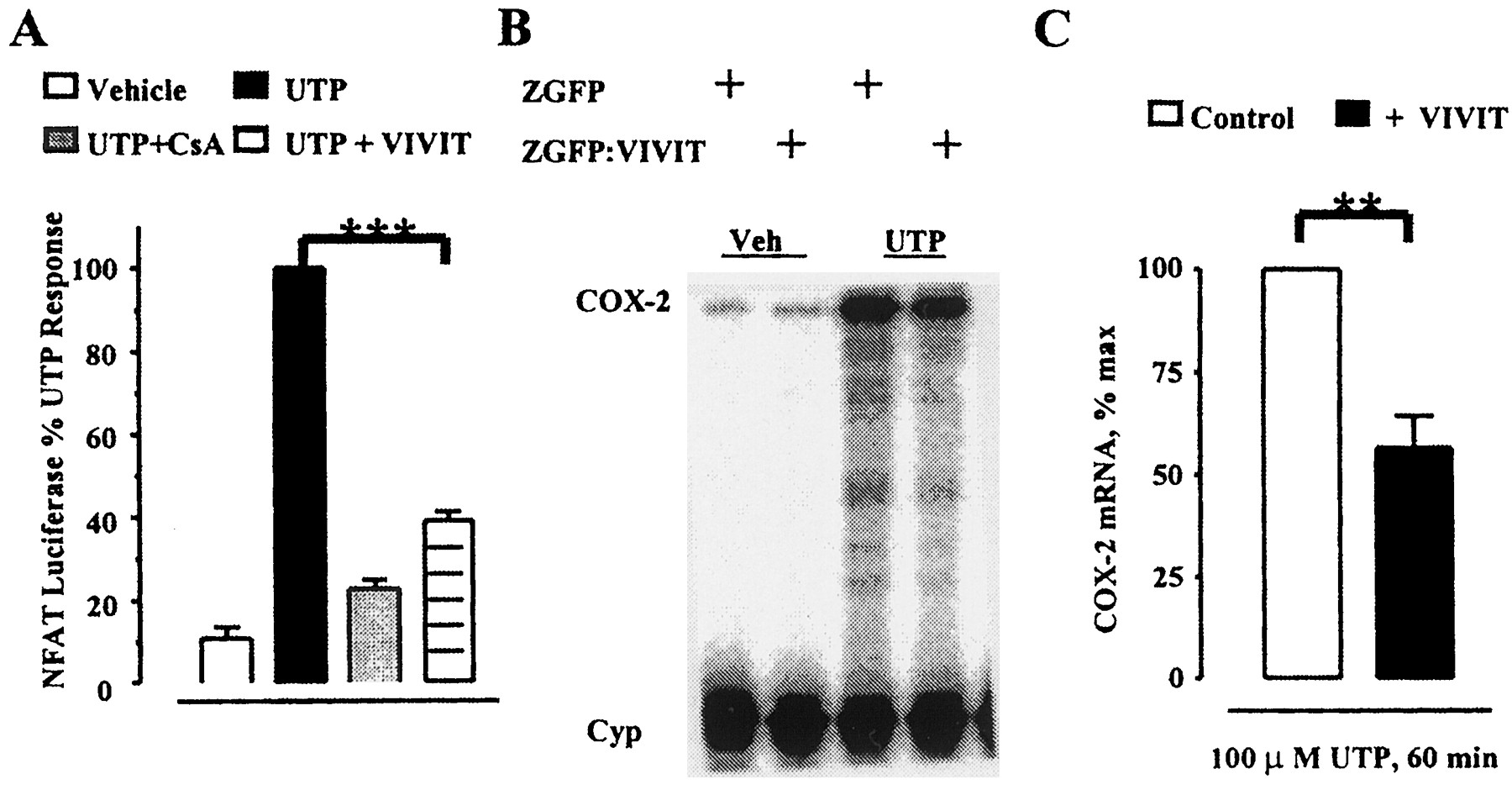
Recently, a specific peptide inhibitor of NFAT, termed VIVIT, has been described (Aramburu et al., 1999). This peptide mimics the calcineurin binding site within NFAT, thereby inhibiting NFAT dephosphorylation and nuclear translocation. Unlike CsA, this occurs without inhibiting calcineurin catalytic activity (Aramburu et al., 1999). VSMC were infected with a retroviral vector containing the VIVIT-coding sequence fused to the carboxyl terminus of a fusion protein made between a zeocin resistance marker and eGFP (Zeo:eGFP). For control, cells expressing Zeo:eGFP were prepared and examined in parallel. Fluorescence-activated cell-sorting analysis of the eGFP signals established that >90% of the cells expressed the recombinant proteins (data not shown). Figure 5A shows that Zeo:eGFP:VIVIT expression reduces significantly (P < .001) UTP-stimulated NFAT-mediated luciferase transcription. This inhibition (60 ± 2% inhibition) was slightly less effective than that caused by CsA (80 ± 2% inhibition). Because the affinity of the VIVIT peptide for calcineurin is > 1 μM (Aramburu et al., 1999), the lower efficacy of VIVIT relative to CsA in our preparation is probably caused by relatively modest expression of the inhibitor from the viral promoter. Nevertheless, expression of Zeo:eGFP:VIVIT also reduces significantly (P < .01) UTP-stimulated COX-2 mRNA expression (44 ± 7%) compared with the level induced in control cells (Fig. 5C).

