


Abstract
The human cannabinoid receptor 1 (CB1) belongs to the G protein-coupled receptor (GPCR) family. Among the members of GPCR family, it has an exceptionally long extracellular N-terminal domain (N-tail) of 116 amino acids but has no typical signal sequence. This poses questions of how the long N-tail affects the biosynthesis of the receptor and of how it is inserted into the endoplasmic reticulum (ER) membrane. Here we have examined the process of membrane assembly of CB1 in the ER membrane and the maturation of the receptor from the ER to the plasma membrane. We find that the long N-tail cannot be efficiently translocated across the ER membrane, causing the rapid degradation of CB1 by proteasomes; this leads to a low level of expression of the receptor at the plasma membrane. The addition of a signal peptide at the N terminus of CB1 or shortening of the long N-tail greatly enhances the stability and cell surface expression of the receptor without affecting receptor binding to a cannabinoid ligand, CP-55,940. We propose that the N-tail translocation is a crucial early step in biosynthesis of the receptor and may play a role in regulating the stability and surface expression of CB1.
G protein-coupled receptors (GPCRs) constitute the largest family of integral membrane proteins in eukaryotic cells. They share a common membrane topology, having seven hydrophobic transmembrane (TM) segments, an N terminus located extracellularly, and a C terminus located intracellularly, indicating structural conservation among GPCRs (Bockaert and Pin, 1999). Despite the central importance of GPCRs, little is known about their folding and membrane assembly. With respect to the mechanism of endoplasmic reticulum (ER) targeting and insertion, members of the GPCR family can be divided in two groups: proteins that have a cleavable signal sequence at the N terminus and proteins that do not (Wallin and von Heijne, 1995). The presence of an N-terminal cleavable signal sequence is predominantly found among GPCRs whose extracellular N-termini form stably folded, ligand-binding domains (Kochl et al., 2002). The majority (about 90%) of GPCRs do not contain a signal sequence. In this case, the first TM domain is thought to function as a targeting sequence (reverse signal anchor sequence) for the protein to initiate the assembly process in the ER membrane via the signal recognition particle (SRP)-Sec61 pathway (Friedlander and Blobel, 1985). Translocation of the extracellular N-terminal tail (N-tail) across the ER membrane may occur after its synthesis, possibly in a C-to-N-terminal direction (Nilsson et al., 2000). It has been suggested that the ER targeting and translocation of opsin can occur either co- or post-translationally, although in both instances, it requires that the nascent peptide chain be attached to the ribosome (Kanner et al., 2002).
The efficiency of N-tail translocation is dependent on several factors. The presence of positively charged residues and rapid folding of the N-tail prevent its translocation (von Heijne and Gavel, 1988; Denzer et al., 1995). Charged residues flanking the first TM segment (von Heijne and Gavel, 1988; Hartmann et al., 1989), and the hydrophobicity and length of this segment are also important determinants (Wahlberg and Spiess, 1997). In some polytopic membrane proteins, downstream TM segments facilitate N-tail translocation, suggesting that the second TM may serve as an ER targeting sequence in some cases (Monne et al., 1999; Nilsson et al., 2000).
Previous studies indicate that the length of an N-tail is not a limiting factor for efficient translocation (Denzer et al., 1995). However, it is conceivable that shorter N-tails may be translocated across the ER membrane more readily than longer ones when no signal sequence is present. Statistical analyses indicate that GPCRs without signal sequences do have considerably shorter N-tails (40 amino acids on average) than the GPCRs with signal sequences (200 amino acids on average) (Wallin and von Heijne, 1995). Nevertheless, there are exceptions to this rule. The human cannabinoid receptor 1 (CB1) has an N-tail of 116 amino acids and yet lacks a signal sequence. This prompts the question of whether the long N-tail of CB1 can be successfully translocated to the luminal side of the ER. This issue must be examined to understand the biological implication of possessing such a large N-terminal domain, in terms of the folding mechanism and the physiological function, of CB1.
CB1 binds to Δ9-tetrahydrocannabinol, the major psycho-active component of marijuana (Matsuda et al., 1990; Goutopoulos and Makriyannis, 2002). It is one of the most abundant GPCRs in the brain and is involved in a wide range of physiological activities (Breivogel and Childers, 1998). At the cellular level, the action of CB1 involves coupling with heterotrimeric G proteins of the Gi/o family. The activation of G proteins induces a series of signaling events, including a decrease in the level of cAMP (Howlett, 1998), activation of inwardly rectifying K+ channels (Pertwee, 1997), inhibition of N- and P/Q-type Ca2+ channels (Mackie and Hille, 1992), and activation of mitogen-activated protein kinases (Bouaboula et al., 1995). CB1 is distributed in many areas of the brain (Tsou et al., 1998). In the hippocampus, it is localized in a subset of presynaptic axon terminals and modulates neurotransmitter release (Katona et al., 1999; Wilson and Nicoll, 2002). Thus, it seems that the expression and subcellular localization of CB1 are well regulated in specific types of cells, suggesting that the process of membrane assembly of CB1 may be an important control point for the function of the receptor. However, this area has not been explored.
As the first step of this investigation, we have examined the effect of the long N-tail of CB1 on its membrane assembly. We have monitored membrane insertion, N-linked glycosylation, and surface expression of the receptor using a cell-free in vitro system and cellular expression systems. We find that the long N-tail inhibits the assembly of CB1 in the ER membrane. This may cause the relatively short half-life of the receptor in the cell and leads to a low level of expression at the plasma membrane. The addition of a signal peptide at the N terminus of CB1 or shortening of the long N-tail greatly increases the stability and cell surface expression of the receptor but has no effect on ligand binding.
