


Abstract
To study behavior of activated Gαs in living cells, green fluorescent protein (GFP) was inserted within the internal amino acid sequence of Gαs to generate a Gαs-GFP fusion protein. The fusion protein maintained a bright green fluorescence and was identified by immunoblotting with antibodies against Gαs or GFP. The cellular distribution of Gαs-GFP was similar to that of endogenous Gαs. Gαs-GFP was tightly coupled to the β adrenergic receptor to activate the Gαs effector, adenylyl cyclase. Activation of Gαs-GFP by cholera toxin caused a gradual displacement of the fusion protein from the plasma membrane throughout the cytoplasm in living cells. Unlike the slow release of Gαs-GFP from the membrane induced by cholera toxin, the β-adrenergic agonist isoproterenol caused a rapid partial release of the fusion protein into the cytoplasm. At 1 min after treatment with isoproterenol, the extent of Gαs-GFP release from plasma membrane sites was maximal; however, insertion of Gαs-GFP at other membrane sites occurred during the same time period. Translocation of Gαs-GFP fusion protein induced by isoproterenol suggested that the internalization of Gαs might play a role in signal transduction by interacting with effector molecules and cytoskeletal elements at multiple cellular sites.
A family of heterotrimeric G proteins transduces chemical and sensory signals across the plasma membrane by sequential interactions with receptor and effectors such as second messenger-generating enzymes or ion channels. Because of the wide array of cellular processes that are mediated by G proteins, the study of G protein function and regulation has provided a cornerstone for research in signal transduction.
Recently, numerous studies have suggested an important function for G proteins at cellular locations other than the plasma membrane. Certain G proteins have been detected at intracellular membranes, such as the Golgi complex (Kehlenbach et al., 1994; Denker et al., 1996; Ugur and Jones, 2000) or light vesicle fractions (Drmota et al., 1999), whereas others associate with cytoskeletal structures, such as microtubules and microfilaments (Wilson et al., 1994; Ibarrondo and Gullin, 1995;Roychowdhury and Rasenick, 1997; Roychowdhury et al., 1999). The mechanisms that govern the cellular destinations of G protein and the relative proportions of the G protein that traffic to subcellular compartment remain unresolved.
Previous studies, using cell fractionation and immunocytochemical assays, suggested that activation by β adrenergic agonist, cholera toxin or direct binding of a hydrolysis-resistant GTP analog resulted in Gαs translocation from plasma membrane to cytoplasm (Rasenick et al., 1984; Ransas et al., 1989; Levis and Bourne, 1992). Gαq/11 has also been reported to transiently translocate to plasma membrane of adrenal glomerulosa cells in response to stimulation by angiotensin II for 1 to 5 min (Cote et al., 1997). However, in other studies, a relocation of activated Gαs was not observed (Jones et al., 1997; Huang et al., 1999). The mechanism by which Gαs is released from the membrane is not yet known, but there are suggestions that activated Gαs is depalmitoylated and released from the membrane (Wedegaertner and Bourne, 1994). This continues to be a subject of some controversy, however, because at least one report demonstrates that Gαs remains associated with the plasma membrane, regardless of palmitoylation state (Huang et al., 1999).
The use of GFP in the study of cellular signaling allows not only the observation of G protein trafficking, but it also provides the opportunity to study the functional dynamics of G proteins in real time. At present, most GFP fusion proteins are constructed by fusing GFP to either the amino or carboxyl terminus of the protein of interest (Barak et al., 1997; Hack et al., 2000; Kallal and Benovic, 2000). For G protein α subunits, this is problematic, because the NH2 region is important for association with G protein β and γ subunits and the COOH terminal is required for interaction with receptor. Recently, functional Gα-GFP fusion proteins were obtained by inserting GFP into an internal loop of Gαq and of Gα2 fromDictyostelium discoideum (Hughes et al., 2001; Janetopoulos et al., 2001). Thus, a biologically active Gαs-GFP fusion protein might need to incorporate GFP at some interior position in Gαs.
This report demonstrates that incorporation of GFP into the internal sequence of Gαs results in a biologically active protein that seems to function identically to native Gαs in the binding of GTP and activation of adenylyl cyclase. This Gαs-GFP displays a heterogeneous distribution on the plasma membrane of the cells in which it is expressed and translocation from plasma membrane to cytoplasm after activation. This study raises the possibility that Gαs might interact with effector molecules and cytoskeletal elements at multiple cellular sites.
Materials and Methods
Construction of Gαs-GFP Fusion Protein Expression Vectors.
Full-length cDNAs encoding Gαs were excised from the pcDNA1 vector by digesting with SamI and XbaI restriction enzymes. The full-length EGFP cDNA obtained by PCR from the pEGFP-N2 using appropriate primers (sense, 5′-GGAATTCATGGTGAGCAAGGGCGAGGAACTG-3′; antisense, 5′-GCTCTAGACGACTTGTACAGCTCGT-3′) and adding restriction sites to its cDNA (EcoRI at initiation codon andXbaI at end of cDNA). To insert the EGFP within the sequence of Gαs, the first fragment of Gαs cDNA (from 1 to 71 amino acids) was amplified by PCR with restriction sites for KpnI at initiation codon and EcoRI at end of the fragment and cloned into pcDNA3 vector. Modified EGFP cDNA was ligated to the first fragment of Gαs by cloning it into pcDNA3 vector at EcoRI and XbaI restriction sites. The second fragment of Gαs cDNA (from 82 to 394 amino acids) was also obtained using PCR with appropriate primers. The sense primer contained sequence overlapping with the 3′ end of EGFP (5′-GAGCTGTACAAGTCGTCTAGAAACAGCGATGGTGAGAA-3′). The anti-sense primer contained an additional XbaI restriction site (5′-CGCTCTAGAGAACATCTAAGCAAG-3′). The second fragment of Gαs cDNA was recombined to 3′ end of EGFP using PCR strategy. Finally, the full-length Gαs-GFP was cloned into pcDNA3 at KpnI and XbaI restriction sites. All DNA manipulations, including ligations, bacterial transformation, and plasmid purification, were carried out using standard procedures. To produce Gαs-NGFP and Gαs-CGFP, full-length Gαs cDNA was subcloned into the pEGFP-C3 vector and pEGFP-N2 vector, respectively. Platinum Taq DNA polymerase (Invitrogen, Carlsbad, CA) was used in PCR. All PCR fragments were identified by DNA sequencing.
