


Abstract
Diltiazem block of Cav1.2 is frequency-dependent and potentiated by Ca2+. We examined the molecular determinants of these characteristics using mutations that affect Ca2+ interactions with Cav1.2. Mutant and wild-type (WT) Cav1.2 channels were transiently expressed in tsA 201 cells with β1b and α2δ subunits. The four conserved glutamates that compose the Ca2+ selectivity filter in Cav1.2 were mutated to Gln (E363Q, E709Q, E1118Q, E1419Q), and each single mutant was assayed for block by diltiazem using whole-cell voltage-clamp recordings in either 10 mM Ba2+ or 10 mM Ca2+. In Ba2+, none of the mutations affected the potency of diltiazem block of closed channels (0.05 Hz stimulation). However, frequency-dependent block (1Hz stimulation) was eliminated in the mutant E1419Q (domain IV), which recovered more rapidly than WT channels from inactivated channel block. Potentiation of diltiazem block of closed Cav1.2 channels in Ca2+ was abolished in the E1118Q, F1117G (domain III), and E1419Q mutants. Frequency-dependent block in Ca2+ was reduced compared with WT Cav1.2 in the F1117G, E1118Q, and E1419Q mutants. The C-terminal tail IQ domain mutation I1627A, which disrupts Ca2+ dependent inactivation, enhanced diltiazem block of closed channels in Ba2+. We conclude that, in Ba2+, E1419 slows recovery from diltiazem block of depolarized Cav1.2 channels, but in Ca2+, E1118, E1419, and F1117 form a Ca2+ binding site that mediates the potentiation of diltiazem block of both closed and inactivated Cav1.2 channels. Furthermore, Ca2+-dependent inactivation, which is impaired in E709Q, E1118Q, E1419Q, and I1627A, is not required for Ca2+ potentiation of diltiazem block.
Ca2+ influx via voltage-dependent L-type Ca2+ channels (α1C, Cav1.2) found in cardiac and vascular smooth muscle initiates contraction and contributes to timing of the cardiac action potential (Bers and Perez-Reyes, 1999). Cav1.2 channels consist of an α1 pore-forming subunit and α2δ, β, and γ auxiliary subunits (Jones, 1998). The α1 subunit is composed of four homologous domains (I–IV), each of which has six transmembrane segments (S1–S6) (Takahashi et al., 1987; Tanabe et al., 1987). Each domain also includes a pore-lining region, between each S5 and S6 segment, each of which contains a conserved Glu residue (EI, EII, EIII, EIV) that contributes to the Ca2+-selectivity filter (Yang et al., 1993). In addition, the Cav1.2 α1 subunit contains an IQ calmodulin binding motif in the intracellular C-terminal tail, which is critical for Ca2+-dependent channel inactivation (Peterson et al., 1999; Zuhlke et al., 1999). Cav1.2 is sensitive to block by three distinct chemical classes of small-molecule drugs: dihydropyridines (DHPs), phenylalkylamines (PAAs), and benzothiazepines (BZPs) (Hockerman et al., 1997b). All three classes are effective vasodilators, and are therefore used to treat hypertension and angina pectoris (Fleckenstein and Fleckenstein-Grun, 1980). The PAA verapamil and the BZP diltiazem are also used to treat some types of supraventricular arrhythmia since they preferentially block channels that are activated at high frequency, a property known as frequency-dependent block (Lee and Tsien, 1983).
Diltiazem blocks Cav1.2 channels at low micromolar concentrations in both primary cardiac myocytes (Lee and Tsien, 1983) and heterologous expression systems (Hockerman et al., 2000). Based on studies using a quaternary amine derivative, BZPs are thought to approach their binding site on Cav1.2 from the extracellular face of the plasma membrane (Hering et al., 1993). Studies using both point mutants and chimeric channels agree that closed-channel block by diltiazem involves specific amino acid residues in segments IIIS6 and IVS6 (Hering et al., 1996; Kraus et al., 1998; Berjukow et al., 1999; Hockerman et al., 2000). Mutation of amino acid residues in IVS6 involved in closed-channel block by diltiazem also decreased the accumulation of block during a 1-Hz train of depolarization (Berjukow et al., 1999). Chimeric channels with clusters of amino acid substitutions in transmembrane segment IVS5 of Cav1.2 prevented frequency-dependent block by diltiazem (Motoike et al., 1999) but also severely disrupted voltage-dependent inactivation.
Mutation of EIII or EIV to Gln in Cav1.2 was reported to decrease affinity of the PAA D888 in Ba2+, suggesting an interaction between the positively charged amino group in the D888 molecule and the negatively charged Glu residues at physiological pH (Hockerman et al., 1997a). Since diltiazem also contains an essential ionizable amino group, we assayed Cav1.2 channels with single-amino acid mutations of each of the conserved pore Glu residues to Gln [E363Q (EIQ), E709Q (EIIQ), E1118Q (EIIIQ), and E1419Q (EIVQ)] for closed-channel block and frequency-dependent block by diltiazem in Ba2+. Diltiazem block of closed channels in Ba2+ was not significantly affected by any of the E to Q mutations. However, frequency-dependent block of Cav1.2 Ba2+ current was virtually eliminated in the mutant EIVQ channel.
The conserved glutamates in the pore region of Cav1.2 bind Ca2+ ions as they traverse the channel pore, thus forming a selectivity filter that largely excludes monovalent cations when Ca2+ is present (Yang et al., 1993). However, other divalent ions such as Ba2+ can pass through this selectivity filter (Hille, 1995). Indeed, the conductance of Cav1.2 is higher for Ba2+ than for Ca2+ since Ca2+ binds more tightly to the pore glutamates (Almers and McCleskey, 1984). Another consequence of Ca2+ binding in the pore is modulation of drug binding sites. All three chemical classes of L-type channel blockers are more potent when Ca2+, rather than Ba2+, is the permeant ion (Lee and Tsien, 1983). The conserved Glu residues in domains III and IV, as well as an adjacent Phe in the pore region of domain III in Cav1.1, mediate the Ca2+ potentiation of DHP affinity (Peterson and Catterall, 1995). In addition, Ca2+ also modulates Cav1.2 channel inactivation via a Ca2+/calmodulin binding domain (IQ motif) in the C-terminal tail (Peterson et al., 1999; Zuhlke et al., 1999). We report here that the EIIIQ, EIVQ, and F1117G mutations each abolish Ca2+ potentiation of both closed-channel and frequency-dependent diltiazem block. In contrast, the IQ-domain mutation I1627A potentiated diltiazem block of closed channels in Ba2+. Furthermore, Ca2+-dependent inactivation, which is markedly reduced in the mutant channels EIIQ, EIIIQ, EIVQ, and I1627A, is not required for the Ca2+ potentiation of diltiazem block.
Materials and Methods
Construction of Wild-Type and Mutant Ca2+Channels. The Cav1.2 (Snutch et al., 1991) and the mutant channels were cloned into pCDNA3 expression vector (Invitrogen, Carlsbad, CA). Mutations were introduced as described (Hockerman et al., 1997a), or using a modification of the QuikChange site-directed mutagenesis procedure (Stratagene, La Jolla, CA). The desired mutations were verified, and the integrity of the clones was confirmed by cDNA sequencing and extensive restriction digest analysis.
Cell Culture. Human tsA-201 cells, a simian virus 40 T-antigen-expressing derivative of the human embryonic kidney cell line HEK293, were maintained in monolayer culture in Dulbecco's modified Eagle's medium/F-12 (Invitrogen) enriched with 10% fetal bovine serum (Hyclone Laboratories, Logan, UT) and incubated at 37°C in 10% CO2.
Expression of Ca2+Channels. tsA-201 cells were cotransfected with WT and mutant Cav1.2 subunits, β1b (Pragnell et al., 1991), α2δ (Ellis et al., 1988), and enhanced green fluorescent protein (GFP) (BD Biosciences Clontech, Palo Alto, CA) such that the molar ratio of the plasmids was 1:1:1:0.8. Cells were transfected using the Gene-Porter reagent (Gene Therapy Systems Inc., San Diego, CA) and cells were replated at low density for electrophysiological recording 20 to 24 h after transfection. Experiments were conducted 20 to 48 h after replating.
Electrophysiology. Transfected cells were recognized by GFP fluorescence at 510 nm with excitation at 480 nm. Barium and calcium currents through Ca2+ channels were recorded using the whole-cell configuration of the patch-clamp technique. Patch electrodes were pulled from VWR micropipettes (VWR, West Chester, PA) and fire-polished to produce an inner tip diameter of 4 to 6 μm. Currents were recorded using an Axon Instruments Inc. (Union City, CA) Axopatch 200B patch-clamp amplifier and filtered at 1 or 2 kHz (8-pole Bessel filter, -3 dB). Voltage pulses were applied and data were acquired using pClamp8 software (Axon Instruments Inc.). Voltage-dependent leak currents were subtracted using an on-line P/-4 subtraction paradigm. (+)-cis-Diltiazem, dissolved in bath saline, was applied to cells using a Biologic RSC 160 fast perfusion system (Molecular Kinetics, Pullman WA) with constant exchange of the bath solution. Barium current was measured in a bath saline containing Tris (150 mM), MgCl2 (2 mM) and BaCl2 (10 mM). Calcium current was measured in the same bath solution, except that Ba2+ was replaced with Ca2+ (10 mM). The intracellular saline contained N-methyl-d-glucamine (130 mM), EGTA (10 mM), HEPES (60 mM), MgATP (2 mM), and MgCl2 (1 mM). (+)-cis-Diltiazem (Fluka Chemical Corp., Ronkonkoma, NY) concentrations were made from a 100 mM stock solution in 70% ethanol. The pH of both solutions was adjusted to 7.3 with methanesulfonic acid. All experiments were performed at room temperature (20–23°C).
