


Abstract
Resistance to multiple, unrelated cancer chemotherapeutic drugs can be mediated by P-glycoprotein, the MDR1 gene product. Numerous substances, including chemotherapeutic drugs, heavy metals, growth factors, activated oncogenes, or changes in temperature increase MDR1 gene expression. Because several of these factors regulate cellular function through the activation of phospholipase C (PLC), we postulated that PLC-mediated signaling could be central to regulating the expression of MDR1. Transfection of NIH 3T3 cells with a pMJ30-PLC-γ1 expression vector increased the activity of the MDR1 promoter by 2- to 10-fold. PLC-mediated activation required a region between −106 and −99 of the MDR1 promoter. Treatment of cotransfected cells with platelet-derived growth factor further enhanced the activity of the MDR1 promoter. The stimulatory effect of PLC on the MDR1 promoter was increased by cotransfection with constitutively active v-raf and was blocked by the dominant-negative mutant, c-Raf-C4. The activity of mitogen-activated protein kinase (MAPK) was also increased in PLC-γ1-transfected cells. Furthermore, PD-98059 and U0126, two MAPK inhibitors, blocked PLC-γ1-induced expression of MDR1. The results of Northern blot analysis showed that activation of PLC by heat shock and growth factors increased expression of endogenous MDR1 mRNA in human renal carcinoma cells. These effects were blocked by inhibitors of the PLC-MAPK pathway. In summary, our results indicate for the first time that activation of PLC by a variety of cellular stimuli can regulate the expression of MDR1 and that the transcriptional modulation of MDR1 expression by PLC is mediated by the Raf-MAPK pathway.
Resistance to multiple chemotherapeutic drugs occurs in cancer and infectious disease. One form of multidrug resistance common to both is characterized by enhanced drug efflux mediated by P-glycoprotein (P-gp), a 150- to 180-kDa plasma membrane phosphoglycoprotein that functions as an energy-dependent drug transporter with broad substrate specificity (Gottesman and Pastan, 1993). Several important characteristics of P-gp include homology with bacterial transport proteins (Chen et al., 1986), ATP binding (Cornwell et al., 1987b), and hydrolysis (Hamada and Tsuruo, 1988), drug binding (Cornwell et al., 1986) and efflux (Kamimoto et al., 1989), and the ability to bind compounds that reverse MDR, such as verapamil and cyclosporin A (Cornwell et al., 1987a; Foxwell et al., 1989).
Regulation of P-gp expression has been studied extensively in normal and malignant tissues, and many factors are now known to increase the expression of MDR1. For example, Chaudhary et al. showed that activation of PKC by the diacylglycerol analog, 12-O-tetradecanoylphorbol-13-acetate, increased MDR1 mRNA and P-gp expression (Chaudhary and Roninson, 1992). Heat shock (Chin et al., 1990), UV irradiation (Hu et al., 2000), certain chemotherapeutic drugs (Chaudhary and Roninson, 1993), heavy metals (Chin et al., 1990), hormones (Altuvia et al., 1993), oncogenes and tumor suppressor genes (Chin et al., 1992), and growth factors such as EGF (Rohlff and Glazer, 1995), were also shown to increase the expression of MDR1. However, it is not known how these diverse agents lead to MDR1 activation.
Several of the stimuli known to increase MDR1 expression utilize signal transduction pathways initiated through PLC. Activation of PLC is one of the most common transmembrane signal transduction mechanisms used by a wide array of extracellular ligands (Rana and Hokin, 1990). PLC produces two second messengers by catalyzing the conversion of phosphatidylinositol 4,5-bisphosphate to Ins(1,4,5)P3 and diacylglycerol. Ins(1,4,5)P3 stimulates the release of Ca2+ from intracellular stores and diacylglycerol activates PKC. PLC has been implicated in regulation of many cellular activities, including cell growth and transformation (Nebigil, 1997). In addition, the activity of PLC is increased in many human tumors (Noh et al., 1994). However, little is known about the role of PLC in drug resistance. Therefore, in this study, we postulated that PLC activation is linked to downstream signaling events that can regulateMDR1 expression in response to diverse stimuli. Our results implicate this pathway in the regulation of MDR1 expression and suggest the possibility of targeting signaling components as a mean to inhibit the expression of this drug resistance gene.
Materials and Methods
Cell Cultures, Expression Vectors, and Reagents.
PLC-γ/3T3 cells were provided by Dr. Mark R. Smith (National Institutes of Health, Bethesda, MD). PLC-γ/3T3 and NIH 3T3 cell lines were maintained in Dulbecco's modified Eagle medium containing 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2/95% air. The human renal carcinoma line HTB-46 was purchased from American Type Culture Collection (Manassas, VA) and grown in McCoy's 5A modified medium containing 10% fetal bovine serum under conditions identical to those described above. Cells were checked routinely and found to be free of contamination by mycoplasma or fungi.