Adherent Cell Culture and Transient Transfection.
COS-1 cells and HEK 293 cells in DMEM (Invitrogen) containing 10% fetal bovine serum, 1% antibiotic (penicillin and streptomycin), and PC12 cells in DMEM medium containing 5% fetal bovine serum, 5% horse serum, and antibiotic were cultured at 37°C with 5% CO2. The cells in the 12-well culture plates or 60-mm tissue culture dishes were transfected using GenePORTER Transfection reagent (Gene Therapy Systems, Inc. San Diego, CA) according to the manufacturer's instruction. Briefly, 60 to 80% confluent cells washed once with Opti-MEM I reduced serum medium (Invitrogen). In each well of 12-well culture plates, 0.5 ml of mixture of plasmid DNA (5 μg) and GenePORTER Transfection reagent (10 μl) in Opti-MEM I medium was applied. In 60-mm tissue culture dish, 2.5 ml of mixture of plasmid DNA (15 μg) and GenePORTER Transfection reagent (50 μl) in Opti-MEM I medium was added. Six hours after incubating, cells were cultivated in complete medium as described above. All experiments were performed in transiently transfected cells.
Microscopy.
Cells were observed 24 h after transfection using fluorescence microscopy or confocal microscopy. Before observation, the medium in 12-well culture dishes was changed to serum-free DMEM containing 20 mM HEPES and placed immediately on the microscope stage. Cells were maintained at 37°C during the entire period of observation.
A microscope (eclipse TE300; Nikon, Tokyo, Japan), equipped with a 100-W mercury arc lamp was used for digital fluorescence microscopy. Images were acquired with an interline charge-coupled device camera (1300YHS; Roper Scientific, Trenton, NJ) driven by IP Lab imaging software (Scanalytics, Inc. Suitland, VA). Images were processed with IP Lab and Adobe Photoshop 5.0 (Adobe Systems, Mountain View, CA). For confocal microscopy, images were collected with a laser scanning confocal microscope (LSM-510; Zeiss, Welwyn Garden City, UK). Equipped with an objective of 60× immersion. Differential interference contrast images of the cells were also acquired. Images of computer-generated cross sections of cells were collected as well (x-z and y-z planes). Images were processed using Adobe Photoshop 5.0. To represent changes in Gαs-GFP localization quantitatively, we evaluated the mean of gray value within the cytoplasm in confocal fluorescence images by having two persons blind to the experimental conditions select an area that corresponded to the maximal cytoplasm area for each cell. Mean gray value before and during treatment was collected using Scion Image (Scion Software, Frederick, MD) and paired Student's t tests were performed using Prism software (GraphPad Software, Inc. San Diego, CA).
Subcellular Distribution of Gαs-GFP Fusion Protein in COS-1 Cells.
COS-1 cells were harvested and resuspended in HEPES-sucrose buffer (15 mM HEPES, 0.5 mM sucrose, 1 mM DTT) containing Complete protease inhibitor tablets (Roche Molecular Biochemicals, Indianapolis, IN). Cell suspensions were homogenized with a Teflon/glass homogenizer. After a low-speed centrifugation to remove unbroken cells and a nuclear pellet, samples were centrifuged for 15 min at 200,000g at 4°C in a centrifuge (TL100; Beckman Coulter, Fullerton, CA). The pellets (particulate fractionation) and the supernatants (soluble fractionation) were separated by 12% SDS-PAGE and transferred to Immobilon-P transfer membranes (Millipore, Bedford, MA). Blots were incubated for 1 h in Tris-buffered saline/Tween 20 (10 mM Tris-HCl, pH 8.0, 159 mM NaCl, and 0.1% Tween 20) containing 5% powdered skim milk and 1% bovine serum albumin. After three washes with Tris-buffered saline/Tween 20, membranes were incubated for 2 h with the primary antibody and for 1 h with horseradish peroxidase-conjugated goat anti-rabbit IgG. Proteins were detected using the enhanced chemiluminescence detection kit (Amersham Biosciences, Piscataway, NJ).
Cell fractionation by sucrose density gradient sedimentation was carried out as described previously (Toki et al., 1999). Briefly, 24 h after transfection with Gαs-GFP, COS-1 cells were harvested into 0.3 ml of HEPES buffer (10 mM HEPES, PH 7.5, 150 mM NaCl, 1 mM DTT) containing 1% Triton X-100 and Complete protease inhibitor tablets. Homogenization was carried out with 10 strokes of a Teflon/glass homogenizer. The homogenate was adjusted to 40% sucrose by the addition of an equal volume of 80% sucrose prepared in HEPES buffer and placed at the bottom of an ultracentrifuge tube. A step gradient containing 30, 15, and 5% sucrose was formed above the homogenate and centrifuged (240,000g) at 4°C for 20 h. From the top of each gradient, 0.05-ml gradient fractions were collected to yield a total of 20 fractions. An equal volume of each fraction was separated by SDS-PAGE and subjected to Western blot analysis for Gαs and Gαs-GFP.
Photoaffinity Labeling.