Data Analysis. Data were analyzed using Clampfit (Axon Instruments Inc.) and SigmaPlot (SPSS Science, Chicago, IL) software. Statistical significance was determined using Student's t test.
Results
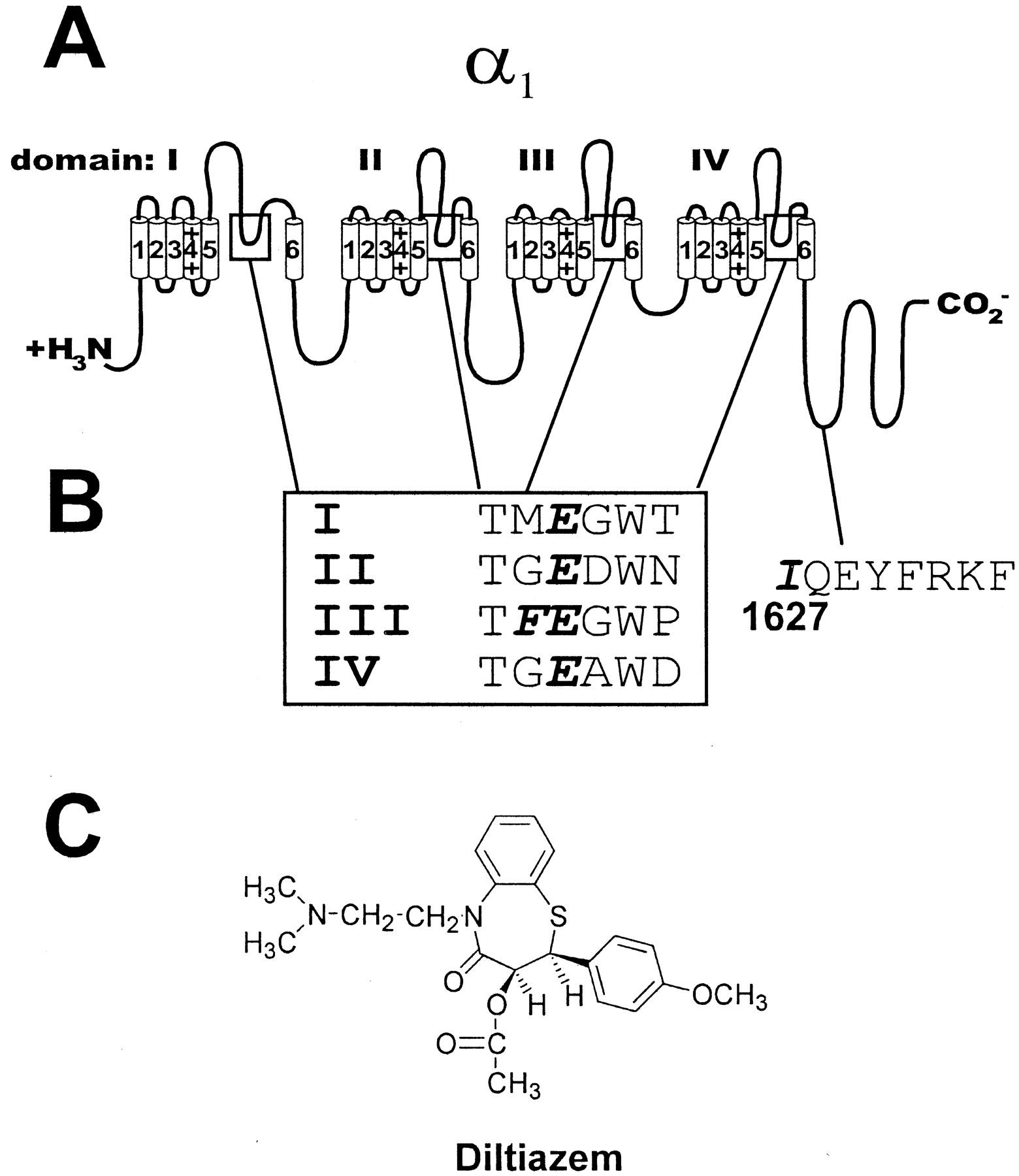
Structural Features and Sites of Mutations in Cav1.2.Figure 1, A and B, illustrates the positions of the amino acids that were mutated for this study. The amino acids E363, E709, E1118, and E1419 were mutated to Gln to create the mutant channels EIQ, EIIQ, EIIIQ, and EIVQ, respectively. As shown in Fig. 1B, the alignment of the pore residues in each domain indicates a high degree of conservation which is also retained across non-L-type channels (Jones, 1998). The structure of diltiazem (Fig. 1C) includes a tertiary amino group that is predominantly charged at physiological pH and could potentially interact with the acidic Glu side chains. The Phe residue at position 1117 in domain III is unique to L-type channels, whereas most non-L-type channels contain a Gly residue at this position. The mutation of the residue corresponding to F1117 to G in Cav1.1 disrupted Ca2+ potentiation of DHP binding (Peterson and Catterall, 1995), as did the EIIIQ and EIVQ mutation. The approximate position of I1627 in the Ca2+/calmodulin-binding IQ motif in the C-terminal tail is indicated. Mutation of I1627 to A disrupts Ca2+/calmodulin binding to the channel and, consequently, Ca2+-dependent acceleration of inactivation (Zuhlke et al., 1999). We examined these Cav1.2 mutants for diltiazem block and potentiation of diltiazem block by Ca2+.

Structural features of the α1 subunit of Cav1.2. A, topology of Cav1.2 (α1C). Cylinders represent transmembrane segments (1–6) organized into four homologous domains (I-IV). The C- and N-terminal domains are intracellular. Boxes highlight the putative pore-lining regions that contain the elements of the Ca2+ selectivity filter. B, the amino acid sequence surrounding the Glu residues in each homologous domain that compose the selectivity filter. The conserved Glu residues are in bold italic type, as is the L-type-specific Phe residue directly adjacent to the conserved Glu residue in domain III (box). The approximate location of Ile 1627 and elements of the IQ Ca2+/calmodulin binding domain are shown. C, the structure of diltiazem. Note the ionizable tertiary alkylamino group.
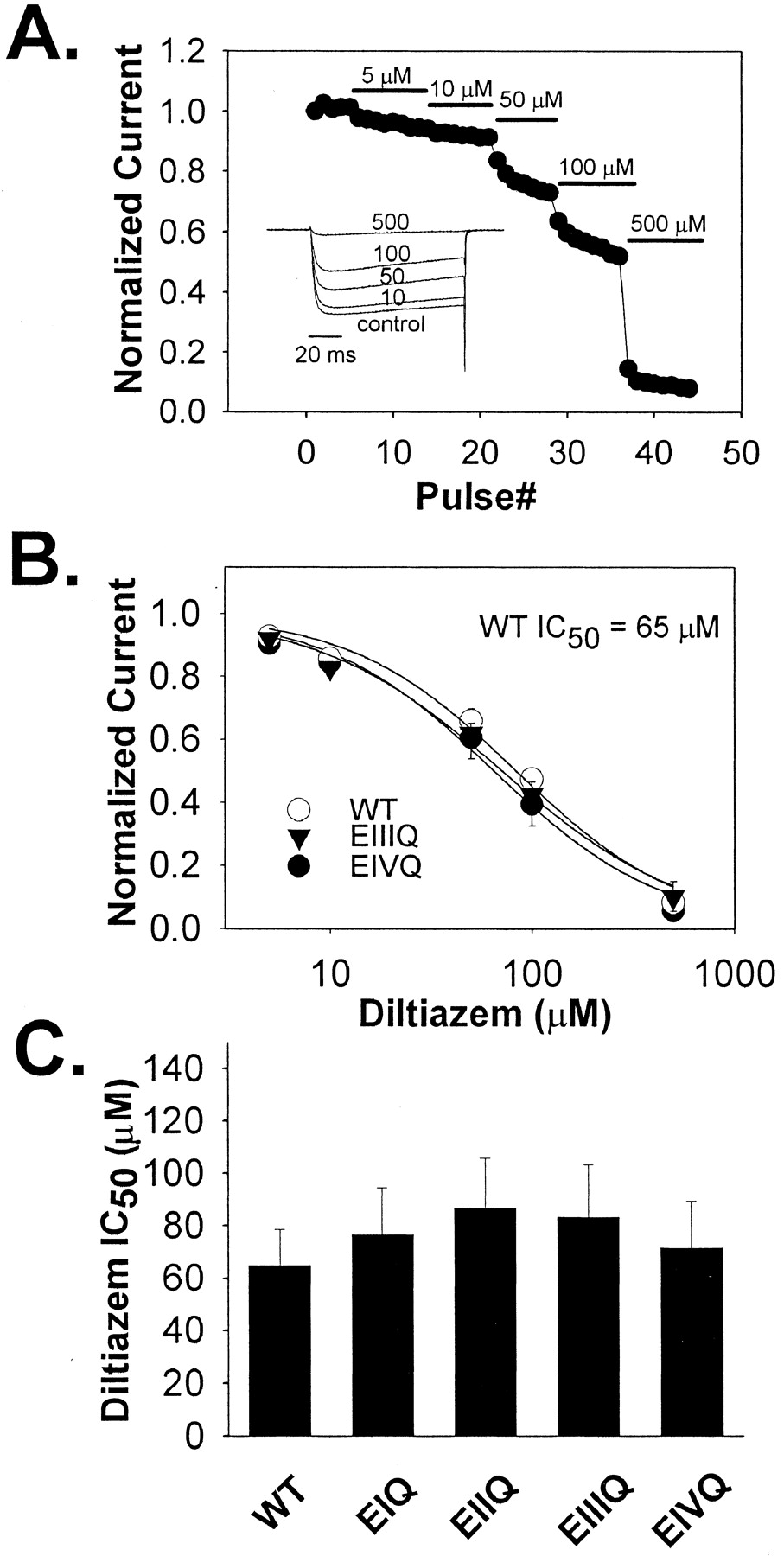
Closed-Channel Block of the E to Q Mutants by Diltiazem in Ba2+ WT and mutant EIQ, EIIQ, EIIQ, EIIIQ, and EIVQ Cav1.2 channels were coexpressed along with α2δ, β1b, and GFP in tsA-201 by transient transfection. Forty-eight hours after transfection, whole-cell Ba2+ currents were measured in the cells expressing the GFP marker. Closed-channel block was measured from a holding potential of -60 mV using 100-ms steps to +10 mV at 0.05 Hz. Increasing concentrations (5, 10, 50, 100, and 500 μM) of diltiazem were applied to cells thus stimulated. Under these conditions, block reached equilibrium rapidly, and little frequency-dependent block was detected (Fig. 2A). The diltiazem IC50 value for WT was 65.04 ± 13.60 μM, and the diltiazem IC50 values for EIQ, EIIQ, EIIIQ, and EIVQ were not significantly different (Fig. 2, B and C).
