


Abstract
Nitric oxide (NO) released under inflammatory and infectious conditions has been implicated in the down-regulation of many cytochrome P450 genes, but its mechanism of action remains unknown. We showed that the expression of the CYP2D6 gene is down-regulated at the transcriptional level by NO in HepG2 cells. The NO donor (±)-N-[(E)-4-ethyl-2-[(Z)-hydroxyimino]-5-nitro-3-hexene-1-yl]-3-pyridine carboxamide (NOR4) decreased the expression of CYP2D6 mRNA in a concentration-dependent manner. Using a CYP2D6 promoter-luciferase construct, we found that NOR4 and another NO donor,S-nitrosoglutathione (GSNO), reduced the luciferase activity in a concentration-dependent manner. A guanylate-cyclase inhibitor failed to prevent suppression of CYP2D6 promoter activity by GSNO, indicating that the activity of the CYP2D6 promoter is suppressed via an NO-guanylate cyclase-independent pathway. Deletion analysis of the CYP2D6 promoter revealed that the −80 to +65 region, which contains the nuclear receptor hepatocyte nuclear factor-4 (HNF4) binding site, was responsible for the suppression of CYP2D6 promoter activity by NO. Therefore, we examined NO responsiveness of the HNF4 binding site by electrophoretic mobility-shift assays and site-direct mutagenesis. The DNA-binding activity of HNF4 was directly inhibited by NO donors, GSNO, andS-nitroso-N-acetyl-penicillamine in a concentration-dependent manner. Mutation of the HNF4 binding site in the CYP2D6 promoter partially restored the suppression of the promoter activity by NO donors. These results demonstrated that NO down-regulates CYP2D6 gene expression, at least in part, by directly inhibiting HNF4 binding to the CYP2D6 promoter.
Cytochrome P450 (P450) enzymes are a major class of heme-containing proteins that participate in the oxidative metabolism of endogenous substrates such as steroid hormones and the biotransformation of xenobiotics. Many studies have shown that the decreases of P450 activity and content caused by lipopolysaccharides (LPS) and cytokines under conditions of inflammation and infection contribute to impaired drug metabolism in vivo and in vitro (Sonawane et al., 1982; Renton, 1983; Morgan, 1989). Lipopolysaccharides and cytokines evoke the excessive production of nitric oxide (NO) via the induction of inducible NO synthase (iNOS) in hepatocytes (Hoffman et al., 1992). Recently, it was reported that NO may be involved in impaired drug metabolism (Khatsenko, 1998).
Nitric oxide has pleiotropic functions, such as the regulation of vascular tone, platelet aggregation, neurotransmission, and the cytotoxic action of activated macrophages (Moncada et al., 1991). Intracellular targets for NO are heme or nonheme iron and thiol-containing proteins. Therefore, the binding of NO to heme of such P450s as CYP1A1/2, CYP2B1, and CYP2E1 inactivated these enzymes (Wink et al., 1993; Gergel et al., 1997). In addition, LPS and inflammatory cytokines inhibit the expression of constitutive and inducible P450 mRNAs in vivo and in vitro, and the suppression of P450 mRNA is prevented by the NOS inhibitorN G-monomethyl-l-arginine (Stadler et al., 1994; Donato et al., 1997; Khatsenko and Kikkawa, 1997). Carlson and Billings (1996) also demonstrated that the suppression of CYP1A2 and CYP2B1/2 by cytokines is largely prevented by NOS inhibitors and that exogenous NO down-regulates these P450s. We reported that 1,25-dihydroxyvitamin D3-inducedCYP3A4 gene expression in human colon carcinoma Caco-2 cells is inhibited by NO donors (Hara et al., 2000). These results suggested that NO plays an important role in the regulation of P450, but the mechanism of this effect is unknown. On the other hand, Morgan and colleagues (Sewer and Morgan, 1997; Iber et al., 2000) reported that NO is not required for the suppression of CYP2C11 by interleukin-1β or LPS and that P450 mRNAs and proteins are down-regulated in iNOS knockout mice under endotoxemia (Sewer et al., 1998). The role of NO in down-regulation of P450s under inflammatory conditions is still controversial.
CYP2D6 is expressed in the liver, intestine, and kidney, where it mediates the oxidative metabolism of such clinically important drugs as tricyclic antidepressants (Dahl and Bertilsson, 1993). CYP2D6 is genetically polymorphic, and the prevalence of poor-metabolizers, which are clinically characterized by a marked deficiency of CYP2D6 activity, is 5 to 10% in white populations (Alván et al., 1990). A direct-repeat element with a one-nucleotide spacer located in the proximal promoter region of the CYP2D6 gene plays an important role in modulating CYP2D6 expression, and hepatocyte nuclear factor-4 (HNF4) interacts with this element (Cairns et al., 1996). HNF4 is a member of the nuclear receptor superfamily that is involved in the liver-specific regulation of many genes, such as those encoding apolipoproteins, coagulation factors, and P450s (Sladek, 1994). Although HNF4 was originally identified as an orphan receptor, fatty acyl-CoA thioesters are actually endogenous ligands for HNF4 (Hertz et al., 1998). The HNF4 binding element is conserved in the proximal promoter regions of more than 20 CYP2 genes (Chen et al., 1994; Ibeanu and Goldstein, 1995). Recently, Jover et al. (2001)demonstrated that HNF-4 plays a general role in the regulation of major P450 genes, including CYP3A4, CYP3A5,CYP2A6, CYP2B6, CYP2C9, andCYP2D6, in human hepatocytes using antisense technique. These findings indicate that HNF4 may act as a common regulator of the liver-specific transcription of many P450 genes.