Materials and Methods
Materials. All restriction enzymes were from New England Biolabs (Boston, MA). The plasmid pcDNA3.1 was from Invitrogen (Carlsbad, CA). QuikChange site directed mutagenesis kit was from Stratagene (La Jolla, CA). T7 RNA polymerase, RNasin, and rabbit reticulocyte lysate were from Promega (Madison, WI). Taq polymerase, [35S]methionine, 14C-methylated marker proteins, ribonucleotides, deoxyribonucleotides, and the cap analog m7G(5′)ppp(5′)G were from Amersham Biosciences (Uppsala, Sweden). Endoglycosidase H (Endo H) and protein G-agarose were from Roche Applied Science (Mannheim, Germany). Monoclonal anti-c-Myc, phenylmethylsulfonyl fluoride (PMSF), and proteinase K (PK) were from Sigma (St. Louis, MO). The proteasomal inhibitor MG132 was from Peptide Institute Inc. (Osaka, Japan). Oligonucleotides were from Cybergene (Stockholm, Sweden). [3H]CP-55,940 (165 Ci/mmol) was from PerkinElmer Life Sciences (Boston, MA). Dulbecco's modified Eagles' medium was purchased from Hyclone Laboratories, Inc. (Logan, UT). Fluorescein isothiocyanate (FITC)-conjugated and unconjugated goat anti-mouse IgG were from Pierce (Rockford, IL).
DNA Techniques.BamHI and EcoRI sites were introduced by PCR at the 5′ and 3′ ends of the human CB1 gene, respectively. The PCR fragment was cloned between BamHI and EcoRI sites of the pcDNA3.1 vector. The N-terminal shortened mutants Δ64CB1, Δ80CB1, and Δ89CB1 were made in the same way. The DNA fragment encoding the signal sequence from bovine preprolactin was inserted between the HindIII and BamHI sites, which placed the signal sequence at the N terminus of CB1. Mutations N77A and N83A of CB1 were made using the QuikChange site-directed mutagenesis kit. The c-Myc epitope was introduced at the N terminus of CB1 or between the signal sequence and CB1, using complementary oligonucleotides encoding the c-Myc peptide and inserted at the BamHI site. The BamHI site was altered after the insertion of c-Myc. DNA constructs based on pcDNA3.1 were used for the cell-free in vitro study and expression in human embryonic kidney (HEK) 293 cells. The coding region of these DNA constructs was transferred into the pSFV1 vector between BamHI and SmaI sites for expression in baby hamster kidney (BHK) cells. PCR primers for the forward and reverse directions were designed so that the BamHI site was followed by a ribosomal binding site (GCCACC) and the ATG start codon, and the TAG stop codon was placed in front of the SmaI site.
Cell-Free in Vitro Expression System. The constructs in pcDNA3.1 were transcribed by T7 RNA polymerase for 1 h at 37°C with the buffer supplied by the manufacturer, essentially following the protocol described previously (Monne et al., 1999). Translation in rabbit reticulocyte lysate in the presence of dog pancreas microsomes was performed as described previously (Liljestrom and Garoff, 1991). Sodium carbonate extraction of the translation mixture in the microsomal membrane was carried out as described previously (Monne et al., 1999). Proteinase K treatment of microsomes was also carried out as described previously (Nilsson et al., 2000).
Cellular Expression Systems. Protein synthesis in BHK cells using the SFV expression system has been described in detail previously (Liljestrom and Garoff, 1991; Liljestrom et al., 1991). Briefly, CB1 constructs under the SP6 promoter in the SFV vector were linearized for in vitro transcription. The resulting RNA was used to transfect BHK cells by electroporation. Six hours after electroporation, cells were prestarved of methionine for 30 min and then labeled with [35S]methionine, followed by chase in media containing 1 mM unlabeled methionine. MG132 was used at a final concentration of 20 μM and added 1.5 h before labeling and during labeling. After pulse chasing, cells were solubilized in lysate buffer containing 1% nonidet P-40. Anti-c-Myc antibodies (1 μl) were added to 100 μl of cell lysate to immunoprecipitate the radiolabeled receptor with 30 μl of protein G agarose, following the protocol from the manufacturer. Samples were then resuspended in 50 μl of Endo H buffer (50 mM sodium citrate, pH 5.5, and 1% SDS) and incubated with Endo H for 16 h at 37°C as described previously (Andersson et al., 1997). For PK treatment, BHK cells were suspended in homogenization buffer (10% sucrose, w/w, 10 mM Tris-HCl pH 7.4, 1 mM EDTA) and passed through a 23-gauge needle 15 times. The homogenates were centrifuged at 5000g for 5 min to remove nuclei, and the supernatants were divided in aliquots and treated with PK, PMSF, and Triton X-100 at final concentrations of 8 μg/ml, 0.46 mg/ml, and 13 mg/ml, respectively, for 30 min on ice. Samples were then lysed in nonidet P-40 at a final concentration of 1%. The lysates were immunoprecipitated and analyzed by SDS-PAGE. For immunofluorescence microscopy, transfected cells were fixed with 3% paraformaldehyde 6.5 hours after electroporation and then processed for indirect immunofluorescence essentially as described previously (Salminen et al., 1992). For surface staining, cells were incubated with the monoclonal antibody c-Myc (diluted 1:200) on ice before fixation, followed by permeabilization with 0.1% Triton X-100 and then incubated with the FITC-conjugated goat anti-mouse IgG as the secondary antibody (diluted 1:100). For internal staining, the primary antibody was diluted 1:1000. To analyze receptor binding, HEK 293T cells were grown in Dulbecco's modified Eagles' medium plus 10% fetal bovine serum with high glucose at 37°C in a 5% CO2 humidified incubator. Partially confluent plates of cultured cells were transiently transfected with pcDNA3.1 containing the wild-type or modified CB1 using calcium phosphate precipitation.
Ligand Binding Assay. Membrane preparations of transiently transfected HEK 293T cells and saturation binding assays to [3H] CP-55,940 were carried out as described previously (Chin et al., 1999).