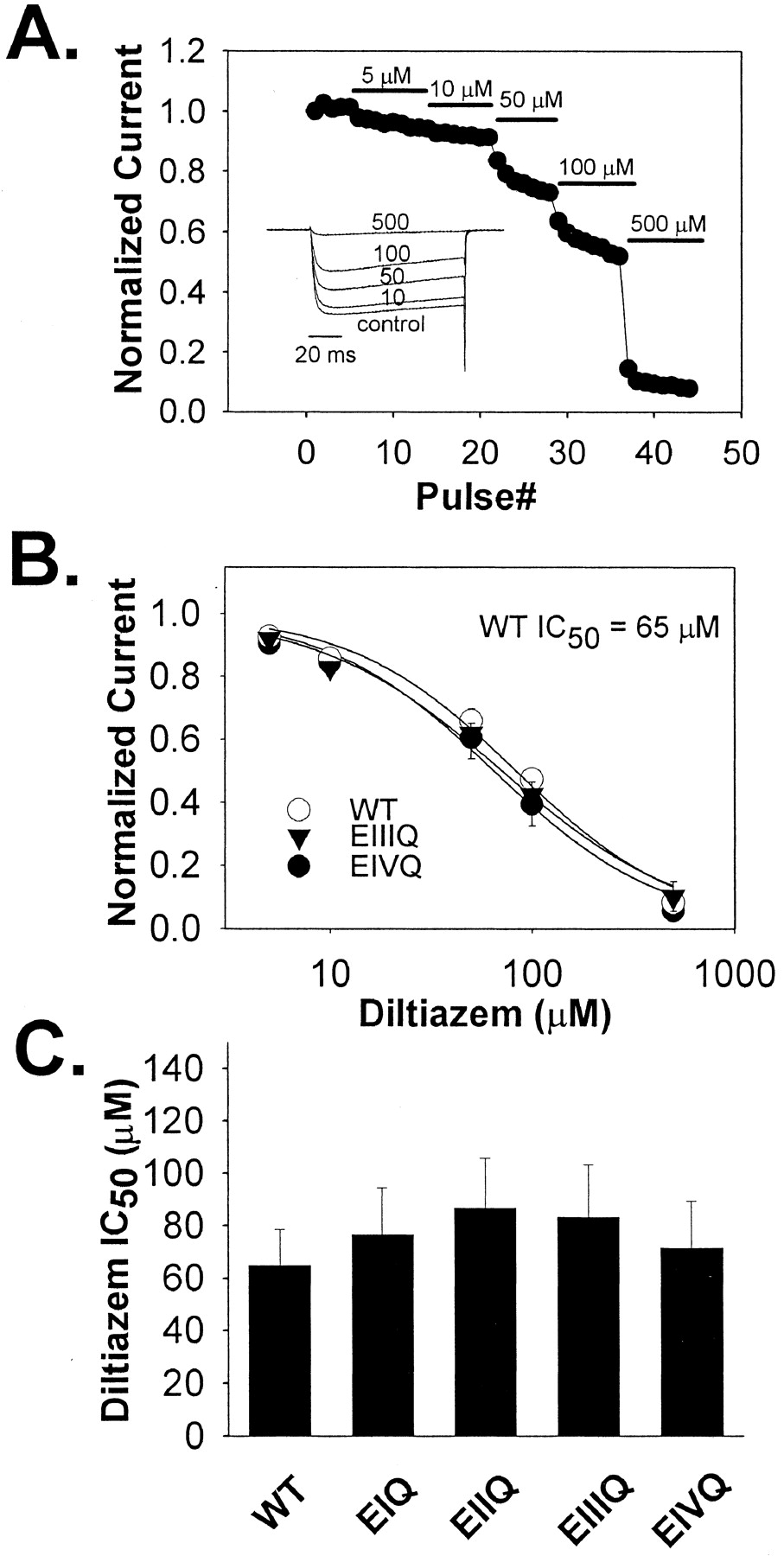
Diltiazem block of WT Cav1.2 and the mutant channels EIQ, EIIQ, EIIIQ, and EIVQ at 0.05 Hz in Ba2+. A, representative Ba2+ current records from a tsA-201 cell expressing wild-type Cav1.2 in the absence (control) or presence of the indicated concentrations of (+)-cis-diltiazem (inset). The time course of diltiazem block of Cav1.2 Ba2+ current is shown. Diltiazem was applied at the pulse number and concentrations indicated. Pulses were from a holding potential of -60 mV to +10 mV every 20 s. B, dose-response relationships for wild-type and the indicated mutant Cav1.2 channels. In each case, the averaged, normalized current amplitudes at 5, 10, 50, 100, and 500 μM diltiazem (symbols ± S.E., n = 3–4) were plotted against the corresponding drug concentration, and the IC50 value was determined by fitting the averaged relative current values at each diltiazem concentration to the equation, relative current = 1 - {1/[1+(IC50/[diltiazem])]} (smooth lines). C, the IC50 values of the indicated mutant channels are shown ± S.E. (n = 3–5). The values were: WT, Cav1.2 = 65.0 ± 13 μM; EIQ = 76.8 ± 17 μM; EIIQ = 86.8 ± 19 μM; EIIIQ = 83.4 ± 20 μM; EIVQ = 71.5 ± 18 μM. None of the mutant channels was significantly different from the WT Cav1.2 channel in sensitivity to block by diltiazem under these conditions.
Frequency-Dependent Block of E to Q Mutants in Ba2+ Since none of the E to Q mutants appeared to affect diltiazem block of closed channels, we next examined the effect of these mutations on frequency-dependent block. After diltiazem block (50 μM) reached equilibrium at 0.05 Hz as described above, we applied a 20-pulse, 1-Hz train of 100-ms depolarizations to +10 mV from a holding potential of -60 mV. The same 1-Hz train of depolarizations was also applied to cells before the application of diltiazem. The results of these experiments with WT Cav1.2 and each of the E to Q mutations are shown in Fig. 3. Frequency-dependent diltiazem block of WT Cav1.2 channels was not different from that of the mutant EIQ and EIIQ channels in 10 mM Ba2+ (Fig. 3A). Frequency-dependent diltiazem block by diltiazem was retained in the EIIIQ channel but was significantly reduced compared with WT Cav1.2 (Fig. 3B). In contrast, frequency-dependent diltiazem block was virtually eliminated in the EIVQ mutant, whereas inactivation at the end of the train of depolarizations in the absence of drug was not significantly different from that of WT (Fig. 3C). Thus, the conserved glutamate residues have distinct effects on the frequency-dependent block of diltiazem. In Fig. 3D, the specific diminution of frequency-dependent, but not closed-channel block in EIVQ is demonstrated in single cells. When 50 μM diltiazem is initially applied to either WT or EIVQ Cav1.2 channels stimulated at 0.05 Hz, the same fraction of current is blocked. However, when a 1-Hz train of depolarizations is applied ∼7 s after the initial application of diltiazem, the further decrease in current is substantially greater in WT Cav1.2 than in EIVQ. These data suggest that the EIV pore glutamate is required for frequency-dependent diltiazem block of Cav1.2 when Ba2+ is used as the permeant ion.

