


Abstract
The tumor suppressor p53 is mutated in more than 50% of all cancers. Importantly, most clinically useful antineoplastic agents are less potent and efficacious in the context of mutant p53. This situation has prompted a search for agents that cause tumor cell death via molecular mechanisms independent of p53. Our recent investigations with electrophilic prostaglandins enabled us to devise a pharmacophore and mechanism of action hypothesis relevant to this problem: a cross-conjugated α,β-unsaturated dienone with two sterically accessible electrophilic β-carbons is a molecular determinant that confers activity among this class of ubiquitin isopeptidases inhibitors, and that inhibitors of ubiquitin isopeptidases cause cell death in vitro independently of p53. Here, we report the use of the National Cancer Institute's Developmental Therapeutics Database to identify compounds to test this hypothesis. Shikoccin (a diterpene), dibenzylideneacetone, and curcumin fit the pharmacophore hypothesis, inhibit cellular isopeptidases, and cause cell death independently of p53 in isogenic pairs of RKO and HCT 116 cells with differential p53 status. The sesquiterpene achillin and 2,6-diphenyl-4H-thiopyran-4-one, which have cross-conjugated dienones with sterically hindered electrophilic β-carbons, do not inhibit isopeptidases or cause significant cell death. Furthermore, we show that a catalytic-site proteasome inhibitor causes cell death independently of p53. Combined, these data verify the p53-independence of cell death caused by inhibitors of the proteasome pathway and support the proposition that the ubiquitin-dependent proteasome pathway may contain molecular targets suitable for antineoplastic drug discovery.
Ubiquitin isopeptidases (ubiquitin specific proteases) are a family of cysteine proteases that salvage ubiquitin for its reuse by the 26S proteasome system (Hochstrasser, 1996; Hershko and Ciechanover, 1998) and regulate the activity of a variety of substrates by altering their ubiquitination status. The ubiquitin salvage activity of the isopeptidases maintains a cellular pool of monomeric ubiquitin by cleaving the isopeptide bond between the C-terminal carboxyl of ubiquitin and the ε-amino group of a lysine residue on an adjacent protein, thereby disassembling ubiquitin oligomers, ubiquitin-protein conjugates, and ubiquitin-peptide conjugates. Few inhibitors of isopeptidases have been identified, other than analogs based on ubiquitin itself. These include nonhydrolyzable ubiquitin dimer analogs (∼16 kDa) (Yin et al., 2001) and ubiquitin aldehyde (∼8.5 kDa) (Dang et al., 1998), which are suitable for investigating isolated enzymes. Recently, we reported that Δ12-prostaglandin J2 (Δ12-PGJ2) is a novel isopeptidase inhibitor with activity in intact cells. Results with cyclopentenone prostaglandins (PG) prompted our hypothesis that isopeptidase inhibition depends on nuances of olefin-ketone conjugation. For example, Δ12-PGJ2, with its cross-conjugated α,β-unsaturated dienone substituent and two sterically accessible β-carbons (Rodriguez et al., 1997), was a potent inhibitor of isopeptidase activity. PGA1, PGA2, and 15-keto-PGs with a simple α,β-unsaturated ketone and only one accessible β-carbon were significantly less potent. PGB1 with an α,β-unsaturated ketone and a sterically hindered β-carbon was inactive (Mullally et al., 2001). In its current formulation, our pharmacophore hypothesis predicts that compounds chemically unrelated to PG, but with a cross-conjugated α,β-unsaturated ketone and two sterically accessible β-carbons, will also inhibit ubiquitin isopeptidases. Although it disrupts ubiquitin salvage and impairs protein turnover, Δ12-PGJ2 induces apoptosis independently of tumor suppressor p53 trans-activation (Mullally et al., 2001). Therefore, our corresponding pharmacological mechanism hypothesis predicts that such isopeptidase inhibitors will also induce cell death independently of tumor suppressor p53 function. Herein, we report the identification of dibenzylideneacetone (DBA), curcumin, and, via the National Cancer Institute's Developmental Therapeutics Database, a diterpene, shikoccin (NSC-302979), as agents that fulfill these predictions and reinforce our pharmacophore and mechanism hypotheses. As isopeptidase inhibitors, DBA, curcumin, and NSC-302979 inhibit the proteasome pathway in a manner chemically and mechanistically distinct from lactacystin (Fenteany et al., 1995), eponemycin (Meng et al., 1999), and peptide-aldehyde or boronate inhibitors (Adams et al., 1999), which all covalently inhibit the 20S catalytic subunit of the proteasome. Our results support the hypothesis that the sterically accessible, cross-conjugated α,β-unsaturated dienone is a pharmacophore that confers inhibitory activity toward isopeptidases. Furthermore, these results lend support to the proposition that the ubiquitin-dependent proteasome pathway contains molecular targets suitable for antineoplastic drug discovery (Kisselev and Goldberg, 2000).
Experimental Procedures
Materials.