Results
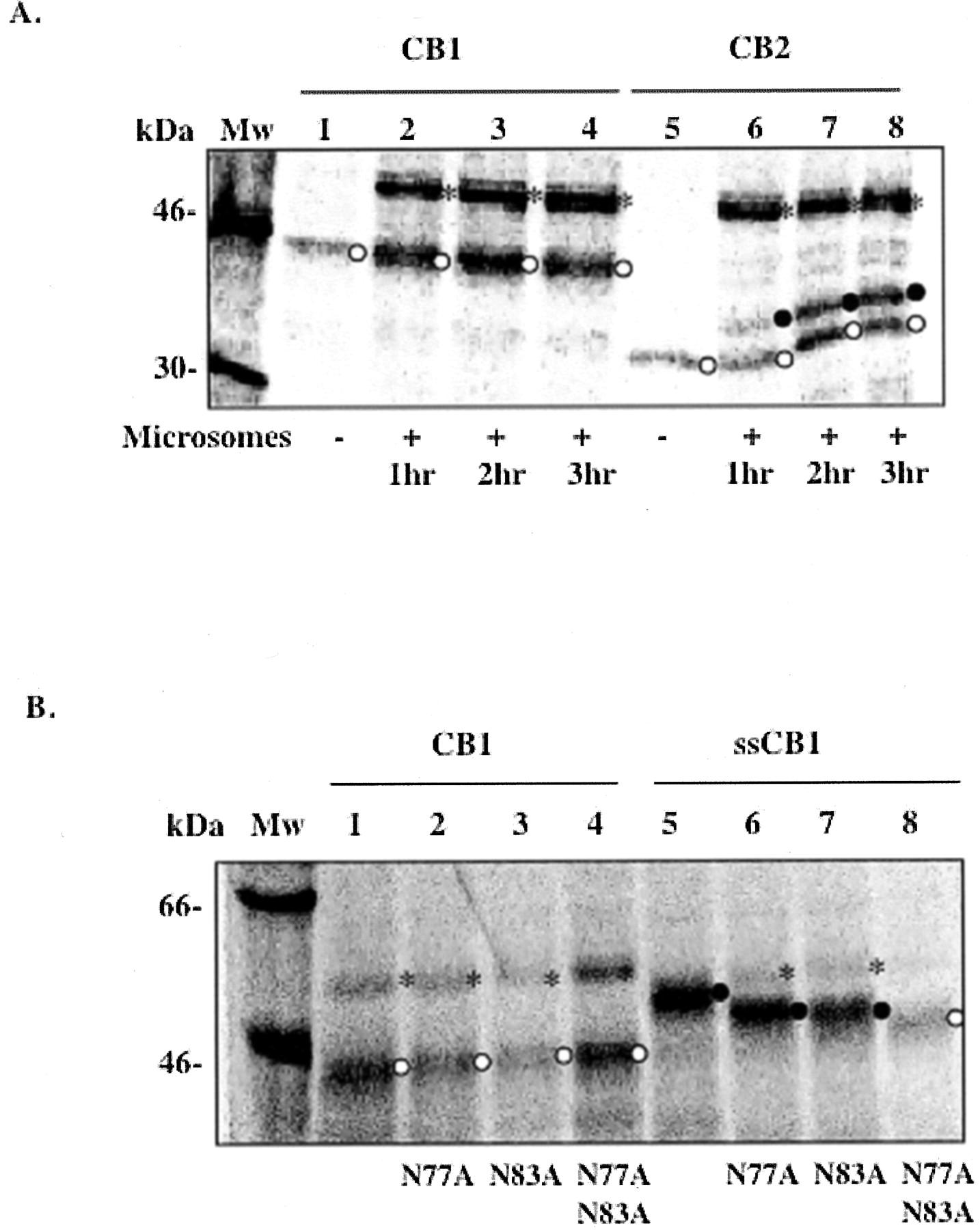
N-Linked Glycosylation of CB1 in the ER Membrane. To study the assembly of CB1 and the translocation of its N-tail across the ER membrane, CB1 gene was expressed and the protein was synthesized using a cell-free in vitro transcription-translation system in the presence of microsomes. As a control, the cannabinoid receptor subtype CB2 was synthesized under the same conditions. CB1 has an estimated molecular mass of 53 kDa and within its long N-tail has two putative N-linked glycosylation sites (Asn-X-Ser/Thr) at positions 77 and 83. CB2 is about 40 kDa, has a relatively short N-tail (33 amino acids), and has only one glycosylation site at position 11. When the N-tail is translocated to the luminal side of the ER membrane, the ER-located oligosaccharide transferase adds high mannose-type oligosaccharides to the Asn-X-Thr/Ser sites. This modification results in an increased size of approximately 2 kDa for each N-linked glycosylation, which can easily be detected by SDS-PAGE. As shown in Fig. 1A, CB1 was not glycosylated when synthesized in the presence of microsomes for 3 h. In contrast, about half of CB2 was glycosylated under the same conditions. Both proteins migrated somewhat faster than anticipated based on their molecular masses. This is not uncommon for membrane proteins and may be attributable to the tightly packed conformation of the protein in SDS micelles or the lower number of micelles associated with the protein (Therien et al., 2001).