The human MDR1 promoter construct, MDRCAT, and deletion constructs, were kindly supplied by Dr. K. V. Chin (The Cancer Institute of New Jersey, New Brunswick, NJ). MDRCAT contains 1.8 kilobase pairs of genomic sequences upstream from the initiation codon (ATG) of the human MDR1 gene cloned directly in front of the CAT gene in pSV00CAT (Chin et al., 1992). The pMJ30-PLC-γ1 and its control vector pMJ30 were provided by Dr. S.-G. Rhee (National Heart, Lung, and Blood Institute, Bethesda, MD). v-Raf and c-Raf-C4 constructs were gifts from Dr. Marilyn M. Cornwell (Fred Hutchinson Cancer Research Center, Seattle, WA).
[14C]Chloramphenicol was purchased from Amersham Pharmacia Biotech, Inc. (Piscataway, NJ). Luciferase Assay System was purchased from Promega Corp. (Madison, WI). Additional reagents were purchased from the following sources: PDGF and EGF (Invitrogen, Carlsbad, CA); PD98059 and U73122 (Biomol, Plymouth Meeting, PA); U0126 (Promega Corp., Madison, WI); doxorubicin (Sigma Chemical Co., St. Louis, MO); LipofectAMINE and TRIzol Reagent (Invitrogen). Antibodies were obtained from the following sources: monoclonal anti-MAPK and anti-β-actin antibodies, Sigma (Saint Louis, MO); monoclonal anti-phospho-MAPK (New England BioLabs, Beverly, MA).
PLC Activity.
PLC activity was measured by assaying the generation of Ins(1,4,5)P3 from phosphatidylinositol 4,5-bisphosphate. Briefly, Ins(1,4,5)P3 was extracted from cell lysates using perchloric acid. Acid extracts were neutralized to pH 7.5 by titration with ice-cold KOH (10 M). Ins(1,4,5)P3was measured by competitive binding to a bovine adrenal Ins(1,4,5)P3-binding protein using the Amersham assay kit (TRK1000; Amersham Pharmacia Biotech, Inc., Piscataway, NJ).
Activation of PKC.
Activation of PKC was determined by measuring the activity and subcellular redistribution of the enzyme. Briefly, 1 × 107 cells were collected and washed twice with PBS/1.0 M sucrose and pelleted in a Microfuge. The cellular pellets were resuspended within 20 s in 50 μl of double-distilled water by passage through a narrow-gauge needle and immediately reconstituted with 1.0 ml of ice-cold buffer A (20 mM Tris-HCl, pH 7.5, 0.5 mM EGTA, 2 mM EDTA, and 2 mM phenylmethylsulfonyl fluoride). Lysates were centrifuged at 100,000g to obtain the soluble and particulate fractions. Particulate fractions were solubilized with buffer A containing 1% Nonidet P40, and both cytosolic and solubilized membrane fractions were separated by DEAE-cellulose chromatography. Aliquots (50 μl) of the DEAE-purified material were assayed for PKC activity in the presence of 10 mM Mg acetate, 0.75 mM CaCl2, 100 μM [γ-32P]ATP, and 25 μg of histone H1. Conditions were adjusted to ensure linearity of the reaction with respect to the time of incubation and concentrations of samples.
Transient Transfections and Reporter-Gene Assays.
NIH 3T3 cells (3 × 105) were plated in 60-mm cell culture dishes then cotransfected with MDRCAT (1 μg) and pMJ30-PLC-γ1 (4 μg) using LipofectAMINE. Forty-eight hours after transfection, CAT activity was measured using equivalent amounts of total protein and quantified by scintillation counting of the acetylated 14C-labeled chloramphenicol. PLC-γ/3T3 cells were used for cotransfection with MDRCAT, v-Raf, or c-Raf-C4. pGL3, a luciferase expression vector, was used as an internal control for transfection efficiency by normalizing CAT activity to the luciferase activity.
Northern Blot Analysis.
RNA was prepared from treated cells using TRIzol Reagent according to the manufacturer's protocol. Twenty micrograms of total RNA from each sample were electrophoresed, blotted onto nitrocellulose, and probed for MDR1 using cDNA 5A probe. An α-32P-labeled β-actin probe was used to determine loading of RNA.
Western Blot Analysis.
Cells were lysed on ice for 30 min with radioimmunoprecipitation assay buffer (10 mM sodium phosphate, pH 7.2, 1% Nonidet P40, 1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl, and 2 mM EDTA) supplemented with fresh 1% aprotinin, 1 mM phenylmethylsulfonyl fluoride, and 50 μg/ml leupeptin and centrifuged at 14,000g at 4°C for 10 min. Proteins were resolved by 7 to 10% SDS-PAGE and transferred to nitrocellulose membranes. The blots were incubated in blocking solution consisting of 5% milk in Tris-buffered saline/Tween 20 (0.1% Tween 20) for 1 h at 25°C, then immunoblotted with monoclonal anti-MAPK, anti-phospho-MAPK, or anti-β-actin antibodies. Detection by enzyme-linked chemiluminescence was performed according to the manufacturer's protocol (Amersham Pharmacia Biotech, Inc., Piscataway, NJ). Experiments were done under conditions of linearity with respect to protein contents.
Results
Effects of PLC-γ1 Transfection on the Activity of PLC, PKC, and the MDR1 Promoter.