The expression of CYP2D mRNA and protein is down-regulated by the inflammatory cytokines in the rat and mouse liver (Kurokohchi et al., 1992; Trautwein et al., 1992). The change of CYP2D6 expression level under inflammatory conditions is believed to be critical for pharmacotherapy. However, whether NO is involved in the suppression of CYP2D6 mRNA is unknown. The present study was conducted to examine the mechanism by which NO decreases P450 gene expression using CYP2D6 expressed in the human hepatoma cell line HepG2 as an experimental model. We found that NO directly suppresses CYP2D6 expression at the transcriptional level and that the suppression might be regulated by the inhibition of HNF4 DNA-binding activity by NO.
Materials and Methods
Chemicals.
(±)-N-[(E)-4-Ethyl-2-[(Z)-hydroxyimino]-5-nitro-3-hexene-1-yl]-3-pyridine carboxamide (NOR4), S-nitrosoglutathione (GSNO), andS-nitroso-N-acetyl-penicillamine (SNAP) were purchased from Dojin Laboratories (Kumamoto, Japan); 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) was purchased from Wako Pure Chemicals (Osaka, Japan), and 8-bromo-cGMP was purchased from Sigma (St. Louis, MO).
Cell Culture.
HepG2 cells were cultured in DMEM supplemented with 10% fetal calf serum, 0.1 mM nonessential amino acids, 100 units/ml penicillin G, and 0.1 mg/ml streptomycin in a humidified 5% CO2/95% air incubator at 37°C. Twenty-four hours before exposure to test agents, the cells were cultured in serum-free DMEM containing 0.1 mM nonessential amino acids, 100 units/ml penicillin G, and 0.1 mg/ml streptomycin. The medium was replaced with fresh medium, and various concentrations of the NO donors (NOR4 or GSNO) were added. In control studies, NOR4 that was decomposed (decomposed NOR4) by prior incubation in serum-free medium for 48 h at 37°C was used.
RNA Extraction and Reverse-Transcription Polymerase Chain Reaction.
Total RNA was extracted from the cells with TRIzol reagent (Invitrogen, Carlsbad, CA). First-strand cDNA was generated from 4 μg of total RNA. Reverse-transcription reaction proceeded for 60 min at 37°C in 25 μl of a solution containing 50 mM Tris-HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2, 10 mM dithiothreitol, 0.5 mM deoxynucleoside-5′-triphosphate, 0.6 μg of random primer, 0.5 units of RNase inhibitor, and 200 units of Moloney murine leukemia virus reverse transcriptase (Invitrogen). Aliquots of the reverse-transcription reaction mixture (1 μl) were amplified with primers specific for human CYP2D6 (forward primer, 5′-CTAAGGGAACGACACTCATCAC-3′; reverse primer, 5′-CTCACCAGGAAAGCAAAGACAC-3′) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; forward primer, 5′-GAAGGTGAAGGTCGGAGTC-3′; reverse primer, 5′-CAAAGTTGTCATGGATGACC-3′). The 25-μl PCR mixtures contained 25 mM Tris-HCl, pH 8.4, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM deoxynucleoside-5′-triphosphate, and 1.25 units of Taq DNA polymerase (Invitrogen). CYP2D6 and GAPDH were amplified by PCR as follows: initial denaturation for 2 min at 94°C, followed by 33 and 27 cycles, respectively, of 40 s at 94°C, 40 s at 60°C, and 1 min at 72°C. There was a linear correlation between the amounts of PCR products and the template CYP2D6 cDNA under our PCR condition. Aliquots of the PCR mixture were separated onto 3% agarose gels (agarose/NuSieve agarose, 2:1) and stained with ethidium bromide. Densitometric measurements of PCR products were obtained using NIH Image Software (http://rsb.info.nih.gov/nih-image/).
Plasmid Construction.
The CYP2D6 promoter-luciferase chimerae were constructed as follows. The 5′-promoter region of theCYP2D6 gene between −870 and +65 was generated by PCR amplification from human genomic DNA using the upstream primer 5′-CAACGCGTAAGGGCCTTCCGGCTACCAA-3′ including an Mlu I site and downstream primer 5′-GTCTCGAGTCCTCTGGACACACCTGGCA-3′ including anXhoI site. The promoter fragment was subcloned into theMluI/XhoI sites of the pGL3 basic vector (Promega, Madison, WI). This construct was designated as −870/+65 pGL3. Deleted promoter fragments generated from the 5′ restriction sites SacI (−429) and HincII (−80) and a common 3′ XhoI site in the downstream primer were subcloned intoSacI/XhoI and blunt-endedMluI/XhoI sites of the pGL3 basic vector, respectively. These deletion constructs were designated as −429/+65 pGL3 and −80/+65 pGL3, respectively. The mut–870/+65 pGL3 was identical to −870/+65 pGL3 except for the replacement of eight nucleotides in the HNF4 binding site. Site-directed mutagenesis was performed by the PCR overlap-extension method (Ho et al., 1989). The forward and reverse of mutagenic primers were 5′-CTCACAGCAGATTGACAATTCACTCATCAGCTCCC-3′ 5′-GGGAGCTGATGAGTGAATTGTCAATCTGCTGTGAG-3′, respectively (mutated sites are underlined). Nucleotide sequences were checked by dideoxynucleotide sequencing. The sequence of CYP2D6 promoter region (−870/+65) used was identical to that of M33388(GenBank).