We used Δ12-PGJ2 and PGB1 (Cayman Chemicals, Ann Arbor, MI); dibenzylideneacetone, etoposide, paclitaxel, Curcumin, and 2,6-diphenyl-4H-thiopyran-4-one (Sigma, St. Louis, MO); NSC-302979 and NSC-156236 (Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute); complete protease inhibitor mixture (Roche Applied Science, Indianapolis, IN); enhanced chemiluminescence reagents (Amersham Biosciences, Piscataway, NJ); antibodies directed against p53 (DO-1), horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA), and ubiquitin (Zymed Laboratories, Inc., San Francisco, CA); MG115 (Peptides International, Louisville, KY); z-LLVY-MCA, z-LRGG-MCA (Biomol Research Laboratories, Plymouth Meeting, PA); (3-(4,5-dimethylthiazo)-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Molecular Probes Inc., Eugene, OR); ubiquitin-PEST (Ub-PEST; a gift of Dr. Martin Rechsteiner, Department of Biochemistry, University of Utah, Salt Lake City, UT). Centricon YM-30 centrifugal filters (Amicon Bioseparations; Millipore, Bedford, MA).
Cell Culture.
We used RKO and RKO-E6 colon cancer cells (gift from Dr. Mark Meuth, Institute for Cancer Studies, University of Sheffield, Sheffield, UK). We used HCT 116 colon cancer cells with varying degrees of p53 haplosufficiency (gift of Dr. Bert Vogelstein, Johns Hopkins School of Medicine, Baltimore, MD). We maintained RKO and RKO-E6 cells in DMEM [supplemented with 2 mM l-glutamine, 1 mM sodium pyruvate, 50 units/ml penicillin and streptomycin, and 10% (v/v) fetal bovine serum] in a humidified incubator with 5% CO2. We maintained HCT 116 cells in McCoy's 5A medium [supplemented with 1 mM sodium pyruvate, 50 units/ml penicillin and streptomycin, and 10% (v/v) fetal bovine serum] in a humidified incubator with 5% CO2.
Immunochemical Detection of Proteins.
We removed the medium and lysed cells in 50 mM Tris, pH 7.4, 100 mM NaCl, 2 mM EDTA with 0.1% SDS, 0.1% deoxycholate, 1× complete protease inhibitor mixture. We measured protein concentration by the method of Bradford (1976). We fractionated equal portions of the total cell lysate from each sample (12.5 μg of protein) by SDS-PAGE. We transferred proteins to polyvinylidene difluoride blocked with 5% (w/v) nonfat dry milk in Tris-buffered saline [20 mM Tris-HCl, pH 7.5, 100 mM sodium chloride, 0.1% (v/v) Tween 20]. We detected proteins immunochemically by using primary antibodies directed against p53 (1:4000) or ubiquitin (1:1000), followed by horseradish peroxidase-conjugated secondary antibodies (1:4000). We detected antigen-antibody complexes with enhanced chemiluminescence reagents. We scanned gels and quantified intensities using Kodak 1D Image Analysis Software (Eastman Kodak, Rochester, NY)
Cell Viability Assay.
We determined cell viability by the MTT assay. Briefly, we incubated 1 × 105cells per well of a sterile, 96-well assay plate with 0–60 μM test compounds for 48 h. We added MTT reagent to each well (final concentration, 0.5 mg/ml) and incubated for an additional 3 h. We aspirated the media and remaining MTT reagent from each well and added 100 μl of HCl/isopropanol (1:24). We measured the absorbance of each sample at 405 nm.
Ubiquitin Isopeptidase Activity Assays.
We measured cellular isopeptidase enzymatic activity with two assays that used different substrates. In one assay we used Ub-PEST, a full-length ubiquitin molecule with an 18-amino acid C-terminal peptide extension (total mass, 10.5 kDa). Ubiquitin isopeptidases specifically cleave the 18-amino acid peptide extension, releasing full-length ubiquitin (8.5 kDa). Briefly, we incubated 6 × 105 cells with 0–60 μM test compounds for 12 h. We lysed cells in 50 μl of 25 mM HEPES, 5 mM EDTA, 0.1% CHAPS, 5 mM ATP, pH 7.5. We adjusted the protein concentration of each sample to 0.3 mg/ml and incubated with 50 μg/ml Ub-PEST for 45 min at 25°C. Under these conditions Ub-PEST hydrolysis occurs at a linear rate. We mixed 20-μl samples with 20 μl of 2× Laemmli buffer, boiled briefly, and fractionated by SDS-PAGE. We monitored isopeptidase activity by determining the amount of product (8.5-kDa ubiquitin) formation.
The second assay used a fluorescent tetrapeptide, z-LRGG-AMC, as a substrate that mimics the carboxyl terminus of ubiquitin. Isopeptidase activity hydrolyzes the bond between the c-terminal glycine and the fluorophore. This tetrapeptide also undergoes slow proteolysis by the catalytic subunit of the proteasome. To minimize this background rate of proteolysis, we incubated cell lysates with 30 μM MG115 for 30 min at 4°C, before substrate incubation (>90% proteasome inhibition). We treated cells as above, lysed them in 250 μl of lysis buffer per sample, and adjusted their protein concentration to 0.5 mg/ml before incubation with MG115. We then added z-LRGG-AMC substrate and quantified fluorescence of the AMC moiety cleaved by isopeptidase action.