[32P]-p3-1,4-Azidoanilido-p1-5′-GTP ([32P]AAGTP) was synthesized as described previously (Rasenick et al., 1994). Membranes (100 μg of protein) from cells transfected with the indicated construct were incubated with [32P]AAGTP for 3 min in binding buffer (20 mM HEPES, 1 mM EDTA, 2 mM MgCl2, 20 mM NaCl, 2 mM β-mercaptoethanol) plus 0.2 μM GDP for 3 min and followed by 50 μM isoproterenol for each sample. After incubation for 10 min at 37°C, the samples were UV-irradiated on ice for 30 s with a 9-W Mineralight (Black Light Eastern Corp, Westbury, NY) at a distance of 3 cm. The samples were washed with binding buffer by centrifugation three times, and the pellets were resuspended in 20 μl of binding buffer containing 1 mM DTT. Proteins were resolved by SDS-PAGE and visualized by autoradiography of dried gels. Densitometric analysis was carried out. Statistical significance was determined by paired Student's t test.
Measurement of cAMP Accumulation in cyc-S49 Lymphoma Cells.
cAMP levels were determined by labeling cyc− cells with [3H]adenine and measuring [3H]cAMP formation from [3H]ATP (Marsh et al., 1998). Gαs-GFP was introduced into cyc- cells (2 × 107 cells in 0.8 ml of 20 mM HEPES-buffered minimal essential medium) by electroporation at room temperature using a BTX Electro Cell Manipulator 600 (capacitance setting, 1600 μF; voltage setting, 250 V; resistance setting, R4 = 72 Ω; BTX Inc. San Diego, CA). After electroporation, the cells were added to 5.0 ml of DMEM containing 10% heat-inactivated horse serum in 25-ml tissue culture flask. Approximately 50% of cells survived electroporation, and about 4% of the cells (revealed by fluorescence microscopy) were expressing Gαs-GFP. At 24 h after electroporation, the cells were labeled with 14 μCi/ml of [3H]adenine. Twenty-four hours after the [3H]adenine addition, cAMP accumulation was measured. The cells first were washed in assay medium (20 mM HEPES-buffered DMEM) and then were suspended in 5 ml of the same medium with 1 mM 3-isobutyl-1-methylxanthine; and transferred to 1.5-ml microcentrifuge tubes (0.8 ml for each) with and without 0.1 mM isoproterenol. Tubes were incubated at 37°C for 20 min and reactions were terminated by addition of 0.2 ml of 25% cold trichloroacetic acid plus 1 mM concentrations of ATP and cAMP. Nucleotides were separated on ion exchange columns (Salomon et al., 1974). cAMP accumulation was expressed as [3H]cAMP / ([3H]ATP + [3H] cAMP) × 1000. Statistical significance was determined by one-way analysis of variance.