Diltiazem block of WT Cav1.2 and the mutant channels EIQ, EIIQ, EIIIQ, and EIVQ at 1 Hz in Ba2+. A–C, Whole-cell Ba2+ currents were recorded in the absence and presence of 50 μM diltiazem using depolarizations to +10 mV from a holding potential of -60 mV, at a frequency of 1 Hz. Current measured in the presence of diltiazem followed equilibration of block by 50 μM diltiazem at 0.05 Hz. Relative peak current (mean ± S.E.) in each successive depolarizing pulse is plotted against pulse number (n = 4–8) in the absence (closed symbols) or presence (open symbols) of 50 μM diltiazem for WT channels and the indicated mutants. Asterisk indicates that the fraction of current remaining at the end of the 20-pulse, 1-Hz train was significantly different from that of WT Cav1.2 channels (p < 0.05). D, demonstration of differential effects of the EIVQ mutation on diltiazem block of Cav1.2 at 0.05 and 1 Hz in single cells. Ba2+ current in cells expressing either WT Cav1.2 or the EIVQ mutant was brought to equilibrium as measured using stimulation at 0.05 Hz as described above. At time 0, 50 μM diltiazem was perfused onto the cells. Approximately 7 s after the initiation of drug perfusion, a 20-pulse, 1-Hz train of depolarization was applied (as described above). The fraction of current blocked at the first depolarization after initiation of drug perfusion is virtually identical in the two cells, but subsequent development of block at 1 Hz is greatly reduced in EIVQ compared with WT Cav1.2.
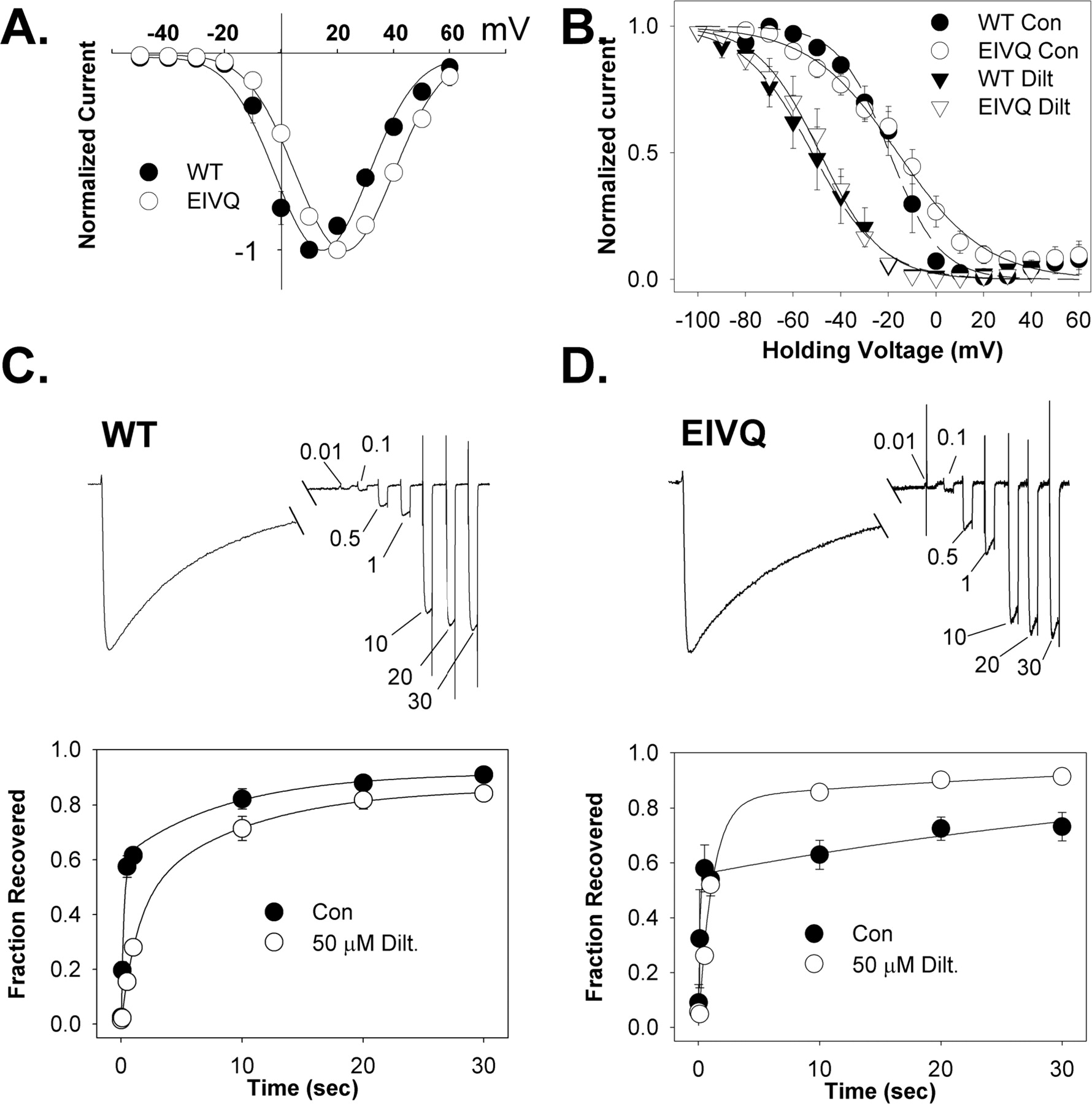
Kinetic Analysis of Depolarized Channel Block of WT and E to Q Mutants. To understand the drastic reduction of frequency-dependent diltiazem block in the EIVQ mutant, we examined some of the voltage-dependent properties of the channel, as well as some kinetic parameters of diltiazem block. In Fig. 4A, the current-voltage relationship for EIVQ is compared with that of WT Cav1.2. Although channel activation and the peak of the IV curve is shifted to more positive potentials for EIVQ, the fraction of channels activated at +10 mV is not greatly different from WT and is not likely to account for the sharp decrease in frequency-dependent block. In Fig. 4B, the voltage dependence of inactivation for both WT Cav1.2 and EIVQ is shown in the absence and presence of 50 μM diltiazem. Despite a slight (∼3 mV) positive shift in the V1/2 inactivation for EIVQ compared with WT, the shift in V1/2 inactivation induced by 50 μM diltiazem is virtually identical in both channels (∼32 mV). Thus, diltiazem interacts with inactivated WT and EIVQ Cav1.2 channels to a similar extent. Recovery from depolarized channel block was also examined for WT and EIVQ channels (Fig. 4, C and D). Whole-cell Ba2+ current was measured by depolarizing cells to +10 mV for 1 s followed by recovery intervals of 0.01, 0.10, 0.50, 1, 10, 20, and 30 s at -60 mV. At the end of each recovery period, recovered current was measured by stepping to +10 mV for 50 ms. This protocol was applied in the absence and presence of 50 μM diltiazem. When performed in the presence of drug, the protocol was applied following equilibration of drug block at 0.05 Hz. As shown in Fig. 4C, recovery of WT channels from inactivation in the absence of drug follows a biexponential time course, reflecting a fast and a slow time course of recovery from inactivation as reported previously for Cav1.2. (Johnson et al., 1996). In the absence of diltiazem, the fraction of current recovering with the fast time constant (fF) was 0.61, with a fast (τf) and slow (τs) time constant of 0.235 s and 8.8 s, respectively. In the presence of 50 μM diltiazem, the fast phase of WT Cav1.2 recovery is substantially slowed (τf = 1.3 s, fF = 0.43) and the time constant of slow recovery is virtually unchanged (τs = 9.1 s). The recovery of EIVQ from inactivation in the absence of diltiazem was also biexponential (fF = 0.56, τf = 0.11 s, τs = 51.9 s; Fig. 4D). However, the recovery of EIVQ from inactivation in 50 μM diltiazem was much more rapid than in WT Cav1.2. Rather than a marked difference in the fast time constant of recovery (τF = 1.1 s; τS = 42.7 s) compared with WT, the acceleration of recovery in EIVQ is likely due to an increase in the fraction of channels recovering with a fast time constant (fF = 0.83). This large increase in fF leads to a “crossover” in the recovery time course, such that after 1 s, the EIVQ channel recovers from inactivation faster in the presence of diltiazem than in its absence.