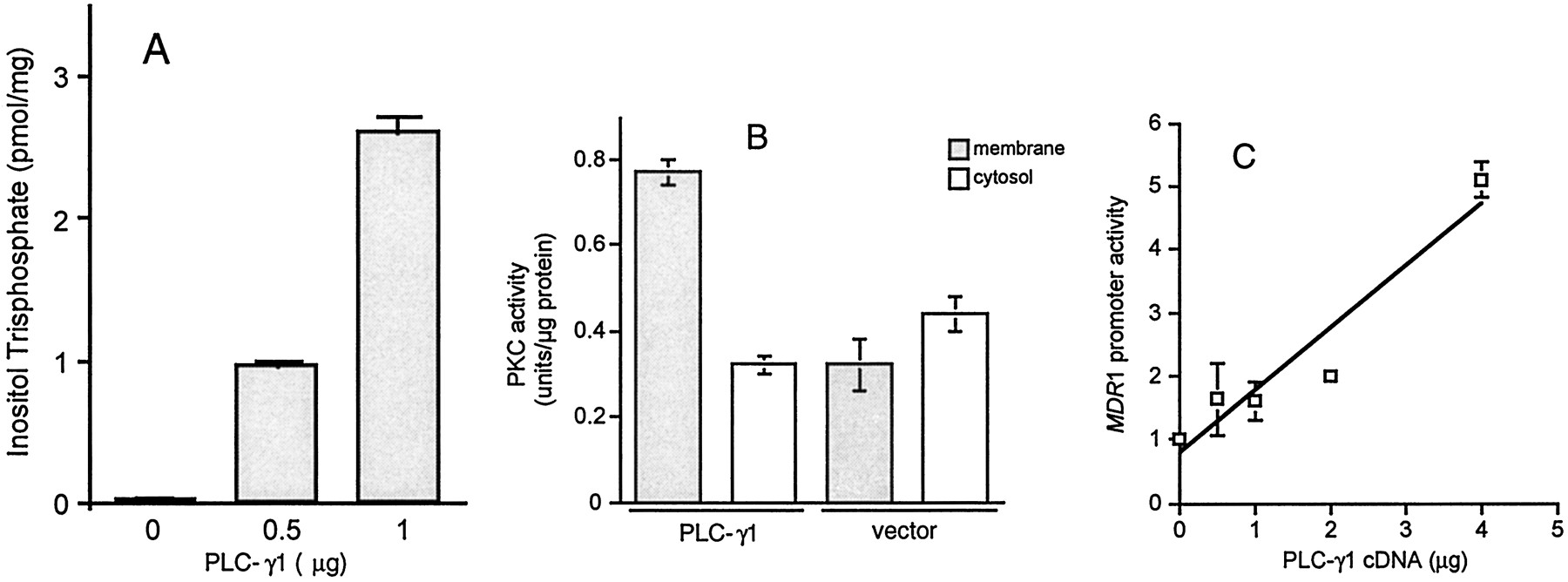
To create a model in which to study the effect of PLC activation on MDR1 expression, we transfected NIH 3T3 cells with a PLC-γ1 expression vector. Figures1, A and B, demonstrate that PLC-γ1 transfection activated PLC-mediated signaling as measured by increased production of Ins(1,4,5)P3 (Fig. 1A) and redistribution of PKC activity from the cytosol to the particulate fraction (Fig. 1B). To determine the effect of PLC on the expression ofMDR1, we next cotransfected NIH 3T3 cells with MDRCAT and PLC-γ1 expression vectors then measured the activity of theMDR1 promoter. Figure 1C demonstrates that PLC-γ1 increased MDR1 promoter activity up to 5-fold in a dose-dependent manner compared with empty vector controls, which had no effect on the expression of MDRCAT (data not shown).

Effect of PLC-γ1 transfection on the activity of PLC, PKC, and MDR1 Promoter. A and B, NIH 3T3 cells were transfected with pMJ30-PLC-γ1 expression vector. PLC activity was measured by assaying the generation of Ins(1,4,5)P3 using Amersham's assay system. Activation of PKC was determined by examining the subcellular redistribution of PKC activity. C, NIH 3T3 cells were plated in 100-mm cell culture dishes, then cotransfected with 1 μg of MDRCAT and various amounts of pMJ30-PLC-γ1 using LipofectAMINE. Forty hours after transfection, the CAT activity was measured with equivalent amounts of protein extracts and quantified by scintillation counting of the percentage of acetylated 14C-labeled chloramphenicol. Each bar or point represents the mean ± S.E. from one of three similar experiments.
To define the region of the MDR1 promoter responsible for the effect of PLC on MDR1 expression, we studied the effect of PLC-γ1 transfection on a series of cotransfected MDR1 promoter deletion constructs. As shown in Fig.2, deletion of sequences from −1073 to −106 or deletion of intron 1 and exon 2 had no effect on PLC-induced activity of MDR1 promoter as well as basal activity. Deletion of GC-rich sequences from −106 to −99 did not affect basal activity of the promoter but abolished the activation by PLC (Fig. 2). These results suggest that the GC-rich region between −106 and −99 was required for PLC-induced activation of the MDR1 promoter. Deletion of regulatory sequences between −99 and −11 resulted in loss of basal activity (greater than 90% decrease; Fig.2), which is consistent with previously reported results (Cornwell and Smith, 1993b; Goldsmith et al., 1993).