Results
Construction and Expression of Gαs-GFP Fusion Proteins.
Gαs exists as short and long splice variants, Gαss and GαsL, as well as a high-molecular-mass Gα splice variant, GαsXL (Kehlenbach et al., 1994). Compared with Gαss, GαsL contains an additional 15 amino acids inserted at position 72 of the polypeptide chain, and there is an exchange of glutamate for aspartate at position 71. Although there has been some indication that subtle differences between short Gαs and long Gαs exist (Seifert et al., 1998; Bourova et al., 1999), the general function of the two forms is similar. Levis and Bourne (1992) modified the long Gαs form at a site (residues 77–81) within the 15-amino-acid insert to confer upon it recognition by antibody directed against a well-defined peptide of the influenza hemagglutinin (HA). Addition of a HA epitope did not alter the ability of wild-type Gαs to mediate hormonal stimulation of adenylyl cyclase or attach to cell membrane. In the study, a Gαs-GFP fusion protein was constructed by replacing the residues (72–81) within GαsLwith the GFP sequence (Fig. 1). Thus, GFP was inserted in linker 1 between the helical and GTPase domains.
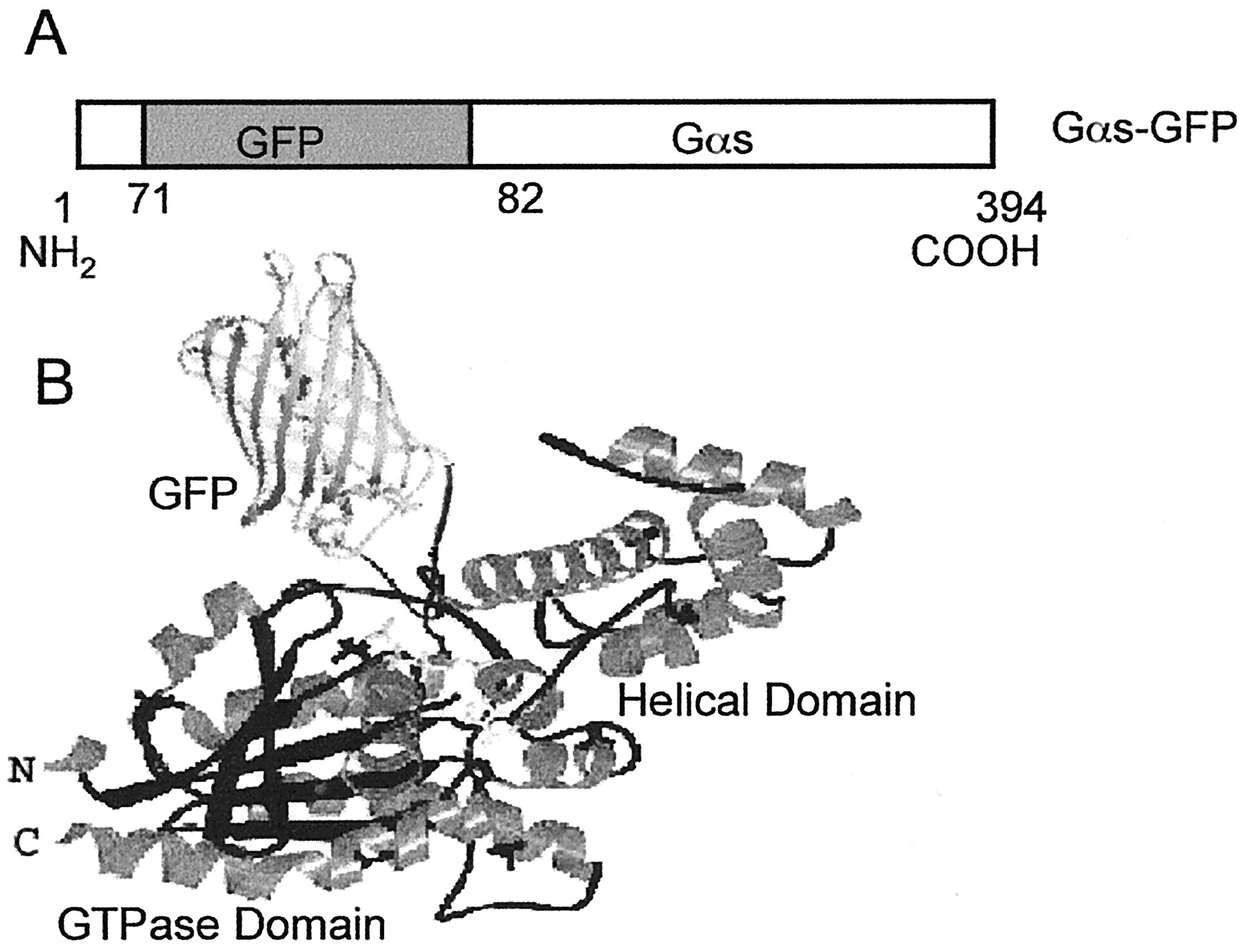
Construction of the Gαs-GFP fusion protein. A, Schematic of Gαs-GFP. Gαs-GFP defines the fusion protein in which GFP was inserted within the NH2-terminal domain of the long form of Gαs. B, model of Gαs-GFP. GFP is depicted as an insert into the Gαs structure described by Sunahara et al. (1997).
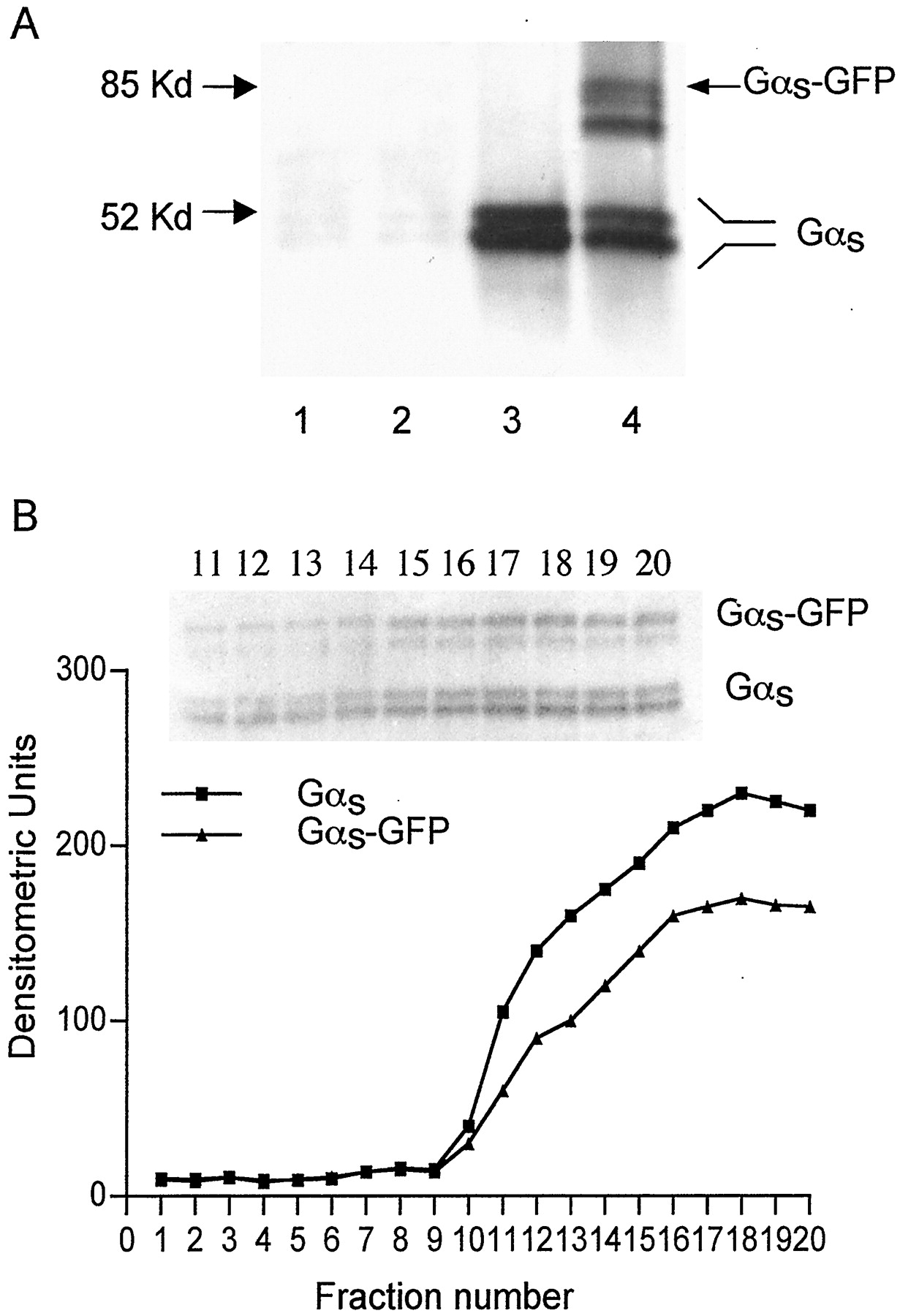
During immunoblot analysis, cell lysates from COS-1 cells transfected with Gαs-GFP were probed with an anti-Gαs polyclonal or anti-GFP monoclonal antibody. Gαs-GFP fusion protein was expressed in about 30% of the COS-1 cells. In cells expressing Gαs-GFP, the protein was present at about 2 times the level of intrinsic Gαs. Gαs-GFP fusion protein was calculated to have an approximate molecular mass of 85 kDa. An extra band of about 81 kDa was found while detecting with antibodies against GFP and Gαs, respectively (Fig.2). The lower band probably corresponds to a proteolytic fragment because expression of HA-Gαs in COS-7 cells shows a comparably sized degradation product (Rondard et al., 2001).