We determined whether Δ12-PGJ2 acted irreversibly. We incubated cell lysates with vehicle or 100 μM test compound and measured isopeptidase activity fluorometrically. We dialyzed a 500-μl portion of each sample through a Centricon filter with a molecular mass cut-off of 25 kDa. After washing each sample with 3 volumes of assay buffer, we measured the isopeptidase activity in the filtrate. If filtration did not reverse inhibition, it implies that Δ12-PGJ2 is an irreversible inhibitor.
Statistics.
We used analysis of variance for statistical calculations.
Results
SubStructure Analysis of the NCI Cancer Screening Database.
The Developmental Therapeutics Program (DTP) of the National Cancer Institute (NCI) has systematically evaluated >70,000 compounds for cytostatic and cytotoxic activity against human cell lines in vitro (Monks et al., 1997). The cell lines typify cancers of the colon, blood (leukemia), brain, breast, kidney, lung, ovary, prostate, and skin (melanoma). Intramural NCI investigators, who have access to the entire database, have applied this information-intensive approach with promising results (Weinstein et al., 1997; Shi et al., 1998). Extramural investigators have access to a restricted portion of the database, last released in August 2000. Using these available data, we conducted substructure searches to test our hypothesis that the cross-conjugated α,β-unsaturated dienone with two sterically accessible β-carbons is the primary molecular determinant that confers the inhibition of isopeptidases. Specifically, we sought nonprostanoid compounds with this feature that varied in the accessibility of their olefinic β-carbons (e.g., β-carbons with -H versus with -CH3 substituents).
Our substructure query, 2-cyclopenten-5-methylene-1-one (Fig.1, i), yielded eight compounds. All have a cyclic (bis) α,β-unsaturated ketone with one endo- and one exo-olefin. They are otherwise chemically unrelated to Δ12-PGJ2 or other PGs. The NCI provided two of the eight compounds we requested for our experimental use: the sesquiterpene NSC-156236 (Fig. 1, ii) and the diterpene NSC-302979 (Fig. 1, v). Like Δ12-PGJ2 (Fig. 1, vi), theendo- and exo-olefins of NSC-302979 have sterically accessible β-carbons that can react with nucleophiles (e.g., cysteine; Rodriguez et al., 1997). Analogous to PGB1 (Fig. 1, iii), NSC-156236 has methyl-substituted β-carbons at the endo- and theexo-olefin of the dienone. These β-carbons are sterically hindered and therefore should not react readily with relevant physiological nucleophiles (Rodriguez et al., 1997). To further reinforce our pharmacophore hypothesis, we also evaluated several commercially available compounds. Two of these compounds, DBA (Fig. 1, vii) and curcumin (Fig. 1, viii), have sterically accessible β-carbons. The final compound, 2,6-diphenyl-4H-thiopyran-4-one (DPTP; Fig. 1, iv), resembles DBA, except that it has a bulky sulfur atom sterically hindering its β-carbons. In summary, our pharmacophore hypothesis predicts that compounds ii through iv will be inactive as isopeptidase inhibitors and compounds v through viii will be active as isopeptidase inhibitors.

Chemical structures of the pharmacophore test panel. We used 2-cyclopenten-5-methylene-1-one (i) as a substructure to search the NCI-DTP database. We identified, and the NCI provided, NSC-156236 (achillin, ii) and NSC-302979 (shikoccin, v). We also identified and used PGB1 (iii), DPTP (iv), Δ12-PGJ2 (vi), DBA (vii), and curcumin (viii) to test our pharmacophore hypothesis.
Electrophilic Cross-Conjugated Dienones Inhibit Cellular Ubiquitin Isopeptidases.
Little is known about the substrate specificity of the individual isopeptidase family members. To investigate total cellular isopeptidase activity, we use two simple substrates, Ub-PEST and z-LRGG-AMC, which most isopeptidases use as substrates. Figure2A shows the effect of the test panel compounds, Δ12-PGJ2 (lanes 3–5), DBA (lanes 6–8), NSC-302979 (lanes 9–11), PGB1 (lane 12), NSC-156236 (lane 13), curcumin (lanes 15–17), and DPTP (lane 18) on cleavage of the Ub-PEST substrate by isopeptidases in HCT 116 colon cancer cell lines. Similar results were obtained for RKO cells (raw data not shown). Consistent with our pharmacophore hypothesis, the compounds with cross-conjugated ketones and sterically accessible β-carbons, Δ12-PGJ2, DBA, NSC-302979, and curcumin, each inhibited isopeptidase activity in a concentration-dependent manner (Fig. 2B). Compounds with sterically hindered β-carbons (PGB1, NSC-156236, and DPTP) did not inhibit isopeptidase activity. The rank order of potency for inhibition of ubiquitin-PEST hydrolysis by isopeptidases was DBA ≈ NSC-302979 ≥ Δ12-PGJ2 > curcumin ≫ NSC-156236 ≈ PGB1 ≈ DPTP.