Biophysical properties of the mutant Cav1.2 channel EIVQ. A, current-voltage relationship for WT Cav1.2 and EIVQ channels. Peak amplitude of Ba2+ current elicited by a 100-ms depolarization to the indicated test potential from a holding potential of -60 mV is plotted against the test potential (mean ± S.E., n = 4–8). B, voltage dependence of inactivation in WT Cav1.2 and EIVQ channels. Peak Ba2+ current elicited by depolarization to +10 mV immediately after 10-s conditioning pulses to the indicated potentials from a holding potential of -80 mV are plotted against the amplitude conditioning pulse voltage (mean ± S.E., n = 3–8) in the presence or absence of 50 μM diltiazem. A 200-s delay was incorporated between each conditioning voltage to allow full recovery of drug-bound channels. The data are fit (smooth lines, WT; broken lines, EIVQ) to the equation, relative current = 1/{1 + exp[(V - V1/2)/k]}, where V is the conditioning potential, V1/2 is voltage at which half of the channels are inactivated, and k is a slope factor (potential required for an e-fold change). For WT Cav1.2, V1/2 in the absence and presence of 50 μM diltiazem was -19.5 ± 1.6 and -51.9 ± 1.5 mV, respectively. For EIVQ, V1/2 in the absence and presence of 50 μM diltiazem was -16.1 ± 2.4 and -48.8 ± 1.3 mV, respectively. Values for k were -10.6 and -14.5 for WT Cav1.2 in the absence and presence of drug, respectively. For EIVQ, k values were -18.6 and -13.0 in the absence and presence of 50 μM diltiazem, respectively. C and D, recovery of WT (C) and EIVQ (D) Cav1.2 channels from inactivation in the presence and absence of 50 μM diltiazem. Recovery from inactivation induced by a 1-s depolarization to +10 mV from a holding potential of -60 mV was measured using 50-ms test pulses to +10 mV after various recovery intervals (0.01, 0.1, 0.5, 1, 10, 20, and 30 s) at -60 mV. Representative traces are shown for both WT and EIVQ (C and D, upper panels). The time points between 300 and 900 ms of the depolarizing pulse are omitted, and the test pulses during the recovery phase are compressed for display purposes. The recovery time before each test pulse is indicated in seconds. The fraction of current recovered is plotted against the recovery interval (mean ± S.E., n = 4) (C and D, lower panels). In both cases, the time course of recovery was well fitted with a double-exponential equation. For WT Cav1.2 channels in the absence of diltiazem, the fast time constant of recovery (τF) was 235 ms, the slow time constant of recovery (τS) was 8.8 s, and the fraction of channels recovering with the fast time constant (fF) was 0.61. In the presence of 50 μM diltiazem, τF = 1.3 s, τS = 9.1 s, and fF = 0.43. For EIVQ in the absence of diltiazem, τf = 110 ms, τs = 51.9 s, and fF = 0.56. For EIVQ in the presence of 50 μM diltiazem, τF = 1.1 s, τS = 42.7 s, and fF = 0.83.
We also examined the effect of the EQ mutations on diltiazem block rate (Fig. 5). WT and mutant channels were depolarized for1sto +10 mV from a holding potential of -60 mV, in the presence or absence of 50 μM diltiazem. The resulting current traces (see Fig. 5A) were normalized to peak current, to facilitate comparison of the rate of current decay in the absence and presence of diltiazem. As shown in Fig. 5B, the rate of current inactivation in individual cells expressing either WT or EIVQ channels was fit to either single- or double-exponential equations. The time constants and fraction of channels inactivating with a given time constant are shown in Fig. 5B. Inactivation of WT channels was biexponential, both in the presence and absence of diltiazem. The fast time constant (τ-fast) for WT channels was not different in the presence or absence of 50 μM diltiazem (0.15 ± 0.01 s and 0.12 ± 0.02 s, respectively); however, the fraction of channels inactivating with the fast time constant was significantly increased in the presence of diltiazem. The slow time constant of WT channel inactivation was accelerated significantly in the presence of diltiazem (τ-slow = 0.66 ± 0.03 s versus 0.42 ± 09 s).
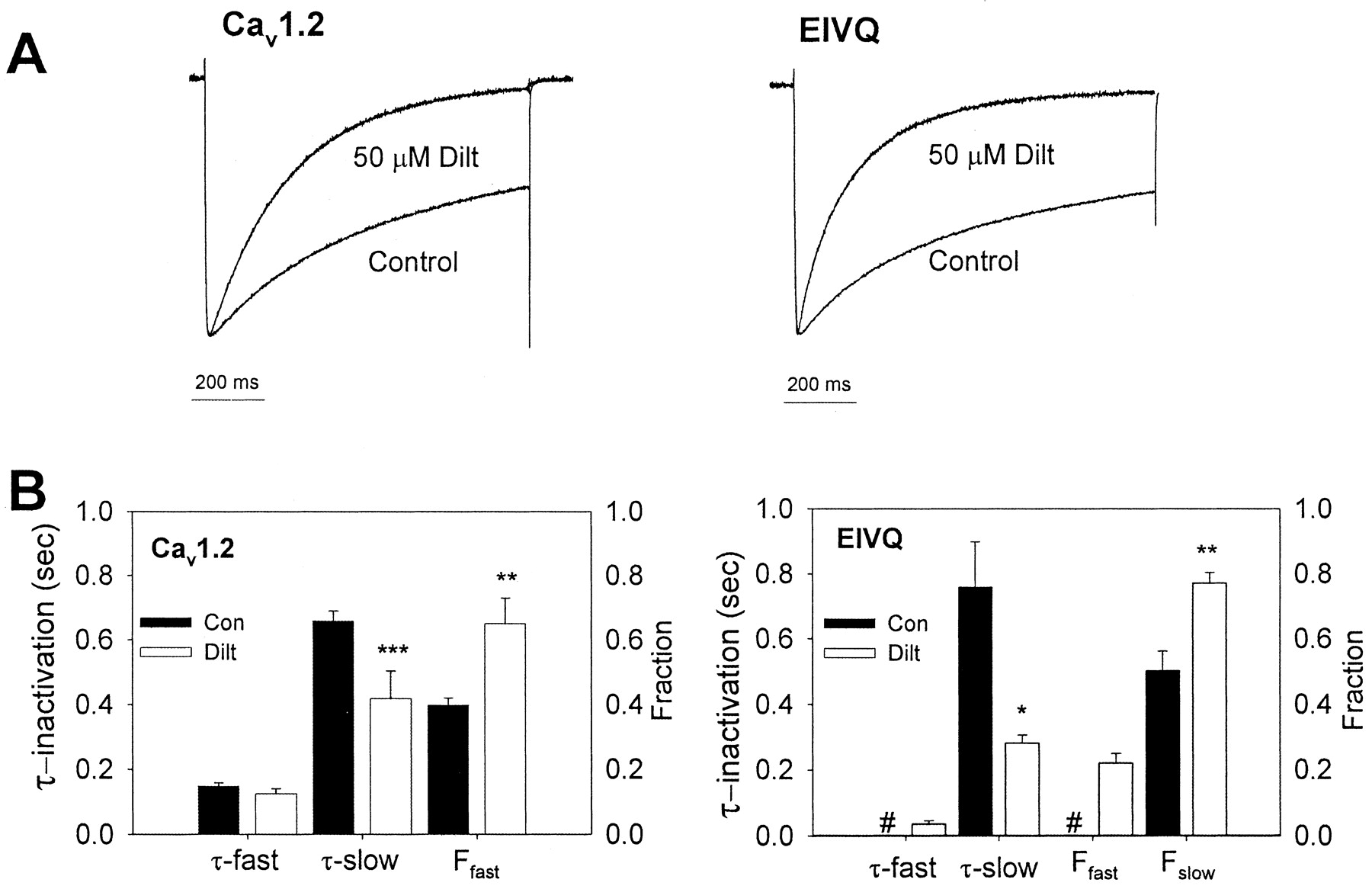
Time course of diltiazem block of depolarized channels. A, representative traces of Ba2+ current through WT Cav1.2 and EIVQ channels measured during a 1-s depolarization to +10 mV from a holding potential of -60 mV in the absence (control) or presence of 50 μM diltiazem (normalized to peak current). B, analysis of inactivation in the absence (Con, filled bars) and the presence (Dilt, open bars) of 50 μM diltiazem for Cav1.2 (left panel) and EIVQ (right panel). In the absence of drug, Cav1.2 inactivation is well fit by a double exponential [τ-fast = 0.15 ± 0.01 s; τ-slow = 0.66 ± 0.03 s; fraction fast (Ffast) = 0.40 ± 0.02]. In the presence of 50 μM diltiazem, τ-fast is not changed (0.12 ± 0.02 s), but τ-slow is accelerated (0.42 ± 0.09 s), and Ffast is increased (0.65 ± 0.08). In the absence of drug, EIVQ channel activation is well fit by a single-exponential function with a time constant similar to τ-slow in WT channels (τ-slow = 0.76 ± 0.14; Fslow = 0.50 ± 0.06). In the presence of 50 μM diltiazem, EIVQ channels display an additional fast component of inactivation (τ-fast = 0.04 ± 0.01 s; Ffast = 0.22 ± 0.03), τ-slow is accelerated, and the fraction channel inactivating with the slow time course is increased (τ-slow = 0.28 ± 0.02; Fslow = 0.77 ± 0.03). Results are means ± S.E. of individual fits (n = 4–6). #, not detected in EIVQ channels. Asterisks indicate significant differences between the indicated parameter in the presence and absence of 50 μM diltiazem (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
In contrast, the inactivation of EIVQ channels followed a single-exponential time course (Fig. 5B) in the absence of diltiazem (τ = 0.76 ± 0.14 s) but a double-exponential time course in the presence of diltiazem (τ-fast = 0.04 ± 0.01 s). The slow time constant was significantly accelerated (τ-slow = 0.28 ± 0.02 s) relative to the time constant of EIVQ channel inactivation in the absence of diltiazem. In addition, diltiazem also significantly increased the fraction of EIVQ channels inactivating with the slower time constant relative to EIVQ channels in the absence of drug. Thus, diltiazem blocks both depolarized WT and EIVQ channels by increasing the fraction of channels inactivating and by decreasing the slow time constant of inactivation.