Inhibition of UTP-stimulated NFAT-mediated transcription and COX-2 mRNA induction by a trans-dominant NFAT inhibitor. VSMC carrying a NFAT-specific luciferase transgene were infected with either the trans-dominant NFAT inhibitor Zeo:eGFP: VIVIT or Zeo:eGFP as a control. A, NFAT-specific luciferase reporter activity after 100 μM UTP stimulation for 4 h in the absence or presence of 1 μM CsA. Data (mean ± S.E.) are from five independent experiments expressed as the percentage of UTP-stimulated, NFAT-specific luciferase expression in Zeo:eGFP control cells within each experiment. ∗∗∗, statistically significant difference, paired t test (P < .001). B, representative PhosphorImage showing basal and 1-h UTP-stimulated COX-2 mRNA levels in cells expressing Zeo:eGFP:VIVIT or Zeo:eGFP as control. C, quantified data (mean ± S.E.) from four independent experiments on two independent cells preps, expressed as the percentage of maximal UTP induction from Zeo:eGFP cells in each experiment. ∗∗, statistically significant difference, paired ttest (P < .01) in VIVIT expressing cells compared with control cells.
Nuclear run-on assays were performed to directly measure the effect of CsA on UTP- and angiotensin II-stimulated COX-2 gene transcription. Forty-five-minute treatments with either UTP or angiotensin II induce COX-2 gene transcription 2- and 4-fold, respectively (Fig.6), which are inhibited significantly in cells treated with CsA (46 ± 10% inhibition for UTP and 38 ± 3% inhibition for angiotensin II). To further examine the effects of CsA on COX-2 gene transcription a 2.7-kb promoter region from the rat COX-2 gene (Sirois et al., 1993) was cloned upstream of a luciferase cDNA in a vector that also expresses CAT constitutively from a second promoter. In cells infected with a promoterless retrovirus, luciferase activity is not induced by any agonist (Fig.7). Treatment of VSMC infected with the full-length COX-2 promoter/luciferase reporter construct using either UTP, angiotensin II, or PDGF, enhances luciferase expression, which is inhibited significantly by CsA. In contrast, forskolin and IL1-β do not stimulate COX-2 promoter driven luciferase activity significantly over basal.

CsA inhibits both UTP- and angiotensin II-stimulated COX-2 gene transcription. After 45-min stimulation with either vehicle, 100 μM UTP, or 100 nM angiotensin II (Ang II) in the absence or presence of 1 μM CsA, nuclear run-on hybridization signals on slot-blots were quantified by PhosphorImaging. Each bar represents transcription over basal levels of activity assessed in vehicle-treated samples, expressed as fold induction ± S.E. COX-2 transcription signals were normalized with those of GAPDH. Each point is the mean ± S.E. from three independent experiments for each agonist. ■, −CsA; ▪, +CsA. ∗, significantly different one-tailed pairedt test (P < .05).

CsA inhibits COX-2 promoter activities. VSMC were infected with retroviral vectors containing either the full-length COX-2 promoter (pAR2) or a promoterless (pCATCUL) vector driving luciferase. A 4-h agonist stimulation with either 100 μM UTP, 100 nM angiotensin II (Ang II), 50 ng/ml PDGF-BB, 10 μM forskolin, or 10 ng/ml IL-1β in the absence or presence of 1 μM CsA was performed. Each bar represents the mean ± S.E. of duplicate determinations from four independent experiments. ∗, statistically significant difference paired t test (P < .05). ∗∗, statistically significant difference paired t test (P < .01).
A region in the distal COX-2 mRNA 3′-untranslated region has been identified that stabilizes COX-2 mRNA decay in response to P2Y receptor stimulation (Xu et al., 2000). To determine whether CsA inhibits COX-2 mRNA expression by interfering with this regulated stabilization mechanism, levels of a tetracycline-suppressible chimeric mRNA containing the COX-2 3′-untranslated region stabilization element fused downstream of a luciferase open-reading frame were measured after 1-h agonist treatments. Chimeric mRNA levels are increased over basal levels by UTP, angiotensin II, PDGF, and IL-1β, most likely because of mRNA stabilization, but CsA treatment does not block this effect (Fig. 8). This finding argues, along with previous data, that CsA inhibits COX-2 mRNA induction predominantly by a transcriptional rather then a post-transcriptional mechanism.