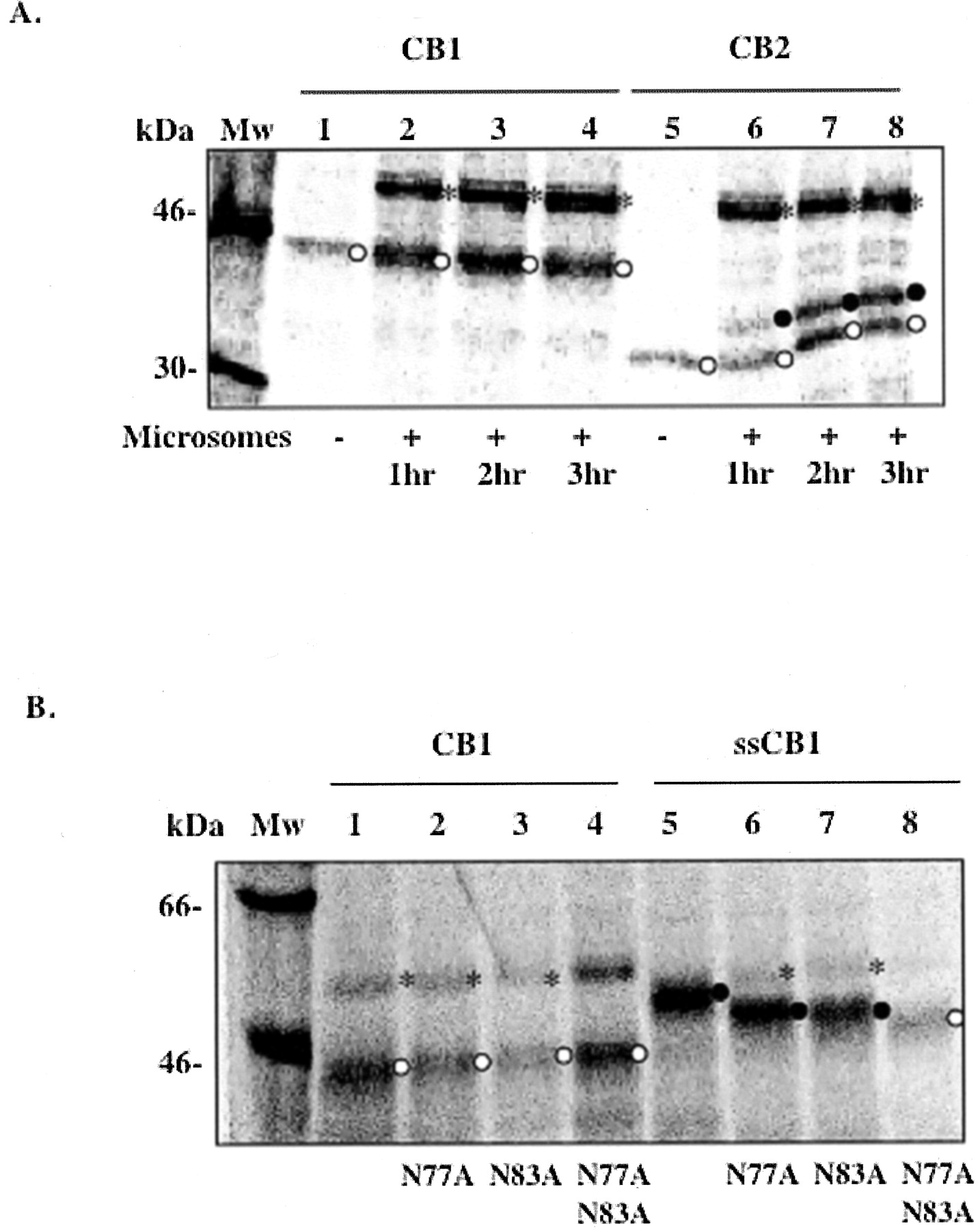
N-linked glycosylation of CB1 and CB2 in the ER membrane. A, CB1 (lanes 1–4) and CB2 (lanes 5–8) were expressed in the absence (lanes 1 and 5) or presence of microsomes incubated at 30°C for 1 h (lanes 2 and 6), 2 h (lanes 3 and 7), and 3 h (lanes 4 and 8). B, glycosylation mutants of CB1 (lanes 2–4) and ssCB1 (lanes 6–8) were expressed in the presence of microsomes at 37°C for 1 h. CB1 (lane 1) and ssCB1 (lane 5) are shown as controls. ○, nonglycosylated form; •, glycosylated form; *, nonspecific band at around 55 kDa, which frequently appeared in reactions with microsomes. Data shown in A are representative of four independent experiments. Data shown in B are from one experiment. Mw, molecular mass.
Three possible scenarios explain the lack of glycosylation of CB1: (1) residues Asn77 and Asn83 are not good substrate sites for oligosaccharide transferase; (2) CB1 fails to integrate into the ER membrane; or (3) the N-tail of CB1 is not translocated across the ER membrane. To assess the first possibility, the signal sequence from preprolactin was attached to the N terminus of CB1 (designated ssCB1). For both CB1 and ssCB1, single or double mutations were made in which residues Asn77, Asn83, or both were mutated to Ala. The N-linked glycosylation of these N-tail variants was examined using the in vitro transcription-translation system in the presence of microsomes (Fig. 1B). The migration of the protein bands on SDS-PAGE indicates that with a signal sequence, CB1 was fully glycosylated at both positions Asn77 and Asn83 (Fig. 1B), ruling out the first possibility.
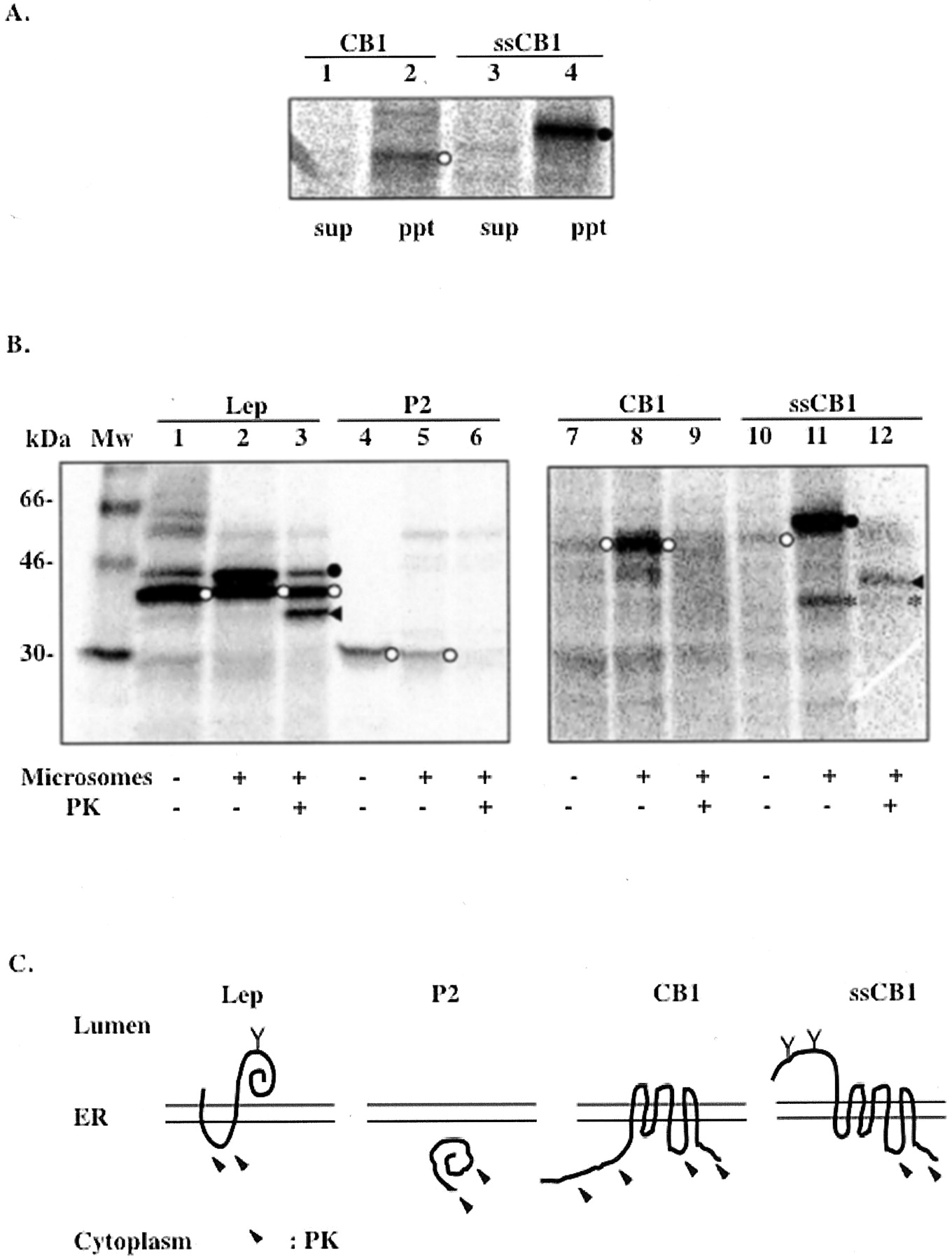
By extracting the unbound, loosely associated proteins from the membrane using 100 mM sodium carbonate, it was demonstrated that the majority of CB1 and ssCB1 remained associated with membrane fractions (Fig. 2A), ruling out the second possibility. To confirm membrane integration and to probe CB1 membrane topology, microsomes containing synthesized proteins were treated with PK. Protein portions translocated into the ER lumen are protected from