Expression of Gαs-GFP in COS-1 cells. COS-1 cells were lysed 24 h after transiently transfection with Gαs-GFP construct. Thirty micrograms of protein was loaded, separated by SDS-PAGE, and Gαs-GFP was detected with polyclonal antibody against Gαs (B) or a monoclonal antibody against GFP (A), as indicated. Lane 1 represents a lysate from cell transfected with Gαs-GFP, lane 2 is the lysate from nontransfected cells.
Images were taken using confocal microscopy in living cells, and computer-generated cross sections show that the expressed Gαs-GFP fusion protein was distributed primarily on the plasma membrane. Gαs-GFP shows a somewhat heterogeneous distribution on the membrane, with regions of strong and weak fluorescence (Fig. 3). COS-1 cell fractionation and immunoblotting with an anti-Gαs polyclonal antibody further showed that distribution of Gαs-GFP fusion protein is similar to endogenous Gαs (Fig.4). These results indicate that insertion of GFP into the sequence of Gαs did not alter the subcellular distribution of Gαs in cells and the fluorescence of GFP in Gαs-GFP protein is stable and readily visible after UV irradiation.
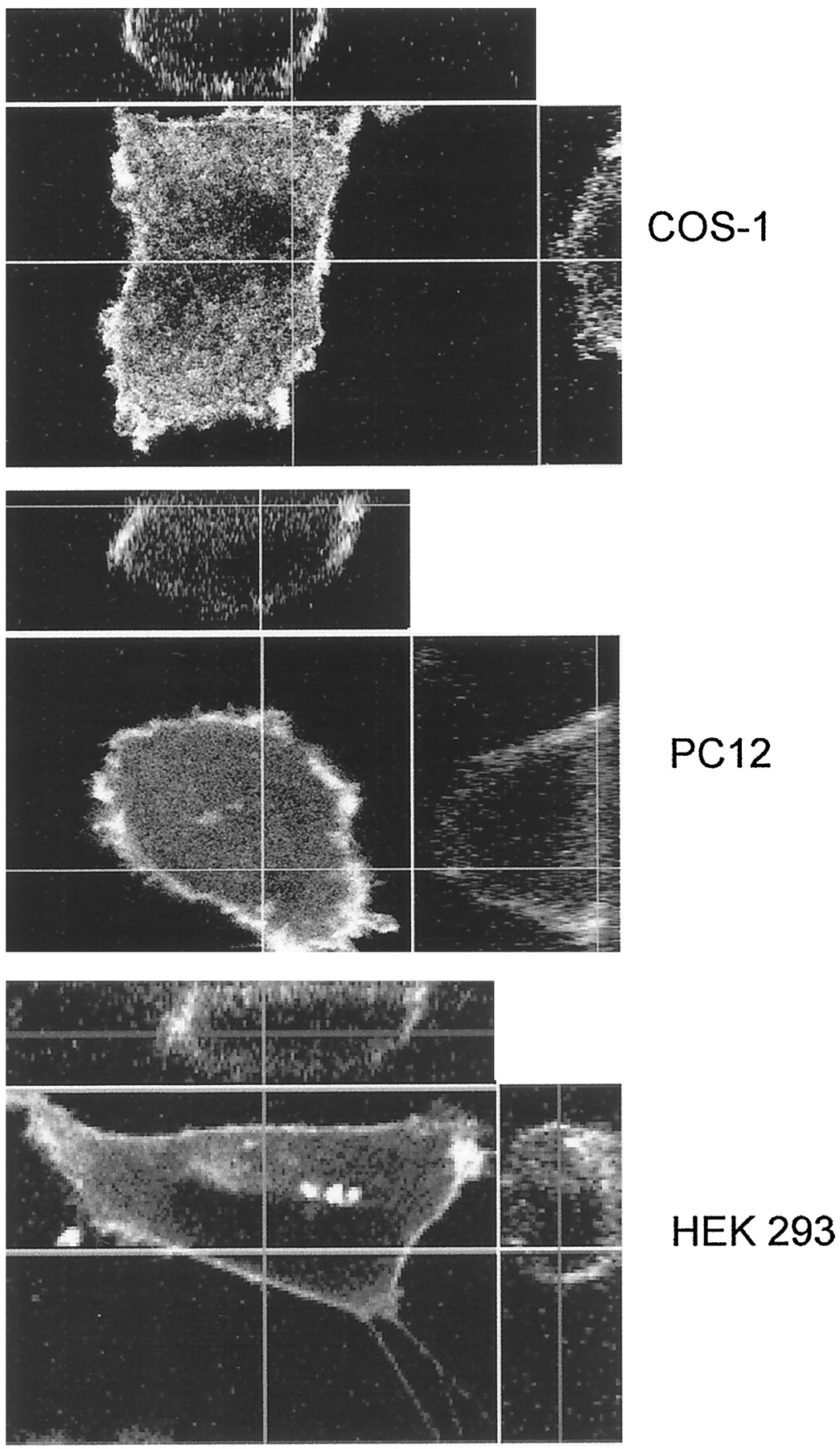
Gαs-GFP is associated with the plasma membrane in COS-1, PC12, and HEK 293 cells. Twenty-four hours after transfection, cells were observed by confocal microscopy at 37°C. A computer-generated cross-section of a typical cell is displayed on the top (x-z plane) and on the right (y-z plane). Each image shown is representative of at least 20 cells observed and subjected to az-scan analysis with similar results.