CsA does not inhibit mRNA stabilization by the COX-2 3′-untranslated region. Experiments were performed in VSMC expressing a tetracycline-suppressible luciferase chimera transgene, which has the most distal 263 bases of the COX-2 mRNA 3′-untranslated region. Cells were stimulated for 1 h with either vehicle, 100 μM UTP, 100 nM angiotensin II (Ang II), 50 ng/ml PDGF-BB, 10 μM forskolin, or 10 ng/ml IL-1β in the absence or presence of 1 μM CsA before RNA isolation. A, representative PhosphorImage of an RPA showing chimera luciferase (LUC) and cyclophilin (Cyp) mRNA levels. B, quantification of normalized LUC/Cyp mRNA levels where each bar represents the mean ± S.E. of the fold increase in LUC mRNA over vehicle treatment from three independent experiments.
Discussion
Immunosuppression with CsA is necessary after organ transplantation and useful in treating some autoimmune disorders, but the therapy is complicated in most subjects by rapidly developing cardiovascular side-effects, including renal toxicity and hypertension (Sander et al., 1996). Although these are reversible on discontinuation of CsA-therapy, this is rarely an option and their consequences remain a major barrier to long-term survival after organ transplantation (Faulds et al., 1993). One implication of this CsA cardiovascular toxicity is it represents a manifestation of interference with essential homeostatic processes, such that understanding the mechanism of action may illuminate important and fundamental cardiovascular control mechanisms. Long-term treatment with nonsteroidal anti-inflammatory agents that inhibit COX enzymatic activity is also associated with progressive renal dysfunction (Sturrock et al., 1994). Inhibition of NFAT-mediated transcription by CsA is well established, and evidence shows that expression of NFAT isoforms is not restricted to the immune system (Hoey et al., 1995; Rao et al., 1997). Therefore, a plausible mechanism for some aspects of CsA cardiovascular toxicity might include interference with nonlymphoid cell NFAT-mediated gene expression. We speculate that, taken together, overlap might exist between the toxic mechanisms of action of CsA and those of nonsteroidal anti-inflammatory drugs; for this reason, we have begun to examine the influence of CsA on COX-2 gene expression in VSMC and a potential role for NFAT.
Several observations reported in this study clearly show that CsA attenuates COX-2 gene expression, which is consistent with the hypothesis that certain agonists use NFAT to induce COX-2 gene expression in VSMC. First, the marked synergistic COX-2 mRNA induction is caused by ionomycin and PMA, which is strongly inhibited by CsA. This pharmacological pattern is typical of NFAT-regulated genes. The synergy presumably involves CsA-sensitive, Ca2+-directed nuclear import of NFAT and PMA-mediated activation of essential NFAT transcriptional partners and are stimuli that mimic the two arms of primary signaling cascades activated by phospholipase C. Second, CsA selectively inhibits COX-2 mRNA induction caused by mitogens acting through cell surface receptors that stimulate phospholipase C, with no effect on the weaker induction caused by agents that do not cause Ca2+mobilization. Third, the direct evidence from nuclear run-on analysis, which is supported further by promoter/luciferase reporter activity studies, shows that CsA inhibits mitogen-induced COX-2 gene transcription. Fourth, UTP-induced COX-2 mRNA expression is blocked by an independently acting inhibitor of NFAT-mediated transcription, the mechanism of action of which does not involve inhibition of calcineurin activity. Fifth, CsA inhibits only transcription mediated by an NFAT-selective enhancer and not that of AP-1, SRE, or NFκB enhancers.
Our findings do not imply that NFAT is the sole transcriptional coactivator linking mitogen signaling to increased COX-2 gene transcription. Clearly, UTP and PDGF are capable of stimulating several different classes of transcription factors (Abbott et al., 2000). CsA seems somewhat selective for inhibiting NFAT-mediated transcription, relative to a handful of other coactivators, and inhibits ∼50% of mitogen-induced COX-2 mRNA, such that NFAT likely plays a prominent role in regulating the gene. We cannot exclude the possibility that unknown CsA-sensitive transcription factors account for this regulation. Future studies to identify the CsA-sensitive, mitogen-inducible promoter elements will be necessary to resolve this uncertainty.