digestion by the microsomal membrane. Two previously characterized proteins derived from Escherichia coli leader peptidase, denoted Lep and P2, were used as the luminal and cytoplasmic markers, respectively (Nilsson and von Heijne, 1998). A large protected fragment of about 35 kDa was observed by SDS-PAGE in the ssCB1 sample after the PK treatment (Fig. 2B, lane 12). The size of this fragment corresponds to a portion of CB1 from the N terminus to the third cytoplasmic loop (ranging approximately from amino acid residue 310 to 345) as depicted in Fig. 2C. The first and the second cytoplasmic loops of CB1 are relatively short (about 8 and 15 amino acids, respectively) and were presumably inaccessible to PK digestion under our assay conditions. However, for the wild-type CB1, no protected fragment was detectable (Fig. 2B, lane 9). This suggested that without a signal sequence, the long N-tail of CB1, along with the first TM domain, were left on the cytoplasmic side of the ER membrane during protein synthesis.
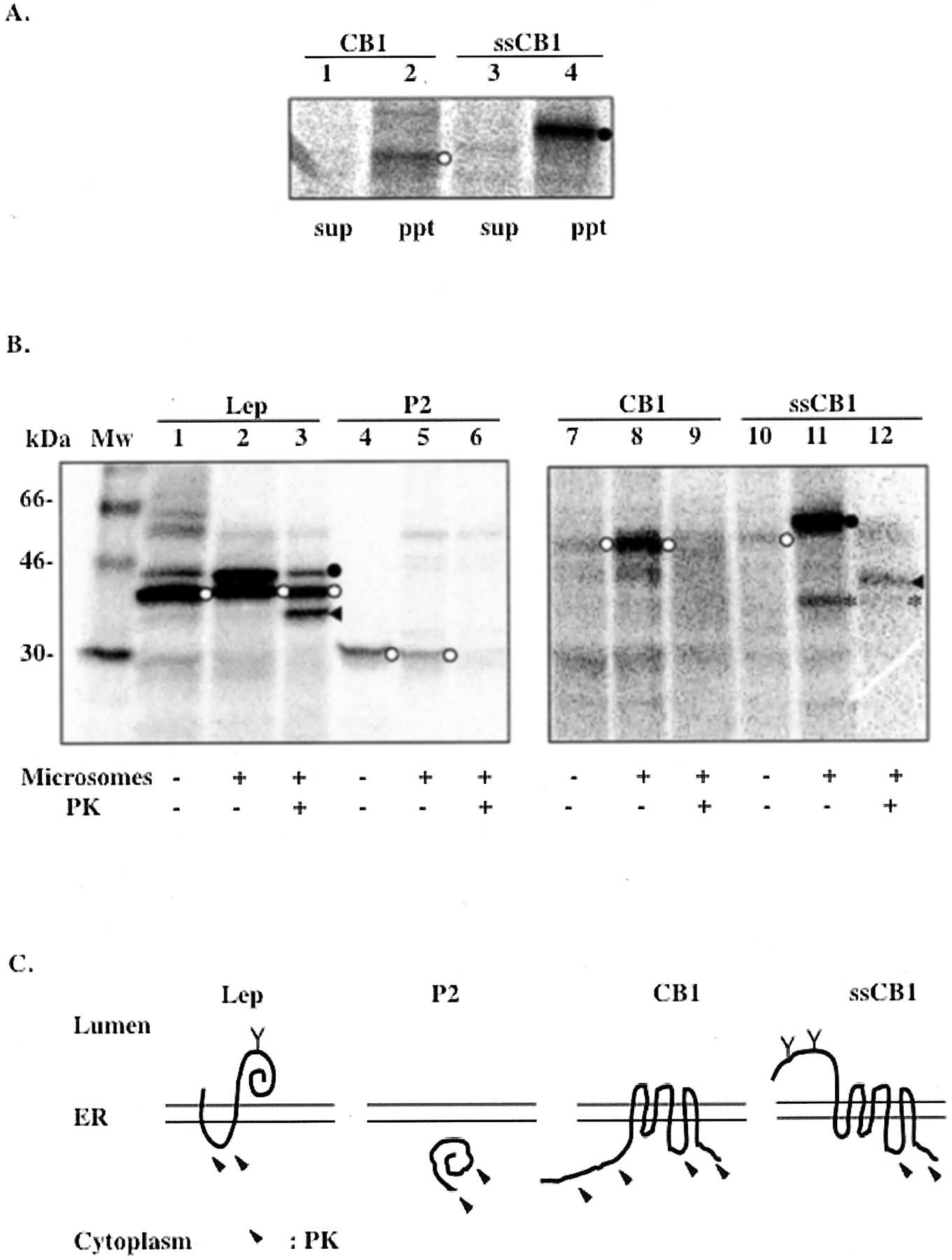
Membrane integration and topology of CB1 and ssCB1 in the ER membrane. A, sodium carbonate extraction of CB1 (lanes 1 and 2) and ssCB1 (lanes 3 and 4) expressed in the microsomal membrane. Lanes 2 and 4, pellets after the extraction of the membrane, which contains membrane-bound proteins. Lanes 1 and 3, supernatants after the extraction. B, PK protection assay. Lep (lanes 1–3), P2 (lanes 4–6), CB1 (lanes 7–9), and ssCB1 (lanes 10–12) were expressed in the absence or presence of microsomes and treated with PK. Lep was used as a control to show that the luminal portions of the protein are protected from digestion by PK. P2 domain of Lep, used as a control for a cytosolic protein. ◂, protein fragments protected from PK digestion; *, a naturally occurring proteolytic fragment (lanes 11 and 12); ○, nonglycosylated form; •, glycosylated form. C, membrane topology in ER deduced from the results of PK treatment. Y, glycosylation site; arrows, regions accessible to PK digestion. Data shown here are representative of two independent experiments.