Transfection Study.
HepG2 cells were cultured to 40 to 50% confluence in 35-mm dishes and transfected using LipofectAMINE reagent (Invitrogen) according to the manufacturer's directions with luciferase-reporter plasmids (1 μg), pGL3 control vector (SV40 pGL3, 0.3 μg) or pGL3 basic vector (pGL3B, 1 μg) (Promega), and the internal reference pCMV·SPORT-βgal (1 μg; Invitrogen). Six hours later, the mixture was replaced with DMEM containing 10% fetal calf serum, and cells were maintained for 18 h. Four hours before exposure to the NO donors GSNO, NOR4, and SNAP, the cells were cultured in serum-free DMEM. The medium was replaced with fresh medium, and the cells were incubated with various concentrations of GSNO or NOR4 for 20 h, washed with phosphate-buffered saline, and lysed. Then luciferase and β-galactosidase activities in the cell lysates were measured. β-Galactosidase expression was used to normalize the results of transfection efficiency. All values represent the means ± S.E. of at least three independent experiments.
Preparation of Nuclear Extract.
HepG2 cells were incubated with NO donors (NOR4 and GSNO) in serum-free medium for 12 h in 90-mm dishes, washed once with ice-cold phosphate-buffered saline, and scraped into phosphate-buffered saline. The suspension was separated by gentle centrifugation, and the cell pellet was suspended in buffer A (20 mM HEPES, pH 7.8, 15 mM KCl, 2 mM MgCl2, 0.5 mM phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin). The suspension was centrifuged for 30 s at 800g, and the pelleted cells were lysed in buffer A containing 0.2% Nonidet P-40. The lysed cells were centrifuged for 30 s at 9,000g,and the pellets were resuspended in 20 mM HEPES, pH 7.8, 0.4 M KCl, 2 mM MgCl2, 0.5 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, and 10% glycerol; gently shaken for 30 min at 4°C; and then centrifuged for 20 min at 20,000g. The supernatant was stored at −80°C. All procedures were performed at 4°C. Protein concentrations in nuclear extracts were determined using a protein assay (Bio-Rad, Hercules, CA).
Electrophoretic Mobility Shift Assay.
Double-stranded HNF4 (5′-tcgaAGCAGAGGGCAAAGGCCATCAT-3′) and specificity protein 1 (Sp1) (5′-agctCGATCGGGGCGGGGCGAGC-3′) oligonucleotides with overhanging tetranucleotides (5′-tcga-3′ and 5′-agct-3′, respectively) were labeled by the Klenow fill-in reaction in the presence of [α-32P]dCTP. This oligonucleotide corresponded to the sequence between −60 and −39 of the human CYP2D6 promoter, which contains the HNF4 binding site. For the DNA-binding reaction, nuclear extract (6 μg) was incubated with a32P-labeled probe in 20 mM HEPES, pH 7.9, 80 mM KCl, 0.1% Nonidet P-40, 2 μg of poly(dI-dC), and 10% glycerol for 30 min at room temperature. In supershift experiments, 1 μg of anti-HNF4 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) was incubated with nuclear extracts for 30 min at room temperature before the binding reaction. Protein DNA complexes were loaded on a 4% polyacrylamide gel in 0.5× Tris borate-EDTA. The gel was dried and visualized using a bioimaging analyzer.
Western Blotting.
Nuclear proteins (8 μg) were separated onto a 12% SDS-polyacrylamide gel, transferred onto nitrocellulose membranes, and sequentially incubated with anti-HNF4 antibody, biotinylated anti-goat IgG, and streptavidin-biotin-peroxidase complex. Proteins were visualized using the enhanced chemiluminescence detection system (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK).
Statistical Analysis.
Data were analyzed using Student'st test. A P value less than 0.05 was considered statistically significant.
Results
NO Suppresses Constitutive CYP2D6 mRNA Expression in HepG2 Cells.
To determine whether NO affects the expression of CYP2D6 mRNA, HepG2 cells that constitutively express CYP2D6 were incubated with the NO donor NOR4. Figure 1 shows that the expression of CYP2D6 mRNA was inhibited by NOR4 in a concentration-dependent manner. The level of CYP2D6 mRNA expression was decreased to approximately 60% of the control level by 300 μM NOR4. The expression of GAPDH mRNA was not affected by NOR4. We then performed control experiments using decomposed NOR4, which is unable to release NO. Decomposed NOR4 was prepared by incubating NOR4 in cell-free culture medium at 37°C for 48 h because NOR4 spontaneously releases NO with a half-life of 60 min. As expected, decomposed NOR4 showed no effect on the expression of CYP2D6 mRNA (Fig.1). These findings suggested that CYP2D6 mRNA is depressed by NO released from NOR4. Similarly, another NO donor, GSNO (1000 μM), decreased the level of CYP2D6 mRNA expression (Fig. 1). To exclude the possibility of cytotoxic effects of NO donors, total protein contents were measured after incubation of HepG2 cells with NOR4 or GSNO for 24 h. However, neither NO donor affected total protein contents at the concentrations used (data not shown).