Ca2+Potentiation of Diltiazem Block. Diltiazem block of Cav1.2 is potentiated by Ca2+ (Lee and Tsien, 1983). This property is demonstrated in Fig. 6, top panel, for WT Cav1.2. Using a concentration of diltiazem (50 μM) near the IC50 for block of Cav1.2 in Ba2+, it is clear that more channels are blocked when Ca2+, rather than Ba2+, is the charge carrier. To determine the molecular determinants of this modulation of diltiazem block by Ca2+, we measured the fraction of current remaining after application of 50 μM diltiazem to WT and mutant Cav1.2 channels in Ba2+ and in Ca2+. Diltiazem block was allowed to reach equilibrium using 100-ms depolarizing steps to +10 mV from a holding potential of -60 mV at 0.05 Hz. Figure 6A shows the averaged values for these measurements in WT Cav1.2. As expected, a greater fraction of WT current was blocked by 50 μM diltiazem in Ca2+ (∼40% remaining) than in Ba2+ (∼70% remaining). Similar potentiation was observed in Ca2+ for the EIQ and EIIQ mutants as well. However, in both EIIIQ and EIVQ, the fraction of current blocked was not significantly different in Ba2+ and Ca2+, and was similar to the fraction of WT current blocked in Ba2+. Similarly, Ca2+ potentiation of frequency-dependent diltiazem block was not different from WT in EIQ and EIIQ, but was significantly reduced in EIVQ and EIIIQ (Fig. 6, B and C). Thus, Ca2+ potentiation of both closed-channel and frequency-dependent block requires the conserved glutamate residues in the pore region of homologous domains III and IV.

Ca2+ potentiation of diltiazem block: contributions of EIII and EIV. Representative traces demonstrating the enhanced block of WT Cav1.2 channels by diltiazem in Ca2+ compared with Ba2+ (top panel). A, Ca2+ potentiation of diltiazem block at 0.05 Hz. Diltiazem (50 μM) was applied to cells expressing the indicated channels under voltage-clamp, as in Fig. 2, using either 10 mM Ba2+ or 10 mM Ca2+ as the charge carrier. The percentage of current remaining (mean ± S.E.; n = 3–10) in each case was: WTBa = 66 ± 3, WTCa = 40 ± 3; EIQBa = 65 ± 3, EIQCa = 47 ± 7; EIIQBa = 66 ± 2, EIIQCa = 47 ± 2; EIIIQBa = 65 ± 3, EIIIQCa = 73 ± 4, EIVQBa = 65 ± 4, and EIVQCa = 58 ± 3. Asterisks indicate significant differences between current remaining in Ba2+ and Ca2+ for each channel (*, p < 0.05; **, p < 0.01; ***, p < 0.001) and plus signs indicate significant differences in current remaining in Ca2+ between WT and mutant channels (++, p < 0.01). B and C, frequency-dependent block in Ca2+. Frequency-dependent block of the indicated channels by 50 μM diltiazem was measured as described in Fig. 3 except that Ba2+ was replaced with 10 mM Ca2+ (open symbols). For comparison, inactivation during an identical stimulus train in the absence of drug is shown for each channel type tested (closed symbols). There was no significant difference in the fraction of current blocked after 20 pulses between WT and the EIQ and EIIQ mutants. Both EIIIQ and EIVQ channels were blocked to a significantly lesser extent than WT channels under these conditions. Data points are mean ± S.E. (n = 3–5) (*, p < 0.05).
We also examined the involvement of two other amino acid residues in the Ca2+ potentiation of diltiazem block. The Phe residue (F1117) immediately adjacent to EIIIQ is required for Ca2+ potentiation of DHP binding (Peterson and Catterall, 1995), and the C-terminal tail Ile 1627 is part of a Ca2+/calmodulin binding site that mediates Ca2+-dependent inactivation (Peterson et al., 1999). Mutation of this Ile residue to Ala disrupts Ca2+-dependent inactivation (Zuhlke et al., 1999). Therefore, we examined Ca2+ potentiation of diltiazem block of both the F1117G and I1627A mutant channels (Fig. 7). Figure 7A shows that for both F1117G and I1627A, the fraction of channels blocked by diltiazem in Ba2+ and Ca2+ at 0.05-Hz stimulation was not significantly different. However, for F1117G, neither the fraction of channels blocked by 50 μM diltiazem in Ca2+ nor the fraction of channels blocked by 50 μM diltiazem in Ba2+ is significantly different from the fraction of WT channels blocked by 50 μM diltiazem in Ba2+. In the I1627A mutant, the fraction of channels blocked by 50 μM diltiazem in Ba2+ was not significantly different from the fraction of WT channels blocked by 50 μM diltiazem in Ca2+. Thus, whereas F1117G disrupts Ca2+ potentiation of diltiazem block of closed channels, the I1627A mutation potentiates diltiazem block of closed channels to the same extent as Ca2+. Neither F1117G nor I1627A differed from WT channels in the extent of frequency-dependent block at the end of a 20-pulse, 1-Hz train of depolarizations in Ba2+ (Fig. 7B). Frequency-dependent diltiazem block of the I1627A mutant in Ba2+ did, however, develop more rapidly than WT or F1117G channels, reaching equilibrium after fewer depolarizations. In Ca2+, frequency-dependent block of WT and I1627A channels by 50 μM diltiazem was not different, although a 20-pulse train of depolarizations in the absence of diltiazem resulted in potentiation of Ca2+ current as previously reported for I1627A (Zuhlke et al., 1999) (Fig. 7C). In contrast, frequency-dependent diltiazem block of F1117G in Ca2+ was significantly reduced compared with WT channels. Thus, the F1117G mutation does not reduce the affinity of closed or inactivated channels for diltiazem in Ba2+, but does disrupt the Ca2+ potentiation of diltiazem block of both closed and inactivated channels. In addition, the I1627A mutation increases the affinity of closed, but not inactivated, channels for diltiazem in Ba2+, mimicking Ca2+ potentiation. However, I1627A does not alter the affinity of diltiazem for either closed or inactivated channels in Ca2+.

Ca2+ modulation of diltiazem block in F1117G and I1627A. A, diltiazem (50 μM) block of WT, F1117G, and I1627A Cav1.2 channels in 10 mM Ba2+ or 10 mM Ca2+ at 0.05 Hz. The percentage of current remaining (mean ± S.E., n = 3–10) in each case was: WT as in Fig. 6A, F1117GBa = 59 ± 4, F1117GCa = 68 ± 5, I1627ABa = 42 ± 5, and I1627ACa = 44 ± 6. Plus signs indicate a significant difference in the indicated value between WT and mutant channels. (++, p < 0.001). Frequency-dependent block (1 Hz) of F1117G and I1627A channels in Ba2+ (B) or Ca2+ (C) by 50 μM diltiazem (open symbols). The protocol is as in Fig. 3A. For comparison, inactivation during an identical stimulus train in the absence of drug is shown for each channel type tested (closed symbols). Relative current (mean ± S.E., n = 3–6) is plotted versus pulse number. Asterisks indicate a significant difference in the relative current elicited by the 20th pulse in the train between WT Cav1.2 channels and the indicated mutant (*, p < 0.05).