Effect of the pharmacophore test panel on ubiquitin isopeptidase activity in colon cancer cells: Ub-PEST as substrate. A, inhibition of isopeptidase proteolysis of Ub-PEST substrate. We treated RKO and HCT 116 cells with DMSO vehicle, 6, 20, or 60 μM Δ12-PGJ2, DBA, NSC-302979, or curcumin, 60 μM PGB1, 60 μM NSC-156236, or 60 μM DPTP for 12 h at 37°C. We incubated cell lysates from each treatment with Ub-PEST as described under Materials and Methods. We fractionated samples by SDS-PAGE and detected proteins with ubiquitin epitopes immunochemically (HCT 116+/+ Western blot shown as example of raw data). Lane 1 shows the substrate before incubation with lysate. B, inhibition of isopeptidase activity by test panel compounds. We determined isopeptidase activity by measuring, via densitometry, the amount of Ub generated by isopeptidase cleavage of Ub-PEST. The amount of Ub generated in the vehicle treated sample (A, lane 2) is arbitrarily designated 100%.
To verify the results with Ub-PEST in Fig. 2, z-LRGG-AMC was used as the substrate for isopeptidases (Fig. 3). NSC-302979, Δ12-PGJ2, DBA, and curcumin each inhibited ubiquitin isopeptidase activity, whereas NSC-156236, PGB1, and DPTP did not. The rank-order of potency for inhibition of z-LRGG-AMC hydrolysis by ubiquitin isopeptidases was NSC-302979 > DBA > Δ12-PGJ2 > curcumin ≫ NSC-156236 ≈ PGB1 ≈ DPTP.

Effect of the pharmacophore test panel on ubiquitin isopeptidase activity in colon cancer cells: z-LRGG-AMC as substrate. We treated RKO (upper) and HCT 116+/+ cells (lower) with DMSO vehicle, 6, 20, or 60 μM Δ12-PGJ2, DBA, NSC-302979, or curcumin, 60 μM PGB1, 60 μM NSC-156236, or 60 μM DPTP for 12 h at 37°C. We incubated cell lysates (0.5 mg/ml) from each treatment with the isopeptidase substrate, z-LRGG-AMC, for 3 h at 37°C. We determined the amount of AMC cleaved by isopeptidase fluorometrically, as described under Materials and Methods.
Consistent with inhibition of the isopeptidases that disassemble ubiquitin polymers and ubiquitin-protein conjugates, protein species with polyubiquitin conjugation accumulated in cells (e.g., in RKO-E6 cells, Fig. 4A) treated with Δ12-PGJ2 (lanes 2–4), NSC-302979 (lanes 5–7), DBA (lanes 8–10) or curcumin (lanes 14–16). PGB1 (lane 11), NSC-156236 (lane 12), and DPTP (lane 17), compounds that did not inhibit isopeptidase activity, did not cause appreciable cellular accumulation of ubiquitin conjugates.

Effect of the pharmacophore test panel on ubiquitin-dependent proteolysis in colon cancer cells. A, accumulation of polyubiquitin. We treated RKO-E6 cells with DMSO vehicle for 24 h, 37°C, or with the concentrations of test compounds that gave 80% cell death (as determined by cell viability assay at 48 h) for 6, 12, or 24 h at 37°C [EC80, Δ12-PGJ2(13.2 μM), DBA (12.0 μM), NSC-302979 (3.5 μM), or curcumin (17 μM)]. For compounds that were inactive in the cell viability assay, we treated with the highest concentrations examined (60 μM PGB1, NSC-156236, and DPTP for 24 h at 37°C). We fractionated cell lysates by SDS-PAGE and detected proteins with ubiquitin epitopes immunochemically. B, accumulation of p53, a protein targeted for proteasomal degradation. i, Schematic depicting p53 degradation via the proteasome pathway in RKO cells versus RKO-E6 cells. The activity of the HPV E6 oncoprotein hastens p53 degradation in RKO-E6 cells. ii, We treated RKO and RKO-E6 cells with vehicle, etoposide (50 μM), MG115 (20 μM), Δ12-PGJ2 (60 μM), NSC-302979 (20 μM), DBA (20 μM), PGB1 (60 μM), NSC-156236 (60 μM), curcumin (60 μM), or DPTP (60 μM) for 6 h, 37°C. We fractionated lysates by SDS-PAGE and determined their p53 content immunochemically. iii, We measured p53 levels by densitometry and calculated the ratio of p53 protein in RKO-E6 cells/RKO cells.