Effects of NO donors on the expression of CYP2D6 mRNA. HepG2 cells were cultured in serum-free medium for 24 h and then incubated for 24 h with or without 150 or 300 μM NOR4, 300 μM decomposed NOR4, or 1000 μM GSNO. Total RNA was prepared, and reverse-transcription polymerase chain reaction was performed as described under Materials and Methods. CYP2D6 mRNA level was normalized with the GAPDH mRNA level in each sample. Values (mean ± S.E., n = 4) are expressed as the percentages relative to untreated cells. * and **, significant differences (P < 0.05 and P < 0.01, respectively) compared with untreated cells.
Effects of NO Donors on CYP2D6 Promoter Activity.
To test whether NO inhibits expression of the CYP2D6 gene at the transcriptional level, we constructed a luciferase-reporter plasmid −870/+65 pGL3 containing the 5′-promoter region from −870 to +65 of the CYP2D6 gene. The luciferase activity of −870/+65 pGL3 was approximately 15-fold higher than that of the pGL3 basic vector in transfected HepG2 cells. Cells transiently transfected with −870/+65 pGL3 were incubated for 20 h with two structurally different NO donors, NOR4 and GSNO; then luciferase activities were measured in cell lysates. The level of luciferase activity was normalized with respect to a cotransfected β-galactosidase plasmid. Figure2 shows that incubation of the cells with NO donors suppressed luciferase activity in a concentration-dependent manner. NOR4 decreased luciferase activity by 67 and 25% at concentrations of 100 and 300 μM, respectively. GSNO also decreased the activity by 70 and 52% at concentrations of 700 and 1000 μM, respectively. These results indicate that NO inhibits expression of theCYP2D6 gene at the transcriptional level. We then conducted control experiments using the pGL3 control vector (SV40 pGL3) containing the SV40 promoter and enhancer. The promoter activity of SV40 was sensitive to NOR4 and decreased its activity by 55% at a concentration of 300 μM, whereas it was insensitive to even 1000 μM GSNO (Fig. 2). Therefore, we treated cells with GSNO in subsequent studies. The cells were not altered morphologically at any concentration of the NO donors tested.

NO donors suppress CYP2D6 promoter activity in HepG2 cells. HepG2 cells were transiently transfected with −870/+65 pGL3 (1 μg) or SV40 pGL3 (0.3 μg) and pCMV·SPORT-βgal (1 μg) and then incubated with the indicated concentrations of NOR4 or GSNO for 20 h. Luciferase activity was normalized to the β-galactosidase activity in each sample. Values (mean ± S.E., n = 3) are expressed as the percentages of the activity of untreated cells. *, significant difference (P < 0.05) compared with untreated cells transfected with −870/+65 pGL3. #, significant difference (P < 0.01) compared with untreated cells transfected with SV40 pGL3.
Effects of Soluble Guanylate Cyclase Inhibitor and Cyclic GMP on CYP2D6 Promoter Activity.
To determine whether cGMP production via the activation of soluble guanylate cyclase by NO is involved in the suppression of CYP2D6 promoter activity, we examined the effects of the guanylate cyclase inhibitor ODQ on suppression of the CYP2D6 promoter activity by GSNO. Cells transiently transfected with the −870/+65 pGL3 reporter construct were incubated with 1000 μM GSNO in the presence or absence of 10 μM ODQ. Figure 3A shows that ODQ did not restore the suppression of luciferase activity caused by NO. These findings indicate that the luciferase activity by NO is suppressed via a guanylate cyclase-independent pathway. However, ODQ alone slightly increased luciferase activity (Fig. 3A). In addition, the cGMP analog 8-bromo-cGMP slightly decreased luciferase activity at a concentration of 500 μM in cells transiently transfected with −870/+65 pGL3 (Fig. 3B). Therefore, it is likely that cGMP contributes in part to the regulation of CYP2D6 promoter.

Effects of a guanylate cyclase inhibitor (A) and cGMP (B) on the promoter activity of CYP2D6. A, HepG2 cells were transiently transfected with −870/+65 pGL3 (1 μg) and pCMV·SPORT-βgal (1 μg), then treated with 1000 μM GSNO, with a combination of 1000 μM GSNO plus 10 μM ODQ, or with 10 μM ODQ for 20 h. B, HepG2 cells were transiently transfected with −870/+65 pGL3 (1 μg) and pCMV·SPORT-βgal (1 μg) and then treated with 200 and 500 μM 8-bromo-cGMP for 20 h. Luciferase activity was normalized to the β-galactosidase activity in each sample. Values (mean ± S.E.,n = 3) are expressed as percentages of the activity of untreated cells. * and **, significant differences (P < 0.05 and P < 0.01, respectively) compared with control.
Analysis of NO-Responsive Element in the CYP2D6 Promoter.