Effect of PLC-γ1 transfection on the activity ofMDR1 promoter deletion constructs. NIH 3T3 cells were plated in 100-mm cell culture dishes, then cotransfected with 1 μg of the MDR1 promoter deletion constructs and 4 μg of pMJ30-PLC-γ1 using LipofectAMINE. Forty hours after transfection, the CAT activity was measured with equivalent amounts of protein extracts and quantified by scintillation counting of the percentage of acetylated 14C-labeled chloramphenicol. Each point represents the mean ± S.E. from one of three similar experiments.
Effect of Raf on PLC-Induced Activation of the MDR1 Promoter.
PKC can phosphorylate and activate Raf kinase (Kolch et al., 1993). Therefore, we determined whether the activation of theMDR1 promoter by PLC involved Raf kinase activity. We undertook transient cotransfection experiments using a PLC-γ1 stable transfectant, PLC-γ1/3T3 and constructs of v-Raf or c-Raf-C4.MDR1 promoter activity was 2- to 5-fold greater in PLC-γ1/3T3 cells than in control cells (Fig.3). Cotransfection of PLC-γ1/3T3 cells with v-Raf, a constitutively activated Raf-1, further increasedMDR1 promoter activity up to 15-fold over that of the control cell line and 3-fold over the PLC-γ1 transfectants (Fig. 3A). Raf-C4, a dominant-negative mutant of Raf-1, markedly reduced the activation of the MDR1 promoter induced by PLC (60 to 70% inhibition) (Fig. 3B).

Effect of wild-type and dominant-negative Raf on PLC-induced activation of the MDR1 promoter. NIH 3T3/PLC-γ1 cells were plated in 100-mm cell culture dishes then cotransfected with MDRCAT (1 μg) and v-Raf (A) or Raf-C4 (B) using LipofectAMINE. Forty hours after transfection, CAT activity was measured using equivalent amounts of protein extracts and quantified by scintillation counting of the percentage of acetylated14C-labeled chloramphenicol. Each bar represents the mean ± S.E. from one of three similar experiments.
Involvement of MAPK in PLC Activation of MDR1.
MAPK is downstream of Raf-1 and is responsible for transcriptional regulation of several genes (Treisman, 1996). We found that the constitutively activated Raf increased the activation of MAPK by 3-fold, and dominant-negative mutant of Raf inhibited the activation of this enzyme to a similar extent as the inhibition of MDR1 promoter activity (60 to 70% inhibition) (data not shown). To assess the role of MAPK in PLC-induced activation of MDR1 expression, we measured the phosphorylation of p42 and p44 MAPK in cells transfected with PLC-γ1. As shown in Fig.4A, the phosphorylation of MAPK, as measured by the ratio of the phosphorylated p42 band (Fig. 4, top) normalized to the total p42 band (Fig. 4, bottom), was increased by 2.3-fold in PLC-γ1-transfected cells compared with cells transfected with empty vector, despite a slight decrease in MAPK protein as measured by Western blot of the total enzyme (Fig. 4A, bottom). These results suggested that increased PLC activity resulted in activation of MAPK.

Effect of PLC, doxorubicin, and MAPK inhibition on the activation of MAPK. NIH 3T3 cells were transfected with the PLC-γ1 expression vector (A) or treated with Dox (100 nM), U0126 (50 μM), or the combination for 48 h (B), then analyzed for phosphorylation of MAPK by Western blot using an anti-phospho-p44/42 MAPK antibody. Results are representative of three similar experiments.
Doxorubicin is a chemotherapeutic agent that activates PLC (Yang et al., 1995) and induces the expression of MDR1 (Abolhoda et al., 1999). Therefore, we investigated whether doxorubicin also activated the MAPK pathway. Figure 4B demonstrates that treatment of NIH 3T3 cells with doxorubicin-activated MAPK as measured by the increased phosphorylation of p42 (top). U0126, a MEK inhibitor, blocked basal MAPK activity and the effect of doxorubicin on MAPK phosphorylation (Fig. 4B, top). Figure 4B, bottom, demonstrates that these treatments had little effects on total MAPK protein.
Effects of Activators and Inhibitors of PLC and MAPK on the Expression of MDR1.