As a consequence of polyubiquitin accumulation, monoubiquitin is initially depleted in cells treated with isopeptidase inhibitors [8.5-kDa band, Fig. 4A, lanes 2, 5, 8, and 14 versus lanes 1 or 13). We predicted that this would affect the rate at which substrates are degraded via the ubiquitin-proteasome pathway. RKO and RKO-E6 cells allow a convenient test of our pharmacophore hypothesis with p53, a single, explicit substrate targeted for ubiquitin-dependent proteolysis. RKO-E6 cells, an isogenic variant of RKO cells, harbor the HPV-E6 oncoprotein that, together with E6-AP ubiquitin ligase, hastens proteasome-mediated degradation of p53 (Fig. 4B, i; Ashcroft and Vousden, 1999). Thus, under most conditions, the levels of p53 are significantly lower in RKO-E6 cells than in RKO cells (e.g., treatment of cells with vehicle or etoposide, a DNA damaging agent; Fig. 4B, ii, lanes 1 and 2, respectively, and iii). One exception is when the ubiquitin-proteasome pathway is inhibited (e.g., treatment of cells with the proteasome inhibitor, MG115; lane 3); then, accumulation of p53 in RKO-E6 and RKO cells equalizes. Accordingly, the ratio of p53 protein (RKO-E6 cells/RKO cells) approached unity in cells treated with the isopeptidase inhibitors, Δ12-PGJ2 (lane 4), NSC-302979 (lane 5), DBA (lane 6), and curcumin (lane 9). Test compounds that did not inhibit ubiquitin isopeptidase activity [PGB1 (lane 7), NSC 156236 (lane 8), and DPTP (lane 10)] had p53 protein ratios (RKO-E6:RKO) significantly less than unity, suggesting that they do not inhibit p53 degradation via the ubiquitin-proteasome pathway. p53 accumulation caused by the pharmacophore test compounds is not a result of 20S proteasome inhibition, because none of the compounds with cross-conjugated dienones inhibited the 20S catalytic subunit of the proteasome under these conditions (Mullally et al., 2001; and data not shown). Note that despite its accumulation in the presence of Δ12-PGJ2, p53 is inactivated as a transcription factor under these conditions (Mullally et al., 2001); furthermore, p53 activation is insufficient to cause its stabilization in RKO-E6 cells, because the p53 protein ratio (RKO-E6:RKO = 0.1) in etoposide treated cells fails to approach unity. Collectively, the data in Figs.2 through 4 affirm that our pharmacophore hypothesis extends beyond the prostanoid family into the diterpene and other chemical families.
The Prototype Isopeptidase Inhibitor, Δ12-PGJ2, Inhibits Ubiquitin Isopeptidases Irreversibly.
Although we have not yet identified a covalent complex between an isopeptidase and one of the isopeptidase inhibitors, this proposed mechanism of action is consistent with data that we have obtained from analyzing cell lysates treated with Δ12-PGJ2. Treatment of cell lysates (versus treatment of whole cells) should exclude the likelihood of transcriptional/translational events due to the nature of the lysate preparation (i.e., sonication of cell lysates probably shears all polynucleotides). Δ12-PGJ2 inhibited isopeptidase activity in treated cell lysates (Fig.5, predialysis). Furthermore, isopeptidase inhibition by Δ12-PGJ2 could not be reversed by dialysis of treated lysates (Fig. 5, postdialysis).

Reversibility of isopeptidase inhibition by Δ12-PGJ2. We treated cell lysates (0.5 mg/ml) with DMSO vehicle or 100 μM Δ12-PGJ2 for 1 h at 25°C. We divided each of the treated lysates into two aliquots. We immediately analyzed one aliquot for isopeptidase activity by incubating with z-LRGG-AMC as described previously (PreDialysis). We dialyzed the second aliquot by washing with 3 volumes of assay buffer on a centricon YM-30 column, followed by analysis for isopeptidase activity by incubating with z-LRGG-AMC (PostDialysis).
Inhibitors of Ubiquitin Isopeptidases Cause Cell Death Independently of Tumor Suppressor p53 Function.
Previously, we showed that electrophilic prostaglandins, typified by Δ12-PGJ2, inhibit p53-mediated transcription under the same conditions in which they cause cell death, suggesting that cell death occurs independently of p53 (Mullally et al., 2001). Therefore, integration of our pharmacophore and molecular mechanism hypotheses predicts that various isopeptidase inhibitors will cause cell death independently of tumor suppressor p53 function. Analysis of the NCI 60 cell line cancer screening data, according to O'Connor et al. (1997), showed that NCI-302979 and curcumin (NSC-32982) act independently of p53 (Table 1). NSC-156236 had no appreciable cytotoxic activity. Thus, data on NSC-302979, NSC-32892, and NSC-156236, available from the DTP public database, fulfill the minimal, initial prediction of our hypotheses.
Mean LC50 of NCI DTP database compounds examined
We confirmed that isopeptidase inhibitors caused cell death independently of p53 by investigating their effects on two pairs of isogenic colon cancer cell lines. Isogenic HCT 116+/+ and HCT 116−/− cell lines have varying degrees of p53 haplosufficiency, p53+/+ and p53−/−, respectively (Bunz et al., 1999). NSC-302979, DBA, Δ12-PGJ2, and curcumin each caused cell death with equal potency (concentration for half-maximal effect) and efficacy (maximal effect) in HCT 116+/+ cells that are homozygous for p53 and HCT 116−/− cells that are null for p53 (Fig.6, right hand). NSC-156236, PGB1, and DPTP, which did not inhibit ubiquitin isopeptidase activity at concentrations <60 μM did not cause significant cell death in HCT 116+/+ or HCT 116−/− cells. Isogenic RKO and RKO-E6 cells accumulate p53 to varying degrees after genomic stress due to the enhancement of p53 ubiquitination by HPV-E6. NSC-302979, DBA, Δ12-PGJ2, and curcumin each caused cell death with equal potency and efficacy in RKO and RKO-E6 cells (Fig. 6, left). NSC-156236 PGB1, and DPTP did not cause significant cell death in RKO or RKO-E6 cells.