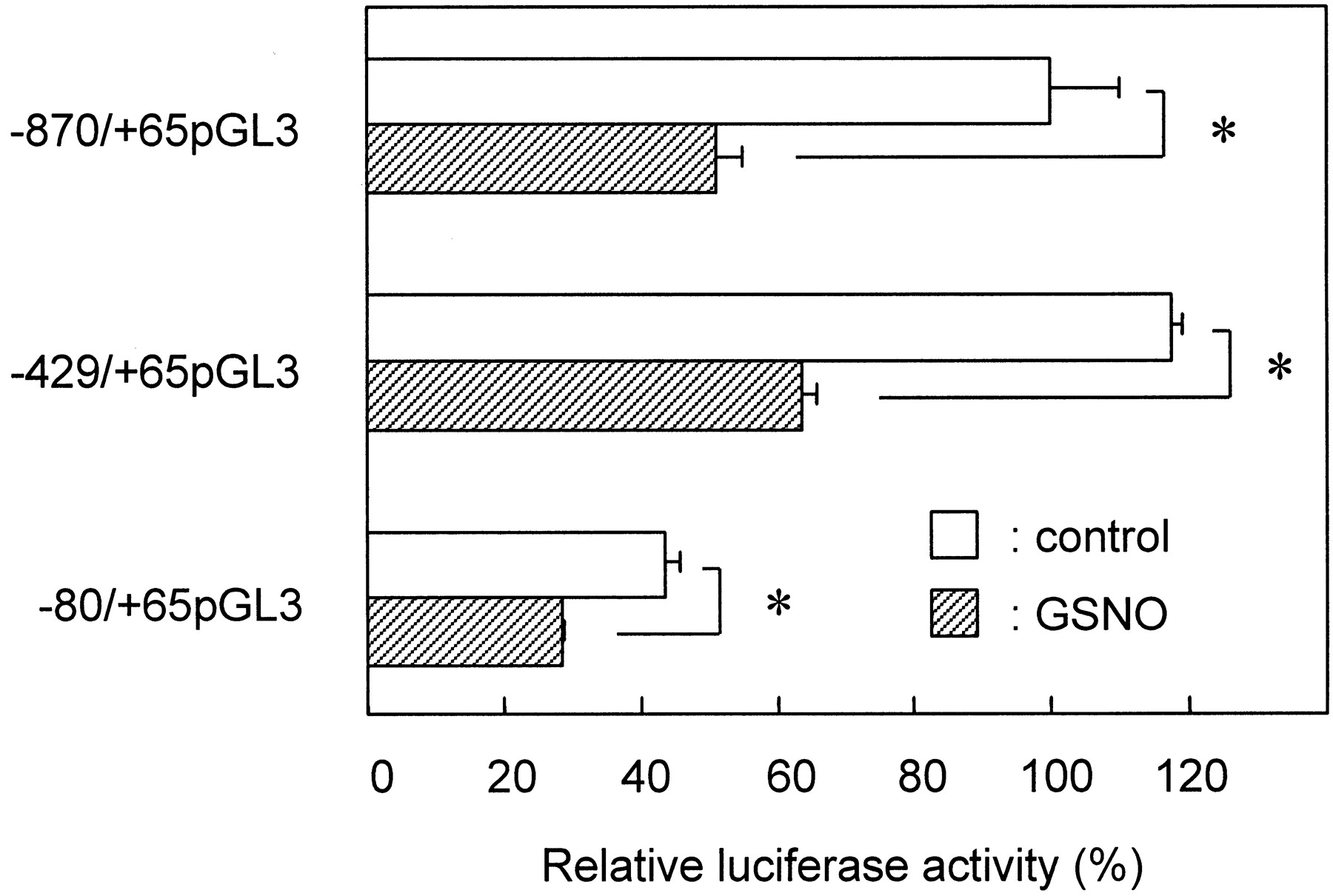
To clarify the regulatory element responsible for the inhibitory action of NO in the CYP2D6 gene-promoter region, we constructed luciferase-reporter plasmids (−429/+65 pGL3 and −80/+65 pGL3) containing 5′-deletion mutants of the CYP2D6 promoter and examined the luciferase activities of these constructs in HepG2 cells in the presence or absence of 1000 μM GSNO. Figure4 shows that GSNO depressed the luciferase activities of −870/+65 pGL3 and −429/+65 pGL3 by 50 and 55% of control, respectively. The luciferase activity of even the shortest construct, −80/+65 pGL3, was decreased by 65% by GSNO. These results suggested that the 5′-promoter region between −80 and +65 of the CYP2D6 gene was responsible for the inhibition of CYP2D6 promoter activity by NO. However, the −429 to −80 promoter region might contribute in part to the reduction of promoter activity by NO because the suppression of luciferase activity of −80/+65 pGL3 by GSNO was somewhat lower (10–15%) than that of −870/+65 pGL3 or −429/+65 pGL3 .

Deletion analysis of NO-responsive element in CYP2D6 promoter. HepG2 cells were transiently transfected with 1 μg of −870/+65 pGL3, −429/+65 pGL3, or −80/+65 pGL3 and 1 μg of pCMV·SPORT-βgal and then incubated for 20 h in the presence (▨) or absence (■) of GSNO (1000 μM). Luciferase activity was normalized to the β-galactosidase activity in each sample. Values (mean ± S.E., n = 3) are expressed as the percentages of activity of untreated cells transfected with −870/+65 pGL3 construct. *, significant difference (P < 0.05) compared with control of designated groups.
NO Inhibits DNA-Binding Activity of HNF4.
According to sequence analysis, the binding site of HNF4, which has been proposed to regulate the transcription of CYP2D6, was located within the proximal promoter region from −80 to +65 of CYP2D6 (Cairns et al., 1996). HNF4 may be important in the transcriptional control of CYP2D6. Therefore, we postulated that the inhibition of DNA-binding activity of HNF4 by NO causes the suppression of CYP2D6 gene transcription. To confirm this hypothesis, we performed an EMSA using nuclear extracts from HepG2 cells treated with NOR4 or GSNO for 12 h. Figure5 shows that the DNA-binding activity of HNF4 to the element located from −55 to −43 of the CYP2D6 promoter was inhibited by NOR4 or GSNO. The DNA-protein complex was specifically reduced with a molar excess of cold competitor (data not shown). HNF4 antiserum supershifted the nuclear extract complexed with the probe (Fig. 5), providing evidence that this complex truly represented HNF4. The HNF4 protein level was not significantly affected by this procedure (Fig. 5). These results suggest that the amount of the HNF4-DNA complex decreased by NO is caused by a loss of DNA-binding activity and not of protein content.