The Effect of the Long N-Tail of CB1 on Cell Surface Expression, Stability, and Ligand Binding to the Receptor. The data above show that the wild-type CB1 did not attain the expected 7TM topology, possibly because of the difficulty in translocating the long N-tail across the ER membrane. This finding prompts several questions. Does this situation reflect what happens in the cell? Can CB1, if incorrectly folded in the ER membrane, be transported to the cell surface? What properties (e.g., length or glycosylation) of the N-tail affect the expression of CB1 in a cell? To address these questions, we characterized the synthesis of CB1 in BHK cells, transiently expressing the receptor using the Semliki Forest virus (SFV) expression system (Liljestrom and Garoff, 1991).
To examine the process of maturation of the receptor from the ER compartment to the plasma membrane, cells expressing either the CB1 or ssCB1 genes were metabolically labeled with [35S]methionine and chased for 2 h. A c-Myc tag was added at the N terminus of the receptor for detection by immunoprecipitation and immunofluorescent staining. The sensitivity of the protein to Endo H was used as an indicator to monitor the state of glycosylation and intracellular trafficking of the receptor. Consistent with the data from the cell-free in vitro system, only a small portion of the wild-type CB1 (less than 10%) was glycosylated (Fig. 3A, lanes 1 and 2). However, with the addition of the N-terminal signal sequence, the protein (ssCB1) was 100% glycosylated and a fraction of the molecules became complex-glycosylated (Endo H-resistant) in 2 h (Fig. 3B).

Expression and maturation of CB1 (A), ssCB1 (B), Δ64CB1 (C), Δ80CB1 (D), and Δ89CB1 (E). Constructs were expressed in BHK cells, pulse-labeled with [35S]methionine for 30 min and chased for 0 h (lanes 1 and 2), 1 h (lanes 3 and 4), and 2 h (lanes 5 and 6). Samples were subjected to immunoprecipitation and Endo H treatment as described previously (Andersson et al., 1997). ○, nonglycosylated form; •, glycosylated form. ◃, Endo H-sensitive forms (derived from core-glycosylated receptors); ◂, Endo H-resistant forms (proteins with complex-type N-linked oligosaccharides). Endo H-resistant forms exhibited a smear on SDS-PAGE because of the heterogeneity of the sugar moieties. Data shown in A and B are representative of three independent experiments. Data shown in C, D, and E are from one experiment.
Most interestingly, we found that the wild-type CB1 was weakly labeled and rapidly degraded in less than an hour, whereas ssCB1 seemed to be more stable, although certain degrees of protein degradation were observed in both samples (Fig. 3, A and B). For the wild-type CB1, after a 1-hour chase, only about 10% of the 35S-labeled receptor remained detectable, whereas for ssCB1, about 50% remained [data obtained from comparing densitometric measurements of total protein in lane 1 (or 2) and lane 3 (or 4) in Fig. 3, A and B]. The above result suggests that, in BHK cells, the newly synthesized CB1 did not attain the correct conformation because of difficulty in translocating the N-tail and possibly was degraded by proteasomes via the ER quality control pathway. The addition of the N-terminal c-Myc tag is unlikely to account for the rapid degradation and lack of glycosylation of the wild-type CB1, because similar results were obtained using proteins without the c-Myc tag with antibodies against the first 14 amino acids of CB1 (data not shown). To rule out the possibility that the inhibition of CB1 processing is caused by overexpression of protein in SFV system, we performed the same pulse-chase experiments at an earlier time-point after electroporation at which the protein expression level is lower. Results similar to those shown in Fig. 3, A and B, were obtained (data not shown).
To test whether CB1 was degraded by proteasomes, the specific proteasome inhibitor MG132 was added to CB1-expressing cells during protein synthesis. We found that the stability of CB1 was greatly enhanced, which resulted in a protein increase of approximately 6-fold (Fig. 4A, lane 1 and 2), whereas ssCB1 was increased by only 2-fold (Fig. 4A, lanes 3 and 4). As shown in Fig. 4A, the enhanced stability by MG132 did not increase the amount of glycosylated CB1, indicating that the N-tail remained cytosolic even when protein degradation was prevented by MG132.