Cytotoxicity of the pharmacophore test panel in HCT 116 and RKO cell lines with different p53 status. We incubated RKO and RKO-E6 cells (left column), and HCT116 p53+/+ and HCT116 p53−/− cells (right column) with vehicle or 0.5–60 μM NSC-302979, DBA, Δ12-PGJ2, NSC-156236, PGB1, curcumin, or DPTP for 48 h at 37°C. We measured cell viability with MTT reagent as described under Materials and Methods. Data are percentage of control viability, mean ± S.D. (n = 4).
As procedural controls and for calibration, we evaluated etoposide and paclitaxel on these same pairs of cells. Etoposide typifies agents that cause cell death via a p53-dependent pathway (Lowe et al., 1993). Accordingly, its potency in HCT 116−/− cells was ∼4-fold less than its potency in HCT 116+/+ cells (Fig.7, top right). Similarly, etoposide potency in RKO-E6 cells was ∼3-fold less than its potency in RKO cells (Fig. 7, top left). Paclitaxel typifies an agent that causes cell death via a p53-independent pathway (O'Connor et al., 1997). Although its efficacy (maximal effect observed) in the HCT 116−/− and RKO-E6 cells with dysfunctional p53 exceeded its efficacy in the corresponding HCT 116+/+ and RKO cells, the potency of paclitaxel (concentration for half-maximal effect) was equivalent in HCT 116−/− compared with HCT 116+/+ cells, as well as in RKO-E6 compared with RKO cells (Fig. 7, middle). Lastly, we evaluated MG115, an inhibitor of the 20S catalytic subunit of the proteasome, to compare its effects with isopeptidase inhibitors. There are conflicting reports on the role of p53 in cell death caused by proteasome inhibitors like MG115 (Dietrich et al., 1996; Shinohara et al., 1996; Lopes et al., 1997;Adams et al., 1999; Wagenknecht et al., 1999; An et al., 2000). MG115 caused cell death with equal potency and efficacy in HCT 116+/+ cells versus HCT 116−/− and in RKO-E6 cells versus RKO cells, analogous to isopeptidase inhibitors (Fig. 7, bottom). Table2 summarizes the potency of all compounds as cell death agonists in both pairs of cell lines.
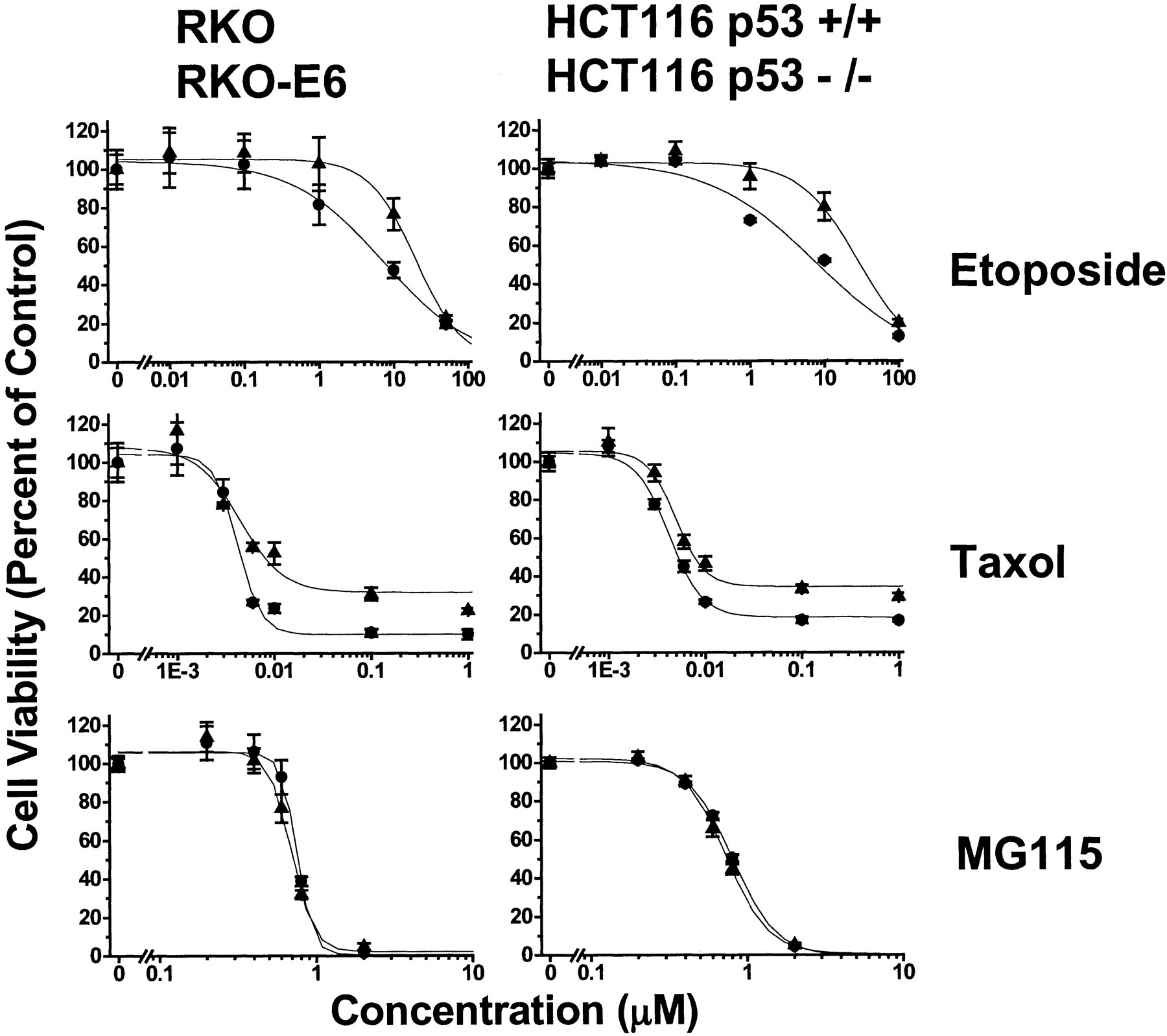
Cytotoxicity of the calibration set in HCT 116 and RKO cell lines with different p53 status. We incubated RKO and RKO-E6 cells (left column), and HCT116 p53+/+ and HCT116 p53−/− cells (right column) with vehicle, 0.01 to 100 μM etoposide, 0.001 to 1 μM paclitaxel, or 0.02 to 2 μM MG115 for 48 h at 37°C and determined their effect on cell viability, as described under Materials and Methods.
Cytotoxic potency of pharmacophore panel and calibration compounds in HCT 116 and RKO cells with different p53 status
Discussion