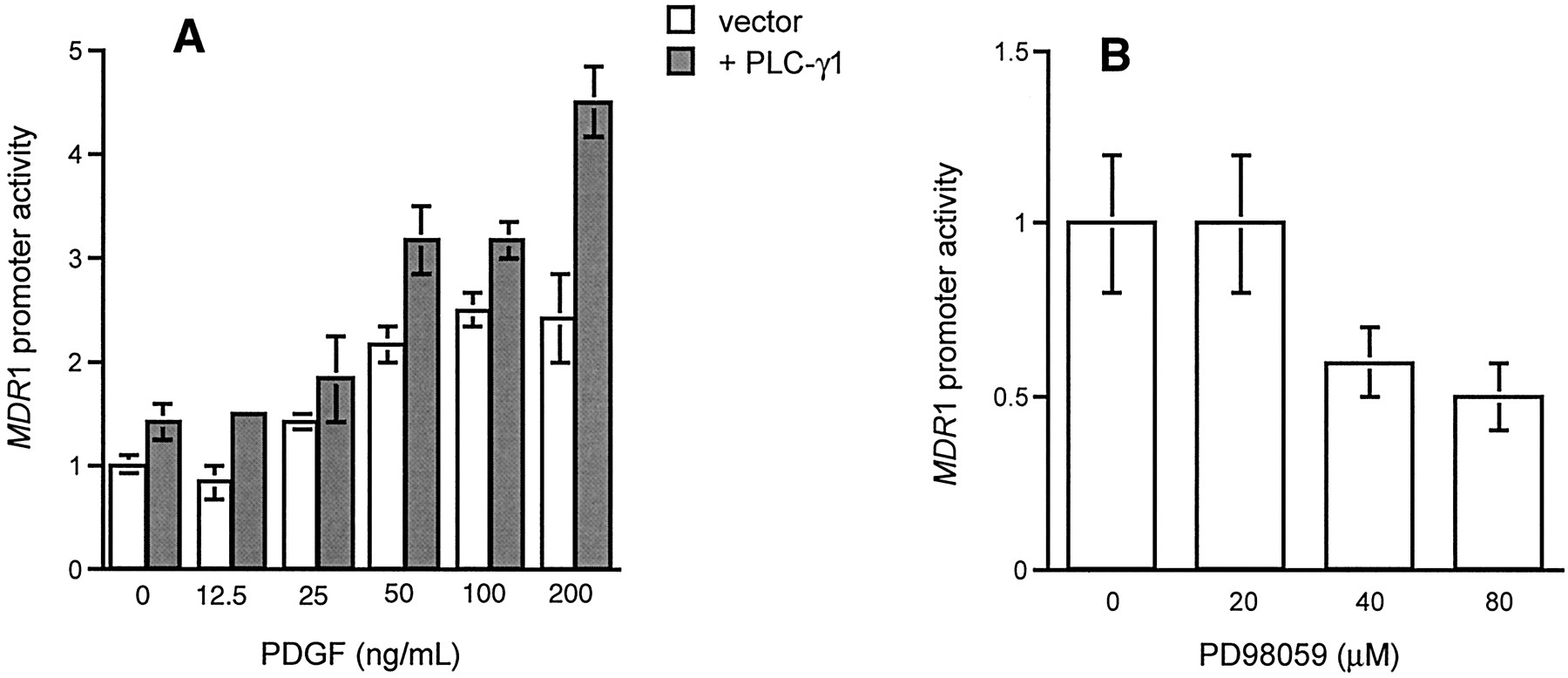
To further delineate the role of PLC-mediated signaling in MDR1 expression, we studied the effects of activators and inhibitors of PLC and MAPK. Figure5A shows that PDGF, an activator of PLC-γ1, increased MDR1 promoter activity in a dose-dependent manner in both control and PLC-γ1 transfected cells. The MEK inhibitor, PD98059, decreased the activity of theMDR1 promoter in NIH 3T3 cells (Fig. 5B). Doxorubicin also increased the activity of the MDR1 promoter (Fig.6), and this effect was blocked by a MEK inhibitor (Fig. 6).

Effect of PDGF and PD98059 on the activity of theMDR1 promoter. A, NIH 3T3 cells were cotransfected with MDRCAT (1 μg) and pMJ30-PLC-γ1 (4 μg), then treated with PDGF (50 ng/ml) for 24 h. B, NIH 3T3 cells were transfected with MDRCAT (1 μg), then treated with different concentrations of PD98059 for 24 h. CAT activity was measured with equivalent amounts of protein extracts and quantified by scintillation counting of acetylated14C-labeled chloramphenicol. Each point represents the mean ± S.E. from one of three similar experiments.