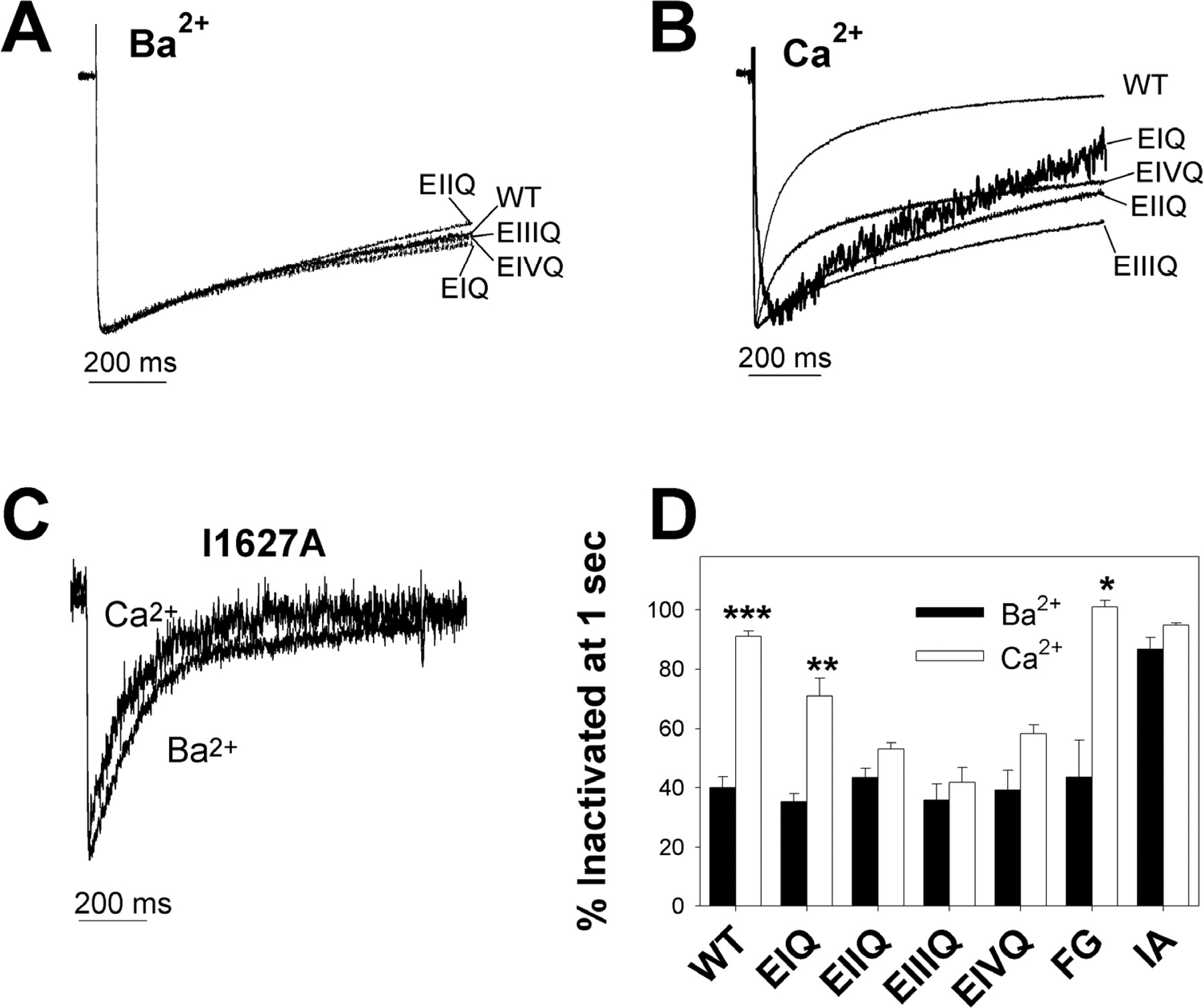
Inactivation in Ba2+and Ca2+ To probe the mechanism that might underlie our observations on the modulation of diltiazem block, we examined the inactivation kinetics of all of the mutations studied in Ba2+ and Ca2+. Figure 8A shows averaged current traces elicited by 1-s depolarizations to +10 mV from a holding potential of -60 mV for several cells expressing the WT, EIQ, EIIQ, EIIIQ, or EIVQ channels recorded in 10 mM Ba2+. The extent of inactivation at the end of the 1-s depolarization is not significantly different among all five channels (Fig. 8D). When the same pulse protocol is applied to WT channels in 10 mM Ca2+, inactivation in the WT channel is markedly accelerated (i.e., Ca2+-dependent inactivation is observed). However, in EIIQ, EIIIQ, and EIVQ channels, the extent of inactivation at the end of the 1-s depolarization is not increased in Ca2+ compared with Ba2+ (i.e., Ca2+-dependent inactivation is disrupted) (Fig. 8, B and D). In the I1627A mutant, Ca2+-dependent inactivation is also disrupted (Fig. 8, C and D). However, with I1627A, the extent of inactivation in Ba2+ is markedly increased such that Ca2+ does not significantly increase it further. Finally, the extent of inactivation at the end of a 1-s depolarization observed in EIQ and F1117G is not different from WT Cav1.2 in Ba2+, and inactivation is also significantly accelerated in Ca2+. Thus, all of the mutant channels that we assayed except for EIQ and F1117G disrupted Ca2+-dependent inactivation. However, in the EIIQ, EIIIQ, and EIVQ mutants, this property was the result of slower inactivation in Ca2+ than in WT channels, whereas in the I1627A mutant, it was the result of faster inactivation in Ba2+.

Inactivation properties of WT, and mutant channels in Ba2+ and Ca2+. Averaged, normalized current traces (n = 2–8) for WT channels, and each of the indicated mutant channels elicited during a 1-s depolarization to +10 mV from a holding potential of -60 mV in extracellular solution containing 10 mM Ba2+ (A) or 10 mM Ca2+ (B) are shown. Note that the percentage of channels inactivated at the end of the 1-s pulse is not significantly different from WT for any of the pore mutants in Ba2+. C, representative current traces for the I1627A channel in 10 mM Ba2+ and 10 mM Ca2+ using the same pulse protocol as in A and B. D, summary of the percentage of channels inactivated at the end of a 1-s depolarization in Ba2+ or Ca2+ (FG = F1117G, IA = I1627A). Whereas WT Cav1.2, EIQ, and F1117G channels display significant increases in the percentage of channels inactivated in Ca2+ versus Ba2+, Ca2+ does not significantly increase the percentage of channels inactivated in EIIQ, EIIIQ, EIVQ, or I1627A mutant channels (values are means ± S.E.). Asterisks indicate a significant difference between the percentage of channels inactivated at the end of a 1-s depolarization in 10 mM Ba2+ versus 10 mM Ca2+ (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Since the F1117G mutant is deficient in Ca2+ potentiation of diltiazem block but retains Ca2+-dependent inactivation that was indistinguishable from that of WT channels, we sought further evidence that F1117G may interact with Ca2+ ions in the pore. The current traces in Fig. 9A were recorded from the same cell, expressing the F1117G mutant, in 10 mM Ba2+ and then in 10 mM Ca2+. Interestingly, we found that the peak current amplitude elicited by depolarization to +10 mV was greater in Ca2+ than in Ba2+ for the F1117G mutant. In contrast, the WT Cav1.2 channel exhibits a marked decrease in peak current when the extracellular solution is switched from 10 mM Ba2+ to 10 mM Ca2+ (Fig. 9C). Despite this apparent increase in permeability for Ca2+ relative to Ba2+, the F1117G mutant maintains robust Ca2+-dependent inactivation (Figs. 9A and 8D). The current-voltage relationship for F1117G in Ba2+ versus Ca2+ (Fig. 9B) shows that the increased permeability for Ca2+ over Ba2+ observed in this mutant is voltage-dependent, occurring only with depolarizations above -10 mV. In Fig. 9C, we compare the change in peak current amplitude, measured at +10 mV, upon switching from 10 mM Ba2+ in the extracellular solution, to 10 mM Ca2+ for WT, EIIQ, EIIIQ, EIVQ, F1117G, and I1627A channels. EIQ and EIIQ show decreases in peak current amplitude upon switching from Ba2+ to Ca2+, similar to WT channels. However, peak current amplitude in EIIIQ, EIVQ, and F1117G channels increased upon switching from Ba2+ to Ca2+. Thus, the Ca2+ binding site formed by EIII, EIV, and F1117 not only modulates the affinity of closed and inactivated channels for diltiazem but also mediates the lower permeability of Ca2+ relative to Ba2+ in Cav1.2 channels.

Permeability of Ca2+ relative to Ba2+ in WT and mutant Cav1.2 channels. A, current traces recorded from a single cell expressing the F1117G mutant recorded in 10 mM Ba2+ or 10 mM Ca2+. Currents were elicited using a 1-s depolarization to +10 mV from a holding potential of -60 mV. Note the increase in peak current in Ca2+ relative to peak current in Ba2+. B, current-voltage relationship of F1117G current in 10 mM Ba2+ and in 10 mM Ca2+. Currents at the indicated membrane potentials were recorded as described in the legend to Fig. 4. C, change in peak current amplitude measured at +10 mV from a holding potential of -60 mV when the extracellular solution is switched from 10 mM Ba2+ to 10 mM Ca2+. The values shown are mean ± S.E. (n = 4–12) for WT Cav1.2 channels and each of the indicated mutants.
Discussion
Selective Effect of EIVQ Mutation on Frequency-Dependent Block of Cav1.2 in Ba2+ We have shown that amino acid residues in the pore region of Cav1.2 can modulate diltiazem block and that this modulation is distinct, depending upon the permeant ion used in the experiments. None of the E to Q mutations significantly affected the IC50 for diltiazem block at low frequency (0.05 Hz) in 10 mM Ba2+ (Fig. 2). This result contrasts with a study using the PAA D888, in which the EIIIQ and EIVQ mutations increased the IC50 for D888 by 15- to 20-fold at a stimulation frequency of 0.10 Hz in 10 mM Ba2+ (Hockerman et al., 1997a). Apparently, closed-channel block of Cav1.2 by diltiazem involves interactions predominantly with amino acid residues from IIIS6 and IVS6 (Hering et al., 1996; Kraus et al., 1998; Hockerman et al., 2000) and does not require an interaction between the alkylamino group and particular pore glutamates as proposed for D888. The near-complete loss of frequency-dependent block by diltiazem in EIVQ in Ba2+ (Fig. 3C) suggests a role for E1419 either in binding of the drug to the inactivated state of the channel or in modulation of the inactivation properties of the channel.