Our results indicate that nonprostanoid classes of compounds, with α,β-unsaturated ketones and two sterically accessible β-carbons, will inhibit ubiquitin isopeptidase activity. We further demonstrate that these compounds cause cell death independently of p53 tumor suppressor function in vitro. Specifically, the diterpene NSC-302979, the synthetic compound DBA, the prostaglandin Δ12-PGJ2, and the curcuminoid curcumin all cause cell death with efficacy and potency that is indistinguishable (p > 0.05) between HCT 116 p53+/+ and HCT 116 p53−/−or RKO and RKO-E6 cells. Furthermore, cell death correlated with inhibition of isopeptidase activity. Regression analysis (IC50 for inhibition of z-LRGG-AMC hydrolysis by isopeptidase versus IC50 for cytotoxicity) yields a straight line with a correlation coefficient r2= 0.93 (n = 7). Regression analysis (IC50 for inhibition of ubiquitin-PEST hydrolysis by isopeptidase versus IC50 for cytotoxicity) also yields a straight line with r2 = 0.73 (n = 13).
Inhibition of ubiquitin isopeptidase activity probably propagates cell death by shifting the polyubiquitin chain length equilibrium to one of greater molecular mass. As a consequence of unfettered polyubiquitin chain growth, the pool of monoubiquitin diminishes. Alteration of monoubiquitin/polyubiquitin dynamics inevitably affects several transcription factors other than p53 (Desterro et al., 2000). Furthermore, with depleted monoubiquitin pools, cells are hampered in their efforts to rid themselves of damaged/toxic proteins, eventually affecting protein-protein or protein-DNA interactions that modulate cell survival and apoptosis. Although our data support a covalent mechanism, we are presently investigating whether α,β-unsaturated dienones covalently inhibit isopeptidases, specifically, via their electrophilic β-carbons (e.g., Michael adduct formation between an isopeptidase cysteine residue and the β-carbon of a dienone). Note that compounds with sterically inaccessible or inert β-carbons (NSC-156236, PGB1, and DPTP) were inactive as isopeptidase inhibitors.
We used the substructure search capabilities of the NCI DTP database (60 cell-line screen) to identify NSC-302979 and NSC-156236, compounds used to test our pharmacophore and mechanism of action hypotheses. We think our results, along with the results by NCI scientists (O'Connor et al., 1997; Weinstein et al., 1997; Shi et al., 1998), exemplify the potential of this database and compound repository and the foresight of the NCI Developmental Therapeutics Branch. Others have suggested that the database content is misaligned with the goal to discover new anticancer drugs, based on a poor correlation between clonogenic survival and the NCI archival antiproliferative activity (Brown, 1997). However, direct extension of data acquired in vitro to clinical situations in vivo is rarely straightforward. Used prudently, to enable or to advance mechanistic and pharmacophore hypotheses, the database supports the quest for anticancer drugs with novel structures and mechanisms of action.