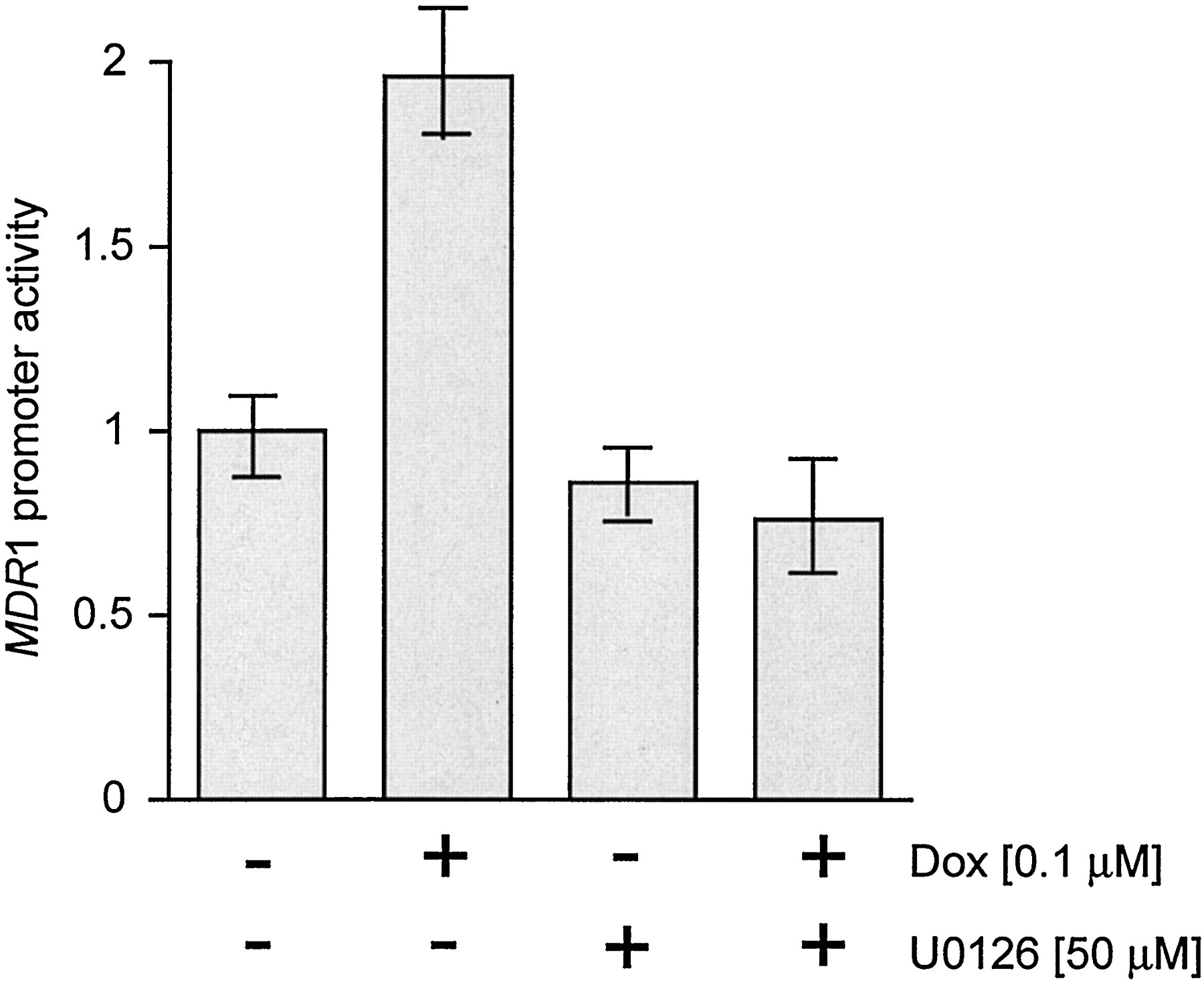
Effect of doxorubicin and U0126 on the activity of the MDR1 promoter. NIH 3T3 cells were transfected with MDRCAT (1 μg), then treated with Dox (100 nM) or U0126 (50 μM), or the combination for 48 h. CAT activity was measured with equivalent amounts of protein extracts and quantified by scintillation counting of acetylated 14C-labeled chloramphenicol. Each bar represents the mean ± S.E. from one of three similar experiments.
To determine the biological relevance of these observations, we next assessed the effects of activators and inhibitors of the PLC signaling pathway on the expression of endogenous MDR1 mRNA using a human renal carcinoma cell line. Figure 7demonstrates that heat shock, PDGF, and EGF increased the expression of MDR1 mRNA by 1.9- to 4.3-fold in HTB-46 cells. Treatment of HTB-46 cells with the PLC inhibitor, U-73122, completely abolished basal and PDGF-stimulated expression of MDR1 mRNA (Fig. 7). The MEK inhibitor, U0126, had little effect on basal MDR1 expression but diminished the heat shock-induced expression of endogenous MDR1 mRNA (Fig. 7).

Effect of activators and inhibitors of PLC-signaling on the expression of the endogenous MDR1 gene. HTB-46 cells were heat shocked for 30 min or treated with PDGF (200 ng/ml) or EGF (500 ng/ml) for 2 h in the presence or absence of U73122 (2.5 μM) or U0126 (5 μM). MDR1 mRNA was analyzed by Northern blot. Results are representative of three similar experiments.
Discussion
Several lines of evidence suggest that PLC-mediated signaling represents one mechanism by which diverse stimuli could activateMDR1 gene expression. For example, heat shock (Chin et al., 1990), growth factors (Rohlff and Glazer, 1995), protein kinase C agonists (Chaudhary and Roninson, 1992), heavy metals (Chin et al., 1990), and certain chemotherapeutic drugs (Chaudhary and Roninson, 1993; Abolhoda et al., 1999) induced MDR1 expression and were also shown to converge on signal transduction pathways downstream of PLC. Yet, the critical role that PLC might play in this regard had not been investigated. In the current studies, we present evidence that supports a central role for PLC in the regulation of MDR1 expression.