Enhanced stability of CB1 by treatment with proteasome inhibitor MG132 (A) and membrane topology of CB1 and ssCB1 in BHK cells (B). A, CB1 and ssCB1 was synthesized in BHK cells, pulse-labeled with [35S]methionine for 30 min in the presence or absence of MG132 and then subjected to immunoprecipitation as described. B, cells expressing CB1 (lanes 1–4) or ssCB1 (lanes 5–8) in the presence of MG132 were metabolically labeled for 30 min before homogenization, followed by PK treatment. Nontreated homogenates are shown as controls (lanes 1 and 5). PK and PMSF (a protease inhibitor) were added to the samples for specific PK activity (lanes 2 and 6). Samples were treated with PK (lanes 3 and 7) or PK and a detergent (Triton X-100) to permeabilize the membrane (lanes 4 and 8) as a positive control for PK activity. ○, nonglycosylated form; •, glycosylated form; ◂, protected fragments from the PK treatment. Data shown in A are representative of two independent experiments. Data shown in B are representative experiments done in duplicate.
To further confirm membrane topology, cells expressing either the CB1 or ssCB1 gene were metabolically labeled for 30 min followed by homogenization. Homogenates (containing inside-out vesicles derived from the ER) were subjected to PK treatment. In good agreement with the observation from the cell-free system (Fig. 2B), a large protected fragment of about 35 kDa was observed in samples containing ssCB1, but no protected fragment was observed in samples containing the wild-type CB1 (Fig. 4B). Can the rapid degradation of CB1 by proteasomes be alleviated by shortening the N-tail? To answer this question, N-tail deletion mutants Δ64CB1, Δ80CB1, and Δ89CB1, with N-tails of 52, 36, and 27 amino acids, respectively, were made and characterized. Two, one, and none of the N-linked glycosylation sites remained in Δ64CB1, Δ80CB1, and Δ89CB1, respectively. The N-tail modified CB1 mutants were all stable and attained their mature forms within 2 h (Figs. 3, C–E). The amount of glycosylation increased as the length of the N-tail decreased from 45% glycosylation in Δ64CB1 to 100% in Δ80CB1 (Fig. 3, C and D). These results substantiate the relation between the stability of CB1 and the efficiency of N-tail translocation.
Simultaneously with the pulse-chase labeling experiments, immunofluorescent staining of receptors expressed at the cell surface was performed using antiserum against the N-terminal c-Myc epitope. The intensity of cell surface staining was consistent with the expression patterns seen in Fig. 3, where strong surface expression was observed for ssCB1, Δ64CB1, Δ80CB1, and Δ89CB1 (Fig. 5, B, C, D, and E, respectively). It is worth noting that Δ89CB1, containing no N-linked glycosylation site, remained stably expressed at the cell surface, suggesting that N-linked glycosylation may not be a requirement for transport of CB1 to the plasma membrane. That little surface staining was detectable for wild-type CB1 could be caused by its instability, low levels of the receptor at the plasma membrane, and/or the lack of extracellularly located N-tail (Fig. 5A). As a control, internal staining was performed on cells expressing the wild-type CB1, ssCB1, and Δ89CB1 (Fig. 6). Weak staining was observed with cells expressing the wild-type CB1, consistent with our earlier notion that the protein was quickly degraded in the cell.

Indirect immunofluorescence analyses of CB1 constructs expressed at the cell surface in BHK cells, Constructs shown are CB1 (A), ssCB1 (B), Δ64CB1 (C), Δ80CB1 (D), and Δ89CB1 (E), cells transfected with control RNA of SFV p62 (F), and ssCB1 incubated with unconjugated secondary antibody before the FITC-conjugated antibody as a control (G). Magnification is 300×. Data shown here are representative of more than two independent experiments.

Indirect immunofluorescence analyses of CB1 constructs expressed in BHK cells (internal staining). Constructs shown are CB1 (A), ssCB1 (B), Δ80CB1 (C), and cells transfected with control RNA of SFV p62 (D). Magnification is 300×. Data shown here are representative of more than two independent experiments.
To evaluate whether these modifications at the N-terminal domain affect interactions of CB1 with cannabinoid ligands, saturation binding analysis was performed using membrane preparations from HEK 293 cells transiently expressing CB1. No significant difference was found between CB1 and the mutants in their binding affinities to [3H]CP-55,940, a representative cannabinoid compound (Table 1). Immunofluorescent staining of HEK 293 cells showed that the levels of both surface and intracellularly expressed receptors were much higher for ssCB1 than for CB1, as seen in BHK cells (Fig. 7). The Bmax values in this particular study did not reflect the intensity of immunofluorescent staining in HEK 293 cells, because these two experiments were performed independently, and empirical factors such as transfection efficiency needed to be taken into account. At this stage, we do not know whether cell surface expression is the prerequisite for ligand binding activity, because the membrane preparation we used in this study is derived from whole cell homogenates, which includes intracellular membranes and plasma membrane.
Binding of [3H]CP 55,940 to the wild-type and N-tail—modified forms of CB1 expressed in HEK 293T cells
Data are presented as the mean ± S.E.M. of two experiments performed in duplicate.

Indirect immunofluorescence analyses of CB1 and ssCB1 expressed in HEK 293 cells. Pictures shown here are surface staining of CB1 (A), internal staining of CB1 (B), surface staining of ssCB1 (C), and internal staining of ssCB1 (D). The analyses were done with a confocal microscope. Magnification is 500×. Data shown here are representative of more than two independent experiments.
CB1 is expressed at the cell surface in several recombinant systems, such as AtT20 cells, Xenopus laevis oocytes and HEK 293 cells (Hsieh et al., 1999; Jin et al., 1999; McAllister et al., 2002), and is not well expressed in other systems, such as Chinese hamster ovary cells and COS-7 cells (D. Kendall, unpublished observations). Our data suggest that CB1, when expressed in BHK cells, is intrinsically unstable; this instability may be caused by the difficulty in translocating the long N-tail across the ER membrane, which in turn causes a low level of expression at the cell surface. The poor surface expression can be improved by adding a signal sequence or shortening the N-tail.