Effects of NO donors on DNA-binding activity of HNF4 in HepG2 cells. Nuclear extracts were prepared from HepG2 cells incubated with or without the indicated concentrations of NOR4 or GSNO for 12 h. Top, EMSA using radiolabeled −60/−39 promoter fragment of CYP2D6 as described under Materials and Methods. In supershift assays, anti-HNF4 antibody was added to the reaction mixture. Bottom, Western blots of HNF4 using nuclear extracts. Similar results were obtained in two separate experiments.
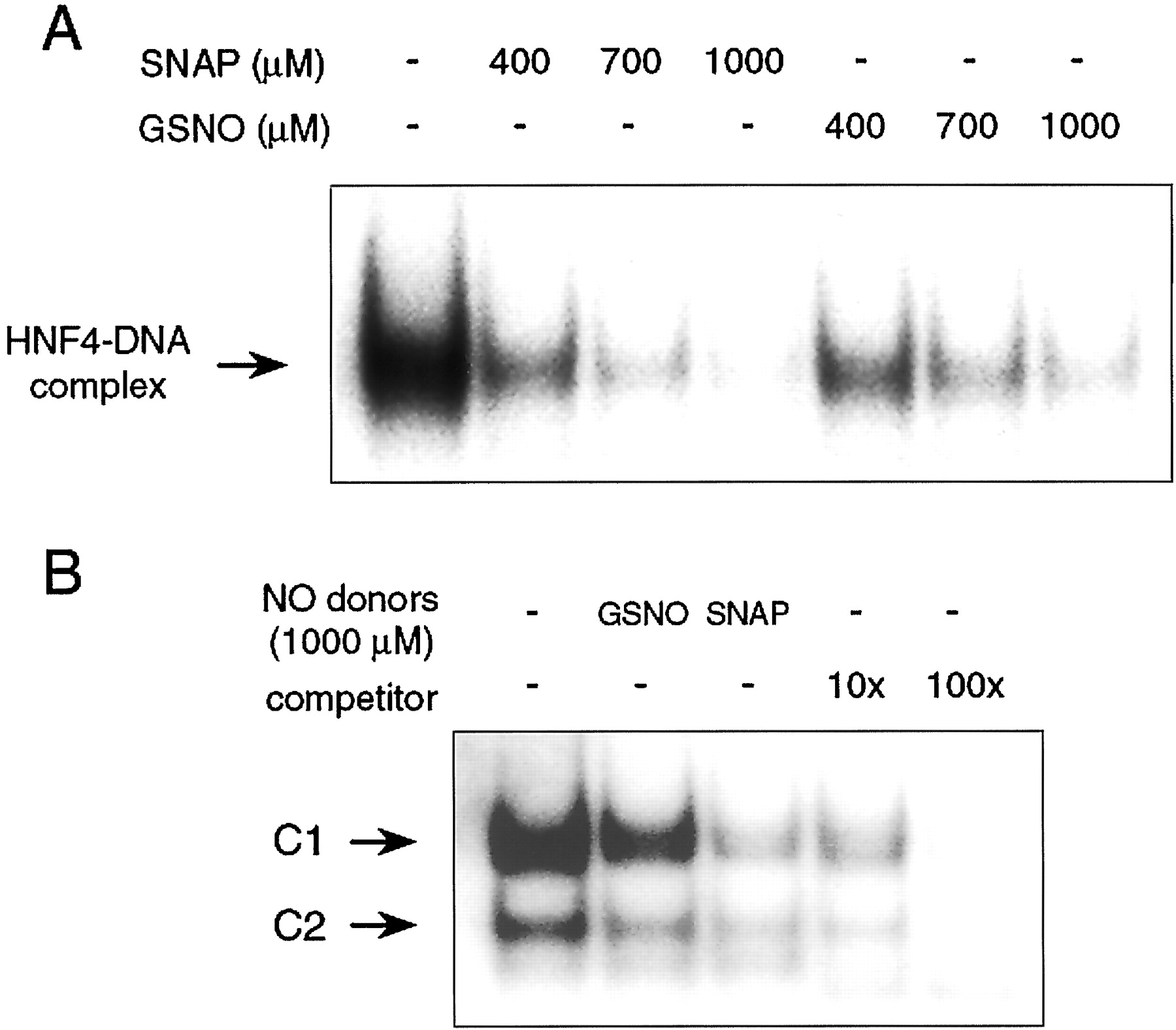
To determine whether NO directly inhibits the DNA-binding activity of HNF4, we performed EMSA using nuclear extracts isolated from untreated HepG2 cells. Nuclear extracts were incubated with various concentrations of SNAP, which is a well-known NO donor, or GSNO for 30 min at room temperature before EMSA. Figure6A shows that the DNA-binding activity of HNF4 was considerably decreased by both SNAP and GSNO. These findings suggested that NO suppresses CYP2D6 promoter activity by inhibiting HNF4 binding to the proximal promoter region of CYP2D6. To monitor other transcription factors in response to NO donors, we also examined the effect of the NO donors, GSNO or SNAP, on the DNA-binding activity of Sp1, which is known to be an NO-sensitive transcription factor, with the Sp1-specific oligonucleotide probe (Berendji et al., 1999). Figure6B shows that the nuclear extract from HepG2 cells formed two specific complexes (C1 and C2). Two distinct complexes were completely blocked by the competition of cold Sp1 oligonucleotide, indicating their specificities. As reported previously (Berendji et al., 1999), both NO donors inhibited the Sp1 DNA-binding activity. This result indicates the possibility that other transcription factors as well as HNF4 might be implicated in the decrease of CYP2D6 promoter activity caused by NO.