Subcellular distribution of Gαs-GFP in COS-1 cells. A, particulate and soluble fractions were isolated from cells transfected with Gαs-GFP constructs as described under Materials and Methods. Twenty micrograms of protein was loaded, separated by SDS-PAGE gel, and detected with a polyclonal antibody against the C-terminal peptide of Gαs. Lanes 1 and 2 represent the soluble portion from the cells transfected with GFP or with Gαs-GFP construct, respectively. Lanes 3 and 4 indicate the particulate fraction from cells transfected with GFP or with Gαs-GFP construct, respectively. B, densitometric analysis of Gαsdistribution after cell fractionation by sucrose density gradient sedimentation. Fractions 1 to 5 correspond to the 5% sucrose layer, fractions 6 to 10 are the 15% sucrose layer, fractions 11 to 15 (lanes 11–15) are the 30% sucrose layer, and fractions 16 to 20 (lanes 16–20) are 40% sucrose layer. The insert represents the immunoblot of fractions 11 to 20.
We also constructed fluorescent fusion proteins by adding GFP at either the NH2 or the COOH terminus of Gαs. When GFP was spliced into the NH2 terminus of Gαs, the fusion protein did not associate with the plasma membrane. When GFP was fixed to the COOH terminus of Gαs, about 50% of this construct was membrane bound, as revealed by Western blotting. Curiously, placement of GFP on the COOH terminus of Gαs resulted in profound quenching of the fluorescent GFP signal. This rendered the construct useless for the purposes of this study.
GTP Binding Properties of Gαs-GFP Fusion Protein.
Based on models derived from the crystal structure of the α-subunit of the retinal G-protein transducin, the sequence of the 15-amino-acid insert localized in GαsL serves as a linker between the GTPase domain and the α-helical domain (Noel et al., 1993; Sunahara et al., 1997). The guanine nucleotide-binding site is embedded between these two domains. Thus, an expansion of this linker sequence might be expected to diminish the ability of Gαs to exchange GDP for GTP in response to agonist. To test this, COS-1 cells were cotransfected with Gαs-GFP and β-adrenergic receptor. Membranes from those cells were then incubated with the photoaffinity GTP analog [32P]AAGTP in the presence or absence of isoproterenol. Gαs from control cells bound [32P]AAGTP and this binding was increased significantly in the presence of agonist. Transfected cells showed agonist-induced [32P]AAGTP binding in both native Gαs and Gαs-GFP (Fig. 5). Thus, it seems that Gαs-GFP fusion protein is coupled to the β-adrenergic receptor and binds GTP in response to agonist. Note that in COS-1 cells, about 30% of cells expressed the Gαs-GFP construct. In the cells expressing the construct, the content of Gαs-GFP is about twice that of the endogenous Gαs. Thus, in a given cell population, endogenous Gαs exceeds Gαs-GFP by a ratio of 3:2. [32P]AAGTP labeling is consistent with this ratio.

Gαs-GFP binding to AAGTP. COS-1 cells were cotransfected with cDNA encoding Gαs-GFP (1 μg) and β adrenergic receptor (4 μg). A, cell membranes were prepared 24 h after transfection and were incubated with [32P]AAGTP in the presence and absence of isoproterenol (as indicated). Proteins were resolved by SDS-PAGE and autoradiography. The result shown is from one of four similar experiments. B, densitometric analysis of Gαs-GFP binding to AAGTP. Densitometric analysis of four independent experiments were carried out and displayed in densitometric units. Shown is the mean ± S.E.,n = 4; **, significant difference in incorporation compared with control (no isoproterenol) (p < 0.01).
Activation of Adenylyl Cyclase by Gαs-GFP Fusion Protein in cyc-S49 Lymphoma Cells.
To further test the ability of Gαs-GFP to couple to β-adrenergic receptor and activate adenylyl cyclase, we measured receptor-dependent accumulation of cAMP after transient transfection of cyc- cells, which lack endogenous Gαs. Basal cAMP levels are identical in these cells transfected with Gαs-GFP or the GFP vector alone. Isoproterenol did not stimulate adenylyl cyclase in cells expressing the GFP construct, but isoproterenol elicited a significant increase in cAMP in cells expressing Gαs-GFP (Fig.6).

Gαs-GFP increases cAMP accumulation in cyc−S49 lymphoma cells after isoproterenol treatment. Cyc cells were transfected with GFP or Gαs-GFP as indicated; cAMP levels were measured in the absence (■) and presence (▪) of 0.1 mM isoproterenol. All values represent the mean ± S.E. of five independent experiments. **, significant difference from cells without isoproterenol (p < 0.01).
Internalization of Cholera Toxin Activated Gαs-GFP.