Our mechanistic and pharmacophore hypotheses are compatible with the structure-activity relationships reported by Kato et al. (1986), Sasaki et al. (1991) and Sasaki and Fukushima (1994). Kato et al. (1986)reported that Δ12-PGJ2 and several related Δ7-PGA1 derivatives (all of which are cross-conjugated dienones) increased the life span of Ehrlich ascites tumor-bearing mice: i.p. doses of 20 to 30 mg/kg/day for 5 consecutive days prolonged survival 66 to 111%. In addition, both Δ12-PGJ2 and Δ7-PGA1exhibit little cross-resistance with cisplatin and doxorubicin in vivo (Sasaki et al., 1991; Sasaki and Fukushima, 1994). Despite these promising results, Δ7-PGA1 is rapidly metabolized to an inactive compound (t 1/2 < 5 min) in serum (Suzuki et al., 1998). Therefore, our discovery of isopeptidase inhibitors among chemical classes other than PG might be advantageous in surmounting any difficulties intrinsic to the antineoplastic development of the PG class.
There exists considerable debate as to whether agents that inhibit the proteasome pathway cause cell death via a p53-independent process (Dietrich et al., 1996; Shinohara et al., 1996; Lopes et al., 1997;Adams et al., 1999; Wagenknecht et al., 1999; An et al., 2000). Our results with inhibitors of ubiquitin isopeptidase activity and with a representative catalytic subunit inhibitor of the 20S proteasome accord with those who conclude that proteasome inhibition causes apoptosis independently of p53. This debate may originate from faulty assumptions about the competence of p53 that accumulates in cells treated with proteasome pathway inhibitors. For instance, genetically wild-type p53 accumulates in the presence of the isopeptidase inhibitor Δ12-PGJ2, but in a conformationally and functionally impaired state (Moos et al., 2000; Mullally et al., 2001).An et al. (2000) have also reported that accumulation of wild-type p53 protein and induction of apoptosis occur as independent markers of proteasome inhibition. Therefore, one must use caution when interpreting the consequences of p53 accumulation without first testing its functionality.
The response to chemotherapy is complex; focus on a single factor, no matter how prominent, may exaggerate its role. However, numerous investigations show that disruption of p53 impairs the potency and efficacy of drugs used in oncology [e.g., 5-fluorouracil (Lowe et al., 1994, 1995; Mueller and Eppenberger, 1996; O'Connor et al., 1997; Bunz et al., 1999; Pich, 1998; Weller, 1998; Karpf et al., 2001)]. It is notable that vinca alkaloids, one of the few drug classes that act independently of p53 (O'Connor et al., 1997; Fan et al., 1998), may target the proteasome in addition to tubulin (Piccinini et al., 2001). LDP-341, the first proteasome inhibitor to enter clinical trials, seems to have a favorable safety and efficacy profile (Dalton et al., 2001). Clinical studies to evaluate proteasome inhibition as an adjuvant to systemic chemotherapy are also currently in development (Cusack et al., 2001). Our results demonstrate that another component of the proteasome pathway, isopeptidase activity, warrants further investigation as a target for antineoplastic drug discovery.
Acknowledgments
We thank Dr. Martin Rechsteiner and Greg Pratt (University of Utah) for helpful advice and ubiquitin-PEST reagent; the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute for compounds NSC-156236 and NSC-302979; Dr. Mark Meuth (University of Sheffield, UK) for RKO and RKO-E6 cells; and Dr. Bert Vogelstein (Johns Hopkins School of Medicine) for isogenic HCT 116 cell lines.
Footnotes
- Received January 14, 2002.
- Accepted May 14, 2002.
-
This work was supported by United States Public Health Service grant RO1-AI26730. F.A.F. is an investigator of the Huntsman Cancer Institute and the Dee Glenn and Ida W. Smith Chair for Cancer Research.
Abbreviations
- PG
- prostaglandin
- DBA
- dibenzylideneacetone
- DMEM
- Dulbecco's minimum essential medium
- NSC-302979
- shikoccin
- NSC-156236
- achillin
- MG115
- carbobenzyloxy-l-leucyl-l-leucyl-norvaline
- z-LLVY-MCA
- succinyl-l-leucyl-l-leucyl-l-valyl-l-tyrosine α-(4-methyl-coumaryl-7-amide)
- z-LRGG-MCA
- carbobenzoxy-l-leucyl-l-arginyl-l-glycyl-l-glycine α-(4-methyl-coumaryl-7-amide)
- MTT
- (3-(4,5-dimethylthiazo)-2-yl)-2,5-diphenyltetrazolium bromide
- PAGE
- polyacrylamide gel electrophoresis
- UB
- ubiquitin
- CHAPS
- 3-[(3-cholamidopropyl)dimethylammonio]propanesulfonate
- NCI DTP
- National Cancer Institute Developmental Therapeutics Program
- DPTP
- 2,6-diphenyl-4H-thiopyran-4-one
- NSC-32982
- curcumin
- DMSO
- dimethyl sulfoxide
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}