NO donors inhibits HNF4 (A) and Sp1 (B) DNA-binding activity. A, nuclear extracts of HepG2 cells were incubated with or without 400, 700, or 1000 μM SNAP or GSNO for 30 min at room temperature before the DNA-binding reaction. EMSAs were performed using radiolabeled −60/−39 promoter fragment of CYP2D6 as described underMaterials and Methods. B, nuclear extracts of HepG2 cells were incubated with or without 1000 μM SNAP or GSNO for 30 min at room temperature before the DNA-binding reaction. EMSAs were performed using radiolabeled Sp1-specific oligonucleotide in the absence or presence of 10-fold or 100-fold molar excess of unlabeled probe. Two distinct DNA-protein complexes are referred to as C1 and C2. Similar results were obtained in two separate experiments.
Analysis of Site-Directed Mutagenesis in CYP2D6 Promoter.
To directly demonstrate whether HNF4 is involved in the down-regulation of CYP2D6 promoter activity by NO, we constructed a mutated CYP2D6 reporter plasmid (mut-870/+65 pGL3) containing an eight-nucleotide substitution in the HNF4 binding site. Figure7 shows that the luciferase activity of mut-870/+65 pGL3 was decreased by 85% compared with that of −870/+65 pGL3, indicating that HNF4 plays an important role in the CYP2D6 promoter activity. Using the mut-870/+65 pGL3 construct, we investigated the effects of GSNO and SNAP on mutated CYP2D6 promoter activity. Figure 7 shows that neither NO donor affected luciferase activity from the promoterless pGL3 basic vector (pGL3B), which served as negative control (98.5% for GSNO and 98.2% for SNAP relative to untreated cells transfected with pGL3B). GSNO and SNAP decreased the wild-type CYP2D6 promoter activity (59.5% and 66.8%, respectively, relative to untreated cells transfected with −870/+65 pGL3). Unexpectedly, NO donors also suppressed the activity of the mutated CYP2D6 promoter (73.5% for GSNO and 83.5% for SNAP relative to untreated cells transfected with mut–870/+65 pGL3). However, the suppression by NO donors of the mutated promoter activity was significantly lower compared with that of the wild-type promoter.

Effects of site-directed mutation of HNF4 binding element on NO responsiveness of the CYP2D6 promoter. HepG2 cells were transiently transfected with −870/+65 pGL3 (1 μg), mut–870/+65 pGL3 (1 μg), or pGL3B (1 μg) and pCMV·SPORT-β-gal (1 μg) and then treated with 700 μM GSNO and 500 μM SNAP for 20 h. Luciferase activity was normalized to the β-galactosidase activity in each sample. Values (mean ± S.E. n = 3) are expressed as the percentages of the activity of untreated cells transfected with −870/+65 pGL3. *, significant difference (P < 0.01) compared with untreated cells transfected with −870/+65 pGL3. #, significant difference (P < 0.05) compared with untreated cells transfected with mut–870/+65 pGL3.
Discussion
Nitric oxide is involved in the suppression of various P450 genes because of the inflammatory cytokines and LPS in the liver and in cultured hepatocytes. However, its mechanism of action remains unknown. In this study, we demonstrated that NO donors reduced the level of CYP2D6 mRNA in HepG2 cells that constitutively express CYP2D6 and that the suppression of CYP2D6 promoter activity by NO donors was caused by the direct inhibition of HNF4 DNA-binding activity by NO.
The concentrations of NO donors used may seem to have been too high to have physiological significance. However, we assumed that the local NO concentrations in the liver, for example, are considerably high when iNOS is induced by endotoxin, because high levels of NO (approximately 600 μM as nitrite/nitrate in plasma) are detected in endotoxemic mice (Komarov and Lai, 1995). The concentration of NO in the culture medium was probably comparable with that of NO generated by iNOS in tissues during sepsis, considering the kinetics of NO release from NO donors together with the low solubility and short half-life of NO itself (Gross and Wolin, 1995). We used NOR4 and GSNO as NO donors in the present study. NOR4 and its analogs are novel NO donors that release NO spontaneously and stoichiometrically (1–1.5 mol/mol NOR analogs) at physiological pH (Fukuyama et al., 1995). NOR4 seems to be more potent than GSNO for the suppression of activities of not only the CYP2D6 promoter but also the SV40 promoter (Fig. 2). The difference in NO release between NOR4 and GSNO would result in this observation because NOR4 releases a larger quantity of NO than SNAP, anotherS-nitorosothiol similar to GSNO (Hara et al., 2000).