G proteins are bound at the inner face of the plasma membrane, where they are positioned strategically to interact with membrane-spanning receptors and appropriate effectors. The Gαs subunit possesses the guanine nucleotide-binding site, intrinsic GTP hydrolytic activity, and an ADP-ribose acceptor site. Cholera toxin activates Gαs by ADP-ribosylating arg201 of Gαs and inactivating the intrinsic GTPase. During real-time observation, it was seen that after 20-min treatment with cholera toxin (3 μg/ml), marked internalization of Gαs-GFP protein occurred. At 1 h, most of Gαs-GFP protein was no longer associated with plasma membrane and was found in the cytoplasm (Fig.7A). A computer-generated cross-section of cells further indicates that almost complete Gαs-GFP internalization occurred at 1 h after cholera toxin treatment (Fig. 7B). Control cells did not show observable internalization of Gαs-GFP during this time period. Quantitative image analysis for seven random cells under treated with cholera toxin established that the effect was statistically significant (Fig. 7C).

Cholera toxin treatment translocates Gαs-GFP fusion protein in living PC12 cells. A, twenty-four hours after transfection with GαS-GFP, media was replaced as described under Materials and Methodsand living cells were viewed by confocal microscopy at 37°C. Cells were initially imaged (0 min), cholera toxin (3 μg/ml) was added, and cells were observed for 1 h. Bar, 10 μm. B, computer-generated cross-section of the whole cell after completion of the hour is displayed on the top (x-z plane) and on the right (y-z plane). C, quantitative image analysis of cells before and during treatment with cholera toxin. Mean gray value were determined and used to quantify the fluorescence intensity within the cytoplasm in cells. **, P < 0.01.
Isoproterenol Activation Rapidly Dissociates Gαs-GFP from the Plasma Membrane.
In COS-1 cells, activation of Gαs is generally caused by activation of the β-adrenergic receptor. The COS-1 cells 24 h after transfection with Gαs-GFP construct were treated with 50 μM isoproterenol. This resulted in a rapid, partial internalization of Gαs-GFP protein. Within 2 min after addition of isoproterenol, the significant release of Gαs from plasma membrane was seen, but it only occurred at some regions of the membrane. It is noteworthy that cell shape changes slightly in response to isoproterenol [see Fig. 8 and videos 1 and 2 (http://www.molpharm.aspetjournals.org)], but this does not account for displacement of Gαs-GFP protein from the membrane. Although minimal changes may occur in focus during the 2-min period, control cells did not show any significant changes of Gαs-GFP protein on the plasma membrane (Fig. 8A and video 1). Figure 8B presents confocal images and results similar to these obtained with the digital fluorescent microscopy in Fig. 8A. It is noteworthy, however, that in some regions of plasma membrane, Gαs-GFP fusion protein fluorescence was increased, suggesting that “naive” Gαs was inserted into the plasma membrane. During the internalization process, clusters of Gαs-GFP protein formed subjacent to the Gαs-GFP “rich” regions of the cell (Fig. 8A and video 2).

Isoproterenol-stimulated rapid internalization of Gαs-GFP fusion protein in living COS-1 cells. Cells were transfected with Gαs-GFP construct and observed 24 h later at 37°C with confocal or digital fluorescent microscopy. A, cells were treated with (top) or without (bottom) isoproterenol (50 μM), and digital fluorescent images were captured every 5 s. Video 1 shows assembled images at 5-s intervals for the control cells and video 2 shows this for the isoproterenol treated cells. The videos are available at http://molpharm.aspetjournals.org. Arrows indicate areas of major release of membrane-bound Gαs-GFP. Clusters of Gαs-GFP protein form subjacent to the plasma membrane (indicated by open arrowhead). B, isoproterenol-induced Gαs-GFP release from plasma membrane visualized by confocal microscopy. Arrows display regions where clusters of Gαs-GFP were released from plasma membrane due to activation of β receptors by isoproterenol. The arrowheads indicate the sites at which clusters of Gαs-GFP were inserted during the response to isoproterenol. Bar, 10 μm. These results are typical of 40 of 58 cells observed during the course of 15 experiments. Eighteen cells did not show Gαs-GFP internalization in response to isoproterenol. C, COS-1 cells transfected with Gαs-GFP and subcultured into 12-well culture plates were treated with isoproterenol for the indicated times. Cells were rapidly harvested and the content of Gαs-GFP in soluble fraction was examined by immunoblot analysis.
Fig. 8C shows the immunoblot analysis of soluble fraction of the Gαs-GFP–transfected COS-1 cells after treatment with isoproterenol for indicated times. One minute after treatment, Gαs-GFP in the soluble fraction was increased by 67.5 ± 13%. At 2 and 3 min, the amount of Gαs-GFP protein in soluble fractions was similar to that observed after 1 min. Quantitative image analysis was also done for determining the effect of isoproterenol on the distribution of Gαs-GFP in the cells. Fluorescence intensity of Gαs-GFP in the cytoplasm was measured for six random cells before and during treatment with isoproterenol. At time 0, the mean gray value was 34.7 ± 2.1; this increased to 45.5 ± 3.5, 47.0 ± 2.4, and 45.5± 2.3 for 1, 1.5, and 2 min, respectively (P < 0.01, compared with that at zero point). Thus, Gαs-GFP in the cytoplasm increased by 35% upon isoproterenol stimulation.
Discussion
This report indicates that GFP can be inserted into the sequence of Gαs, without substantially altering the normal function of that G protein. Gαs-GFP exhibits properties associated with Gαs, suggesting that insertion of GFP at position 71 was transparent with regard to function. We have successfully tracked the behavior of Gαs in living cells and investigated the effect of activation by receptor and cholera toxin on the localization of Gαs. This study confirms previous studies that indicated translocation of activated Gαs(Ransas et al., 1989; Levis and Bourne, 1992; Wedegaertner et al., 1996) and provides insights into the cellular location and behavior of Gαs in cells.