Transient transfection of NIH 3T3 cells with a PLC-γ1 expression vector increases intracellular signaling as measured by the production of Ins(1,4,5)P3 (Fig. 1A) and activation of PKC (Fig. 1B). Augmentation of PLC-mediated signaling activates theMDR1 promoter (Fig. 1C). These data are consistent with the role of PLC signaling in the transcriptional regulation of other genes. For example, Schalasta and Doppler (1990) demonstrated that PLC activity is necessary for transcriptional activation of c-fos and inhibitor of PLC inhibited c-fostranscription.
Transcriptional regulation of MDR1 expression has been under intense investigation. Numerous transcription factors including NF-IL6 (Combates et al., 1997), YB-1 (Bargou et al., 1997), p53 (Chin et al., 1992; Goldsmith et al., 1995), NF-Y (Jin and Scotto, 1998), Sp1 (Cornwell and Smith, 1993b; Rohlff and Glazer, 1998), and EGR-1 (McCoy et al., 1995) are known to bind to several canonical sequences within the MDR1 promoter. For example, the sequences involved in stimulation of the MDR1 promoter by wild-type p53 were contained within the region between −39 and +53 (Goldsmith et al., 1995), and the −50 G-box is involved in the activation of basal promoter activity by Sp1 (Goldsmith et al., 1995). In addition, the sequences between −134 and +286 and the Y-box that is located between −82 and −73 are reported to be required for basal promoter activity (Goldsmith et al., 1993; Madden et al., 1993). Using a series of nested deletion constructs, we found that a GC-rich region between −99 and −106 is essential for MDR1 promoter activation after PLC transfection (Fig. 2). This region was shown by Miyazaki et al. (1992)to produce MDR1 activation by heat-shock, an effect mediated by the heat-shock factor (Kim et al., 1997). We previously demonstrated that heat shock activates PLC in MDR cell lines (Yang et al., 1995), and Chin et al. (1990) demonstrated that heat-shock activatesMDR1 gene expression. These results suggest that the effect of heat shock on MDR1 expression is mediated through PLC. In contrast, Cornwell and Smith (1993a) found that the transcriptional response to v-Raf required the MDR1 promoter region between −69 and −41. Because there are multiple responsive elements in theMDR1 promoter, these data suggest that v-Raf might act on more than one site depending on the cellular context and that sequences from −99 to −106 may provide an additional required signal for PLC activation. Deletion of other GC-rich regions of the promoter had no effect on PLC-stimulated activation of MDR1 (Fig. 2). Several sites between the basal promoter and −99 to −106 contain elements that control basal transcription but not stimulated transcription of the MDR1 promoter. These include a consensus Inr sequence extending from −13 to +23 that was shown to be required for accurate initiation of transcription from this promoter; a NF-R1 site between −56 and −45 believed to contain a repressor binding site, and an inverted Y box at −82 to −73. However, these elements are believed to affect basal rather than stimulated transcription. (Scotto and Egan, 1998).
PDGF, an activator of PLC-mediated signaling (Kim et al., 1991), increased MDR1 promoter activity in both control and PLC-transfected NIH 3T3 cells (Fig. 5A). At low PDGF concentrations, we did not see augmentation of this response in the PLC-γ1 transfectant. This may not be unexpected because NIH 3T3 cells express abundant PDGF receptors (data not shown) and at low ligand concentrations PLC is unlikely to be important. At high PDGF concentrations, we find a significant augmentation of MDR1 promoter activity in the PLC-γ1 transfectant, suggesting that during ligand-receptor occupancy PLC becomes important. In addition, the activation of MDR1 is inhibited by PD98059 (Fig. 5B), an inhibitor of MEK activity.
The activation of PLC initiates signals that converge on Raf-dependent pathways. We found that activation of Raf signaling by v-raf, a constitutively active form of the enzyme (Cornwell and Smith, 1993a), increases PLC induced-MDR1 promoter activity (Fig. 3A) and that Raf-C4, a dominant-negative raf mutant (Cornwell and Smith, 1993a), blocks this effect (Fig. 3B). These data support those ofCornwell and Smith (1993a) who found that Raf kinase is involved in the activation of the MDR1 promoter by serum and growth factors. Furthermore, these data indicate that the effect of certain growth factors on MDR1 expression is mediated by PLC, and that Raf is an important downstream component of this PLC-mediated signal transduction pathway.