The nuclear receptor HNF4 is expressed in a restricted manner in the liver, intestine, kidney, and pancreas, and it plays a critical role in the transcriptional regulation of many liver-specific genes (Sladek, 1994). It is also involved in the expression of several but not all P450 genes, and the binding sites for HNF4 are located in the regulatory regions of CYP2C and -2D subfamilies (Chen et al., 1994;Ibeanu and Goldstein, 1995). Cairns et al. (1996) showed that coexpression with an HNF4 expression plasmid significantly increases the induction of CYP2D6 promoter activity in HepG2 and COS-7 cells and that HNF4 plays an important role in the regulation ofCYP2D6 gene expression. Deletion analysis of the CYP2D6 promoter showed that an important element responsible for transcriptional suppression of CYP2D6 by NO is located between positions −80 and +65 in the proximal promoter region of theCYP2D6 gene in which the HNF4 binding site is included (Fig.4). In addition, we found that the DNA-binding activity of HNF4 to the proximal region of the CYP2D6 promoter was inhibited when HepG2 cells were exposed to exogenous NO (Fig. 5). These results suggested that HNF4 plays a key role in the suppression of CYP2D6 gene expression by NO.
Nitric oxide has been reported to increase gene expression from activator protein 1 (AP-1) responsive promoters (Pilz et al., 1995) or decrease hypoxic induction of the vascular endothelial growth factor gene (Liu et al., 1998b) via an NO-guanylate cyclase-dependent pathway. However, the guanylate cyclase inhibitor ODQ failed to prevent the reduction of CYP2D6 promoter activity by GSNO in HepG2 cells in our experiments. In contrast, the promoter activity was slightly increased by ODQ alone and decreased by 8-bromo-cGMP (Fig. 3). From these results, we postulated that NO would mainly down-regulateCYP2D6 gene expression via the direct inhibition of HNF4 DNA-binding activity caused by NO (discussed below), but the NO-guanylate cyclase pathway may be involved in part in the suppression of CYP2D6 promoter activity.
Nitric oxide inhibits the DNA-binding activity of various transcription factors including nuclear factor-κB (Matthews et al., 1996), Ap1 (Tabuchi et al., 1996), octamer transcription factor 1 (Liu et al., 1998a), and Sp1 (Berendji et al., 1999) via a guanylate cyclase-independent pathway. Nitric oxide induces the nitrosylation of cysteine residues (thiol groups) within or near the DNA binding domain and/or disruption of the zinc finger structure, which is a DNA-binding motif, resulting in the inhibition of the DNA-binding activity of transcription factors (Kröncke et al., 1994; Nikitovic et al., 1998). We found that the DNA-binding activity of HNF4 is inhibited in a concentration-dependent manner in nuclear extracts from HepG2 cells exposed to exogenous NO, and the inhibition is caused by a direct action of NO itself (Figs. 5 and 6). The DNA-binding activity of HNF4 that has two zinc-finger DNA-binding motifs (Sladek et al., 1990) is believed to be inhibited by the same mechanism. Therefore, the direct inhibition by NO of HNF4 DNA-binding activity would account for the suppression of CYP2D6 promoter activity. However, mutation of the HNF4 binding site on the CYP2D6 promoter could not completely abolish the suppression of promoter activity by NO. This result indicated that it is more likely that the promoter region between −870 and −80 is also partially involved in the suppression of the CYP2D6 promoter activity by NO. Our results of the deletion analysis (Fig. 4) suggest that the full transcriptional activity of the CYP2D6 promoter requires the participation of regulatory elements other than the HNF4 binding element, which is similar to the result reported previously (Cairns et al., 1996). Therefore, these observations suggested that other NO-sensitive transcription factors such as Sp1 (Fig. 6), which is responsible for the regulation of CYP2D5 (Lee et al., 1994), might be involved in the suppression of CYP2D6 by NO.
In conclusion, our findings indicate that the inhibition of the ability of HNF4 to bind to the CYP2D6 promoter caused by NO contributes, at least in part, to the down-regulation of CYP2D6 gene expression. Because HNF4 plays an important role in the transcriptional control of other P450 genes (Jover et al., 2001), the inhibition of DNA-binding of HNF4 might also participate in the suppression of other P450 genes caused by NO. However, it has been reported that some P450 isoforms are down-regulated by cytokines or LPS in an NO-independent manner (Sewer and Morgan, 1997; Sewer et al., 1998; Iber et al., 2000). Therefore, the modulation of transcription factors by NO might be one of the mechanisms by which the expression of P450 genes is down-regulated under conditions of inflammation and infection.
Footnotes
- Received May 3, 2001.
- Accepted October 2, 2001.
Abbreviations
- P450
- cytochrome P450
- LPS
- lipopolysaccharide
- NO
- nitric oxide
- iNOS
- inducible nitric-oxide synthase
- HNF4
- hepatocyte nuclear factor-4
- NOR4
- (±)-N-[(E)-4-ethyl-2-[(Z)-hydroxyimino]-5-nitro-3-hexene-1-yl]-3-pyridine carboxamide
- GSNO
- S-nitrosoglutathione
- SNAP
- S-nitroso-N-acetyl-penicillamine
- ODQ
- 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- DMEM
- Dulbecco's modified Eagle's medium
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PCR
- polymerase chain reaction
- SV40 pGL3
- simian virus 40 pGL3 control vector
- pGL3B
- pGL3 basic vector
- EMSA
- electrophoretic mobility shift assay
- mut–870/+65 pGL3
- mutated CYP2D6 reporter plasmid
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}