Images from unstimulated living cells indicate that although most Gαs-GFP are associated with the plasma membrane, some of this protein is in the cytosol. Cell fractionation studies were consistent with this (Fig. 4A). Similar findings were made with a Gαq-GFP construct (Hughes et al., 2001). Immunolocalization studies had suggested that Gαs might be highly concentrated within such limited membrane regions (Aoki et al., 1992; Aronin and DiFiglia, 1992). Our results provide evidence that distribution of Gαs-GFP on the plasma membrane is not uniform (see Fig. 2). The functional significance of this clustering remains to be determined.
Previous studies, using cell fractionation or observations of fixed cells, have suggested that activated Gαs may be released from plasma membranes (Rasenick et al., 1984; Ransas et al., 1989; Levis and Bourne, 1992). In this study, cholera toxin-activated Gαs-GFP was released from plasma membrane during the course of activation and were gradually distributed through the cytosol. This result coordinates well with observations of HA tagged Gαs in HEK 293 cells (Wedegaertner and Bourne, 1994; Wedegaertner et al., 1996). It provides the first real-time evidence from living cells for translocation of activated Gαs from plasma membrane into cytoplasm.
The β-adrenergic receptor signals primarily through Gαs and agonist binding to this receptor causes activation of Gαs. We tested receptor-mediated activation of Gαs-GFP fusion protein. Within 2 min after addition of isoproterenol, Gαs-GFP was translocated from the plasma membrane to cytoplasm. The extent of this release was maximal by 1 min and did not increase significantly after that point. The observation is basically consistent with immunohistochemistry and cell fractionation studies, suggesting that the significant redistribution of activated Gαsoccurred 5 min after isoproterenol treatment (Levis and Bourne, 1992). It is noteworthy, however, that in some regions of the plasma membrane in COS-1 cells, the fluorescence density was actually increased in the presence of isoproterenol (Fig. 8B). Levis and Bourne (1992) suggested that epitope-tagged Gαs, released from the membrane after β-receptor activation and might be returned to membrane fraction. Although data in this study might be consistent with this, it is also possible that the Gαs-GFP recruited to the membrane during the course of isoproterenol treatment was not identical to the Gαs-GFP that was released.
The activated Gαs translocation from plasma membrane into cytoplasm might be explained by rapid depalmitoylation of αs, as the activation of αs mediated by both receptor and other mechanisms (e.g., cholera toxin), causes depalmitoylation of αs (Wedegaertner and Bourne, 1994). Moreover, some studies have shown that both mutational activation and removal of the palmitoylation sites reduce association of Gα subunits with plasma membrane (Levis and Bourne, 1992; Wedegaertner and Bourne, 1994;Wedegaertner et al., 1996; Hughes et al., 2001). Nevertheless, one recent report demonstrated that Gαs remained associated with the plasma membrane, regardless of palmitoylation state (Huang et al., 1999). Adding GFP to the NH2terminal of Gαs, a modification likely to block palmitoylation, yielded a protein that did not associate with the membrane.
It is possible that Gαs is unique with regard to agonist-induced membrane dissociation. Experiments with Gαq labeled with AAGTP did not reveal that this G protein was released into the cytosol upon activation (Popova et al., 1997; Popova and Rasenick, 2000) and agonist activation did not evoke relocalization of GFP-modified Gαq (Hughes et al., 2001). It is noteworthy in this regard that Gαs may have an association with specialized membrane domains that is not seen with Gαqproteins (Oh and Schnitzer, 2001).
The role and fate of the Gαs released from the plasma membrane are unknown at this time. Gα released from the membrane in response to agonist might interact with cytoskeletal components (Cote et al., 1997). Gα binds to tubulin and has been shown to regulate microtubule polymerization and microtubule dynamics (Roychowdhury et al., 1999). Gαs seems to activate the intrinsic GTPase of tubulin and, in doing so, removes the “GTP cap” that confers microtubule stability. This results in rapid microtubule depolymerization. Changes in cell shape subsequent to agonist activation have been demonstrated (Popova and Rasenick, 2000;Witt-Enderby et al., 2000) and such changes may be particularly relevant in the central nervous system, where it seems that cellular stimulation may result in a rapid reorganization of dendritic spines (Maletic-Savatic et al., 1999). Slight shape changes of COS-1 cells in response to β-adrenergic agonist were seen in this study. Thus, it is possible that Gαs, released from the membrane subsequent to agonist activation, could influence rapid changes in synaptic shape.
In summary, this report reveals that insertion of GFP into Gαs yields a functional protein that can be used to track the behavior of this G protein in living cells. Gαs-GFP displays a heterogeneous distribution on the plasma membrane of the cells in which it is expressed and translocates from plasma membrane to cytoplasm after activation. It is suggested that the release of Gαs from plasma membranes may play a specific role in the process of cellular signal transduction.
Acknowledgments
We thank Drs. Richard Green and Robert Donati for advice and discussion.
Footnotes
- Received May 23, 2001.
- Accepted October 24, 2001.
-
This work was supported by United States Public Heath Service grants MH39595, MH57391, and AG15482.
-
The online version of this article (available athttp://molpharm.aspetjournals.org) contains two QuickTime videos.
Abbreviations
- Gαs
- α subunit of stimulatory G protein
- GFP
- green fluorescent protein
- EGFP
- enhanced green fluorescent protein
- PCR
- polymerase chain reaction
- HEK
- human embryonic kidney
- DMEM
- Dulbecco's modified Eagle's medium
- DTT
- dithiothreitol
- PAGE
- polyacrylamide gel electrophoresis
- [32P]AAGTP
- [32P]P3(4-azido-anilido)-P1-5′-GTP
- HA
- hemagglutinin
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}