Activation of Raf stimulates the MEK-MAPK (ERK1/ERK2) cascade, which phosphorylates many transcriptional factors and regulates transcription of a variety of genes (Treisman, 1996). Therefore, we studied the effects of PLC-γ1 on MAPK activity. As shown in Fig. 4, transient transfection of NIH 3T3 cells with PLC-γ1 increases MAPK activity as measured by phosphorylation of p42 and p44 (Fig. 4A). The MEK inhibitor, U0126, inhibits the phosphorylation of p42 and p44 (Fig.4B). Our results demonstrate that MAPK (p42/p44) is an important component of PLC-mediated regulation of MDR1 expression. These data provide a previously missing link between extracellular stimuli and transcription factors that are known to be involved in the regulation of MDR1. Although the roles of those factors inMDR1 expression and their binding sites on theMDR1 promoter have been studied extensively, the upstream regulators of these factors was poorly understood. Our studies demonstrate that activation of PLC provides a route to MDR1 transcription through the PKC-Raf-MAPK and may shed light on how transcription factors are activated by various stimuli. Because PLC is activated by a variety of environmental stresses and theMDR1 gene product is required for cell survival, the activation of MDR1 by PLC may represent a cellular adaptation mechanism. In fact, a role of Ras-MAPK signaling pathway in promoting cell survival was recently reported by Bonni et al. (1999), who demonstrated a dual mechanism comprising post-translational modification and inactivation of a component of the cell death machinery and increased transcription of prosurvival genes. In addition, Osborn et al. previously reported that stress-activated/c-Jun NH2-terminal protein kinase, a member of the MAPK family, was activated in response to several chemotherapeutic agents such as doxorubicin, vinblastine, and VP-16 (Osborn and Chambers, 1996). However, they also found that induction of MDR1 by phorbol ester occurred independently of the MAPK pathway (Osborn et al., 1999). This difference may reflect the complexity of the signal transduction pathways involved in regulation of MDR1 expression and/or the cellular context under investigation.
The effects of cellular manipulations on promoter-reporter constructs do not necessarily reflect an effect on the endogenous gene. However, we found evidence that supports the role of PLC in expression of MDR1 mRNA in a human renal carcinoma cell line (Fig. 7). Heat-shock, a potent inducer of both MDR1 (Chin et al., 1990; Miyazaki et al., 1992) and PLC (Calderwood and Stevenson, 1993), increases MDR1 mRNA in HTB-46 cells; this effect is abrogated by a MEK inhibitor, U0126 (Fig. 7). Similarly, activators of PLC such as PDGF and EGF increase MDR1 mRNA and the PLC inhibitor U73122 blocks this effect (Fig. 7). We also observed that U73122 completely abolished basal expression of MDR1 mRNA (Fig. 7); however, we cannot conclude with certainty that this is fully attributable to inhibition of PLC, because the PLC inhibitor may not be totally selective.
Attempts to overcome MDR with P-gp modulators have had limited success. Several groups have turned to alternative approaches. For example, Futscher et al. found that cotreatment of P-gp(+) cells with P-gp substrates and modulators can prevent the emergence of P-gp(+) cells in the surviving population, suggesting that earlier use of modulators may produce a meaningful therapeutic gain (Futscher et al., 1996). Others have concentrated on the transcriptional regulation of MDR1 and on post-transcriptional modification of P-gp as alternative approaches to overcoming drug resistance (Glazer and Rohlff, 1994;Rohlff and Glazer, 1995). It seems now that signal transduction pathways central to cell growth and differentiation can regulate the expression and post-transcriptional modification of MDR1 and its gene product, P-gp. The current studies help elucidate the signaling mechanisms that regulate the expression of MDR1 gene and identify several targets to potentially prevent emergence of P-gp-expressing cells in tumor populations. In fact, Jin et al. (2000)demonstrated that pharmacologic inhibition of MDR1 expression through targeting transcription might be possible. They demonstrated that ecteinascidin 743, a transcription-targeted chemotherapeutic, could abrogate transcriptional activation of both the endogenous MDR1 gene and MDR1 promoter (Jin et al., 2000). Similarly, one could envision the application of PLC inhibitors to block MDR1 activation in response to chemotherapeutic agents.
In summary, our results suggest that a variety of extracellular factors modulate the expression of MDR1 through a common PLC-mediated signaling pathway. Inhibition of the component(s) of this pathway might represent an approach to preventing P-gp-mediated drug resistance.
Acknowledgments
We thank Mr. Michael Cho for technical assistance. We thank Dr. Sue-Goo Rhee (National Heart, Lung, and Blood Institute, Bethesda, MD) for providing us with pMJ30-PLC-γ1 and its control vector pMJ30, Dr. K.V. Chin (The Cancer Institute of New Jersey, New Brunswick, NJ) for human MDR1 promoter construct, MDRCAT, and deletion constructs, Dr. Marilyn M. Cornwell (Fred Hutchinson Cancer Research Center, Seattle, WA) for v-Raf and c-Raf-C4 constructs, and Dr. Mark R. Smith (National Institutes of Health, Bethesda, MD) for PLC-γ/3T3 cells. We also thank Drs. Arnold Rabson, Celine Gelinas, and Cory Abate-Shen for their critical reading of this manuscript.
Footnotes
-
This work was supported by grants from the US Public Health Service NCI CA72720 and CA66077.
- Abbreviations:
- P-gp
- P-glycoprotein
- MDR
- multidrug resistant or multidrug resistance
- EGF
- epidermal growth factor
- PLC
- phospholipase C
- Ins(1,4,5)P3
- inositol 1,4,5-trisphosphate
- MAPK
- mitogen-activated protein kinase
- PDGF
- platelet-derived growth factor
- MEK
- MAP/ERK kinase (mitogen-activated protein kinase kinase)
- PKC
- protein kinase C
- CAT
- chloramphenicol acetyl transferase
- Received April 9, 2001.
- Accepted June 20, 2001.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}