A notable discrepancy is that although COX-2 mRNA levels are profoundly induced in this preparation by mitogenic stimuli, the stimuli have lesser effects on direct measurements of COX-2 gene transcription. We have recently investigated the possibility that the full mRNA response to mitogen involves simultaneous activation of transcriptional and post-transcriptional mechanisms. UTP activation of the p42/44 MAP kinase causes stabilization of a chimera mRNA composed of a luciferase coding sequence and the full-length COX-2 mRNA 3′-untranslated region, which has been delimited to a response element in the most distal AU-rich segment of the mRNA (Xu et al., 2000). However, just as transcriptional induction alone cannot completely explain the magnitude of UTP-stimulated COX-2 mRNA expression, neither can this post-transcriptional response, because it also is only on the order of 3-fold. Yet the fact that it occurs forced us to address the possibility that CsA might block it. However, the data shown in Fig. 8indicate that CsA does not inhibit this distal 3′-untranslated region-mediated mRNA stabilization mechanism. This implicates the proven transcriptional mechanism as most likely responsible for the effect of CsA on COX-2 mRNA induction. However, at this time we have not ruled out the possibility that additional regulated stability determinants exist in other regions of the COX-2 mRNA that may be sensitive to CsA.
By showing that CsA affects transcriptional activity, and by establishing potential connections through NFAT, the data provided in this study establishes, to our knowledge for the first time, a mechanistic basis for several previous observations showing that CsA interferes with COX-2 protein expression and/or prostaglandin metabolism. Some studies have suggested CsA-mediated effects reflect post-translational mechanisms (Stahl et al., 1989), whereas others have also described inhibition of stimulus-evoked COX-2 mRNA expression (Martin et al., 1994; Stroebel and Goppelt-Struebe, 1994). Our finding that IL-1β-mediated induction is not inhibited by CsA differs from earlier reports demonstrating that IL-1β-stimulated COX-2 mRNA expression in mesangial cells is attenuated by CsA (Martin et al., 1994). The discrepancies may be caused by cellular differences; however, effects of CsA on IL-1β-induced mesangial cell COX-2 mRNA are most prominent several hours beyond the immediate-early time points measured in the present study. The mesangial cell response may thus reflect secondary compensatory effects; for example, a delayed but increasing contribution of putative IL-1β-regulated autocrine/paracrine factors that signal through CsA-sensitive mechanisms.
Our study shows differences between mitogens and IL-1β in terms of CsA sensitivity, magnitude of expression, and kinetics of induction that indicate the two classes of agonists use very distinct mechanisms to induce the COX-2 mRNA in this preparation. The robust but transient effects of mitogens compared with the slowly activated, longer-duration response to IL-1β may have several explanations. Our data show that mitogens are capable of activating several classes of transcription factors in this preparation on which IL-1β has little effect, which more robustly and selectively induces NFκB-mediated transcription (Abbott et al., 2000). NFκB may function in the nucleus for longer periods, whereas mitogen-stimulated factors may be inactivated quickly. Alternately, it is also possible that IL-1β and mitogen receptors desensitize or their agonists degrade at different rates.
It is likely that the COX-2 gene has capacity for regulation by many different mechanisms that function in cell-type and stimulus-dependent contexts. The extent to which NFAT contributes to responses elicited by phospholipase C-coupled receptors in other cell phenotypes is unclear at present, but CsA has been reported recently also to inhibit COX-2 mRNA induction in T lymphocytes by ionomycin and PMA, which seems to involve NFAT (Iniguez et al., 1999, 2000). The COX-2 mRNA is induced in our preparation by 80-fold and yet transcriptional induction is at least an order of magnitude less than this. Yet the relative magnitude of CsA inhibition of steady-state mRNA (∼50%) approximates its effect on transcriptional induction. This raises the possibility that other mechanisms, perhaps those raised here that function coordinately at the post-transcriptional level, work to magnify responses acting at the level of transcriptional control. In this way, modest direct transcriptional effects of NFAT and other transcriptional coactivators might by amplified substantially.
Acknowledgments
We thank R. N. Dubois and J. S. Richards for the generous gift of plasmids containing the COX-2 cDNA and 2.7-kb COX-2 promoter region, respectively.
Footnotes
- Received March 3, 2000.
- Accepted June 21, 2000.
-
Send reprint requests to: T. J. Murphy, Ph.D., Department of Pharmacology, Emory University School of Medicine, 5031 O.W. Rollins Research Bldg., Atlanta, GA 30322. E-mail:medtjm{at}bimcore.emory.edu
-
Supported by Grants HL52810 and HL56107 from the National Heart Lung and Blood Institute. T.J.M. is an established investigator of the American Heart Association. A.M.R. is supported by a predoctoral training grant (GM08602-03).
Abbreviations
- COX-2
- cyclooxygenase-2
- CsA
- cyclosporin A
- NFAT
- nuclear factor of activated T-cells
- VSMC
- vascular smooth muscle cells
- PDGF-BB
- platelet-derived growth factor-BB
- RPA
- RNase protection assay
- AP1
- activator protein 1
- SRE
- serum response element
- NFκB
- nuclear factor-κB
- kb
- kilobase pairs
- PMA
- phorbol 12-myristate 13-acetate
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}