To distinguish these two possibilities, we investigated the mechanism by which this mutation exerted such a strong effect on frequency-dependent block in Ba2+. The voltage dependence of activation and inactivation for WT Cav1.2 and EIVQ was not greatly different. Moreover, the shift in V1/2 inactivation induced by 50 μM diltiazem was not different for WT Cav1.2 and EIVQ channels (Fig. 4B), suggesting that the EIVQ mutation did not disrupt the ability of diltiazem to bind to the inactivated state of the channel (Li et al., 1999). This characteristic of the EIVQ mutation contrasts with mutations in IVS5 of Cav1.2 that disrupted frequency-dependent block but also markedly disrupted voltage-dependent inactivation (Motoike et al., 1999; Bodi et al., 2002). Thus, neither the small changes in the voltage dependence of activation and inactivation nor changes in the ability of diltiazem to bind the inactivated state of EIVQ likely explain the marked loss of frequency-dependent block accumulation in EIVQ.
Recovery from inactivation in Cav1.2 is biexponential (Johnson et al., 1996; Kraus et al., 1998), and diltiazem slowed recovery of WT Cav1.2 channels from inactivation by increasing τF and decreasing the fraction of channels recovering with the fast time constant (fF) (Fig. 4C). The time constant for slow recovery was not different in the presence and absence of drug for WT Cav1.2. Our results differ from those of Kraus et al. (1998), who found that diltiazem decreased fF but did not appreciably change either τF or τS. This discrepancy may be due to the differences in the α1 subunits and expression system, since Kraus et al. (1998) used chimeric channels that were predominantly Cav2.1, with L-type sequence inserted into IIIS6 and IVS6, expressed in Xenopus laevis oocytes. Our results with diltiazem are consistent with studies of PAA block of Cav1.2, since PAA binding also decreases the fraction of channels recovering with the fast time constant (Johnson et al., 1996; Hering et al., 1997).
EIVQ channels also demonstrated biexponential recovery from inactivation (Fig. 4D). The fF for EIVQ in the absence of drug was similar to that of WT Cav1.2, but τF was decreased, and τS was increased 5-fold. In the presence of 50 μM diltiazem, τF increased and τS was not changed for EIVQ, as in WT channels. In contrast to WT channels, 50 μM diltiazem increased fF in EIVQ channels such that, after 1s of recovery, EIVQ channels actually recovered faster in the presence of diltiazem than in the absence of the drug. This unexpected result suggests that, in Ba2+, diltiazem binding prevents EIVQ channels from entering the slowly recovering, inactivated state. A similar result was observed for PAA block with a mutant channel in which two amino acids in IIIS6 (corresponding to amino acids I1163 and F1164) were changed to alanine (Hering et al., 1997).
The association of diltiazem with depolarized channels also suggests that diltiazem binds to and modulates the inactivated state of Cav1.2 channels. Figure 5B shows that diltiazem increases the rate of inactivation of WT channels by increasing the fraction inactivating with the fast time constant, and by accelerating the slow time constant of inactivation. In EIVQ channels, diltiazem induces a small fraction of channels to inactivate with a very rapid time constant not observed in the absence of drug (Fig. 5B). However, the major effect of diltiazem on EIVQ channels is the acceleration of the slow time constant of inactivation and an increase in the fraction of channels inactivating with the slow time constant. Thus, it appears that diltiazem can bind to the inactivated state of EIVQ but induces rapid recovery from inactivation that disrupts the accumulation of frequency-dependent block in Ba2+.
Ca2+Modulation of Diltiazem Block. Our results indicate that, as for DHP binding, Ca2+ potentiation of diltiazem block of closed channels is lost in the EIIIQ, EIVQ, and F1117G mutations, whereas it is retained in the EIQ and EIIQ mutants. These mutations do not affect the IC50 for diltiazem in Ba2+ at 0.05 Hz but selectively abolish the increase in diltiazem potency observed for WT Cav1.2 channels in Ca2+. These data suggest that EIII and EIV may cooperate to form a Ca2+ binding site. The role of F1117G may involve participation in Ca2+ binding via a π-cation interaction (Heginbotham and MacKinnon, 1992), or may involve the transduction of a conformational change to the diltiazem binding site. However, the observation that Ca2+ permeability relative to Ba2+ is altered in F1117G (Fig. 9) argues that F1117 likely interacts directly with Ca2+ ions in the pore. The extent of frequency-dependent block accumulation in Cav1.2 is also greater in Ca2+ than in Ba2+(Figs. 6 and 7). As we observed for closed-channel block, the Ca2+ potentiation of frequency-dependent diltiazem block is retained in EIQ and EIIQ but is lost in the EIIIQ, EIVQ, and F1117G mutants. In sharp contrast to our observations in Ba2+, frequency-dependent diltiazem block of the EIVQ mutant in Ca2+ was similar to that of EIIIQ. This result suggests that Ca2+ binding in the pore of the EIVQ channel prevents the diltiazem-induced acceleration of recovery from inactivation observed in Ba2+.
In contrast to all the other mutant channels used in this study, the I1627A mutation is not in the putative pore region of the channel but, rather, in the intracellular C-terminal tail. However, this mutation potentiated diltiazem block of closed channels in Ba2+ compared with WT channels, to an extent similar to that in Ca2+, such that the extent of closed-channel diltiazem block in I1627A was not further increased in Ca2+. Frequency-dependent diltiazem block of I1627A was not different from WT in either Ba2+ or Ca2+. To understand this observation, we examined the inactivation rates of the channels used in this study in Ba2+ and Ca2+ by measuring the percentage of peak current inactivated at the end of a 1-s depolarization. We found that all of the mutations except I1627A were not different from WT channels in the percentage of inactivated channels in Ba2+. In the I1627A mutation, inactivation was accelerated in Ba2+ such that Ca2+ did not appreciably increase the percentage of inactivated channels at the end of a 1-s depolarization. The V1/2 inactivation of I1627A in 10 mM Ba2+ (measured as described for Fig. 4B) is not different from that of WT Cav1.2 (N. Dilmac and G. H. Hockerman, unpublished data). Thus, it appears that the conformational change that induces faster inactivation of I1627A may also increase the affinity of diltiazem for closed, but not inactivated, I1627A channels. We have observed that this acceleration of inactivation in Ba2+ of the I1627A mutation is β subunit-dependent. In addition to the β1b subunit used in this study, we observed similar acceleration with the I1627A subunit coexpressed with the β3 but not the β2 subunit (G. H. Hockerman and N. Dilmac, unpublished data; also see Peterson et al., 1999). A recent study that examined the modulation of (-)-gallopamil (a PAA) block of Cav1.2 by different β subunits (Sokolov et al., 2001) concluded that channels with an accelerated voltage-dependent inactivation rate were also more sensitive to gallopamil block. Furthermore, Sokolov et al. (2001) proposed that PAA block of Cav1.2 depends upon the fast voltage-dependent inactivation observed in Ba2+ and not the further acceleration of inactivation observed in Ca2+. Thus, our data fit well with the model of Sokolov et al. (2001), since the main effect of the I1627A mutation is to shift >80% of channels into the fast inactivating mode in Ba2+ (i.e., the fast voltage-dependent inactivation of Sokolov et al., 2001; Fig. 8C). Comparing the extent of inactivation in Ba2+ and Ca2+ (Fig. 8), it is clear that EIIQ, EIIIQ, and EIVQ are all deficient in Ca2+-dependent inactivation compared with WT. However, since the EIIQ mutant shows normal Ca2+ potentiation of both closed-channel and frequency-dependent diltiazem block, we conclude that Ca2+-dependent inactivation does not play a critical role in the potentiation of diltiazem block by Ca2+. Furthermore, our results suggest that Ca2+-dependent inactivation of Cav1.2 may involve Ca2+ binding in the pore that is modulated by conformational changes in the C-terminal tail.
In conclusion, our results support the notion that Ca2+ potentiates diltiazem block of both closed and inactivated Cav1.2 channels via a Ca2+ binding site composed of F1117, E1118, and E1419. Thus, our results suggest that diltiazem binds to the same site between transmembrane segments IIIS6 and IVS6 in both the closed and inactivated states, with the inactivated state having a higher affinity for diltiazem. In Ba2+, the disruption of frequency-dependent diltiazem block in the mutant EIVQ results from a diltiazem-induced shift of channels into a rapidly recovering, inactivated state. Furthermore, it appears that Ca2+-dependent inactivation per se is not required for Ca2+ potentiation of diltiazem. However, the same conformational change in I1627A that accelerates inactivation in Ba2+ increases the affinity of the closed channel for diltiazem. It will be interesting to determine whether Ca2+ potentiation of block of Cav1.2 by PAAs such as verapamil occurs via the same or a distinct mechanism.
Footnotes
-
This work was supported by Scientist Development Grant 9930016N from the American Heart Association (G.H.H.).
-
ABBREVIATIONS: DHP, dihydropyridine; PAA, phenylalkylamine; BZP, benzothiazepine; GFP, green fluorescent protein; V½, voltage at which half of the channels are inactivated; D888, desmethoxyverapamil.
- Received December 10, 2002.
- Accepted May 12, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}