Discussion
We have examined the effect of the long N-tail of CB1 on receptor synthesis using both a cell-free in vitro system and cellular expression systems. Data from the cell-free system suggest that the large size of the N-tail of CB1 hampers its translocation across the ER membrane. We have also found that, when synthesized in BHK cells, the majority of the receptor is misfolded and subsequently degraded by proteasomes. Numerous studies indicate that attaining the correctly folded conformation in the membrane is a requirement for exiting from the ER and represents the rate-limiting step in the biosynthesis of polytopic membrane proteins. Examples can be found in mutations of the cystic fibrosis transmembrane conductance regulator and several other GPCRs (Kopito, 1999; Petaja-Repo et al., 2000). Here, we demonstrate that CB1 follows this general mechanism; in this case, translocating the N-tail and acquiring the correct topology in the ER membrane is a key control point for receptor biogenesis. The inefficient processing of CB1 but not CB2 in the cell-free translation system shown in Fig. 1 has provided the initial indication that CB1 may be intrinsically unstable, but CB2 may not. Indeed, later we found that CB1 was rapidly degraded by proteasomes when expressed in BHK cells. This result coincides with the observation that CB1 failed to be expressed as a fusion protein in E. coli because of severe proteolytic degradation, whereas CB2 was expressed in a functional form at a high level (Calandra et al., 1997). The addition of a cleavable signal sequence at the N terminus enhances the stability and surface expression of the receptor. This has been also observed for other GPCRs, such as the β2 adrenergic receptor and the endothelin B receptor (Guan et al., 1992; Kochl et al., 2002). In this study, we show that the advantage of adding a signal sequence is to facilitate targeting of the receptor to the ER membrane and to promote N-tail translocation. Without a signal sequence, which occurs in the 90% of GPCRs, the insertion of the N-tail may happen after its synthesis; in this case, the first or the second TM may serve as the ER targeting signal. The topological model of CB1 in the ER membrane shown in Fig. 2C suggests a possible scenario that the second TM directly targeted the receptor to the ER membrane and initiated partial assembly of the receptor; subsequently, the correct topology of the first TM and translocation of the N-tail could be achieved. As in the case of CB1, the latter step was hindered by the long N-tail. Detailed mechanism of GPCR assembly in the ER membrane is not clear; however, a recent cross-linking study on the assembly of opsin and neurotensin receptor showed that the first and the second TMs associate with distinct components of the ER translocon during the receptor biosynthesis (Meacock et al., 2002).
The hydrophobic nature of cannabinoid ligands suggests that their binding site is localized within the 7TM bundle of the receptor (McAllister et al., 2002). Our data show that the N-tail of CB1 can be deleted up to 89 amino acids without affecting the receptor binding to the ligand CP-55,940. This suggests that the long N-tail of CB1, which accounts for more than 25% of the protein, may not be relevant to ligand binding or activation of the receptor. This notion is supported by the existence of an alternatively spliced isoform, CB1A, that has a shorter N-tail of 55 amino acid and differs from CB1 in the first 28 amino acids, yet displays pharmacological properties similar to those of CB1 (Gerard et al., 1991; Rinaldi-Carmona et al., 1996). In native tissues, the expression of CB1A amounts to about 20% of CB1 at the mRNA level (Shire et al., 1995). According to the results presented in this study, one may speculate that, with a shorter N-tail, CB1A would be more stable during its synthesis and would be expressed at the cell surface more readily than CB1.
Cellular factors may be required to facilitate surface expression of CB1. In hippocampal neurons, strong labeling of CB1 was found in presynaptic terminal plasma membranes but not in other plasma membranes (Katona et al., 1999). The presynaptic localization of CB1 at the plasma membrane is essential for its quick response as a neuromodulator. Agonist-induced internalization of CB1 has been found to be an important mechanism to regulate the availability of the receptor at the plasma membrane (Jin et al., 1999; Coutts et al., 2001). Nonetheless, it is reasonable to speculate that the process of biosynthesis can also be a control point to modulate the expression, subcellular location, and thus function of CB1. Many specific GPCR-associating proteins have been identified to play a role as molecular chaperones to regulate the cell surface expression of GPCRs (Brady and Limbird, 2002). In addition, a recent study on the δ-opioid receptor has demonstrated that biosynthesis of a GPCR can be regulated at the ER level pharmacologically, where hydrophobic ligands act as chemical chaperones to promote the correct folding and maturation of the receptor (Petaja-Repo et al., 2002). Interestingly, it has been shown that the cell surface expression of CB1 was increased by adding a CB1-specific inverse agonist, SR141716 (Rinaldi-Carmona et al., 1998). A chaperone-mediated mechanism may exist that regulates the synthesis, degradation, folding, and trafficking of CB1 in which the long N-tail may play a role.
Acknowledgments
CP-55,940 was kindly provided by Pfizer Inc. (Groton, CT). The human CB1 cDNA was kindly provided by Dr. Marc Parmentier (University Libre de Bruxelles, Bruxelles, Belgium). Dog pancreas microsomes were a kind gift from Dr. M. Sakaguchi (Kyushu University, Fukuoka, Japan). We thank Sharyn Rusch for helpful discussions during manuscript preparation. We thank Dr. Donald M. Engelman. We are deeply grateful to Dr. Henrik Garoff for providing excellent laboratory facilities.
Footnotes
-
This research was supported in part by grants from the Swedish Research Council and the Swedish Cancer Foundation (to G.v.H.) and National Institutes of Health grants GM37639 (to D.A.K.) and GM54160 (to Donald M. Engelman).
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; TM, transmembrane; ER, endoplasmic reticulum; Endo H, endoglycosidase H; PMSF, phenylmethylsulfonyl fluoride; PK, proteinase K; MG132, carbobenzoxy-leucyl-leucyl-leucinal; CP-55,940, (1α,2β)-(R)-5-(1,1-dimethylheptyl)-2-[5-hydroxy-2-(3-hydroxypropyl)cyclohexyl]phenol; FITC, fluorescein isothiocyanate; PCR, polymerase chain reaction; HEK, human embryonic kidney; PAGE, polyacrylamide gel electrophoresis; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; SFV, Semliki Forest virus; SR141716, N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboximide hydrochloride; BHK, baby hamster kidney; Lep, leader peptidase.
-
↵1 Current address: Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut.
- Received November 5, 2002.
- Accepted May 20, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}