


Abstract
The cytoplasmic side of the voltage-dependent Na+ channel pore is putatively formed by the S6 segments of domains I to IV. The role of amino acid residues of I-S6 and II-S6 in channel gating and local anesthetic (LA) block was investigated using the cysteine scanning mutagenesis of the rat skeletal muscle Na+ channel (Nav1.4). G428C uniquely reduced sensitivity to rested state or first-pulse block by lidocaine without alterations in the voltage dependence or kinetics of gating that would otherwise account for the increase in the IC50 for block. Mutations in I-S6 (N434C and I436C) and in II-S6 (L785C and V787C) increased sensitivity to first-pulse block by lidocaine. Enhanced inactivation accounted for the increased sensitivity of N434C to lidocaine and hastening of inactivation of I436C in the absence of drug could account for higher affinity first-pulse block. Mutations in I-S6 (I424C, I425C, and F430C) and in II-S6 (I782C and V786C) reduced the use-dependent lidocaine block. The reduction in use-dependent block of F430C was consistent with alterations in inactivation gating; the other mutants did not exhibit gating changes that could explain the reduced sensitivity to lidocaine. Therefore, several amino acids (I424, I425, G428, I782, and V786), in addition to those previously identified (Yarov-Yarovoy et al., 2002), alter the sensitivity of Nav1.4 to lidocaine, independent of mutation-induced changes in gating. The magnitude of the change in the IC50 values, the isoform, and LA dependence of the changes in affinity suggest that the determinants of binding in I-S6 and II-S6 are subsidiary to those in IV-S6.
Voltage-dependent sodium (Na+) channels are key mediators of cellular excitability and targets of local anesthetic (LA) antiarrhythmic and anticonvulsant drugs. Each Na+ channel is formed by an α subunit that is a large membrane-spanning glycoprotein composed of four homologous domains (I–IV) and, depending on tissue type, one or more smaller accessory β subunits (Catterall, 2000). Each domain of the α subunit contains six transmembrane segments (S1–S6). A portion of the cytoplasmic mouth of ion-conducting pore of the Na+ channel is thought to be formed by the carboxyterminal portion of the S6 of each domain.
Important insights into the structure and function of the outer mouth of the Na+ channel pore have been revealed by site-directed mutagenesis (Chiamvimonvat et al., 1996; Favre et al., 1996; Perez-Garcia et al., 1996; Sunami et al., 1997; Tsushima et al., 1997; Yamagishi et al., 1997). However, the constituents of the inner mouth of the pore are far less certain. Clues to the structure of the inner pore mouth have come from examination of the structure of a potassium channel from Streptomyces lividans, KcsA (Doyle et al., 1998) and from studies of LA block of voltage-dependent Na+ channels (Gingrich et al., 1993; Ragsdale et al., 1994, 1996; Nau et al., 1999; Wang and Wang, 1999; Wang et al., 2000; Yarov-Yarovoy et al., 2001, 2002).
Local anesthetics are amphiphilic molecules that have been useful molecular probes of the structure of the Na+ channel (Gingrich et al., 1993; Ragsdale et al., 1994, 1996; Nau et al., 1999; Wang and Wang, 1999; Wang et al., 2000; Yarov-Yarovoy et al., 2001, 2002). Alanine scanning mutagenesis of IV-S6 of the rat brain (Nav1.2) Na+ channel has identified amino acid residues that form the hydrophobic receptor subsite for LA binding (Ragsdale et al., 1994, 1996). F1764A abolished and I1760A and Y1771A reduced use-dependent etidocaine block. F1764A was the only mutant that significantly reduced rested state block (Ragsdale et al., 1994). F1764A and Y1771A also reduced use-dependent lidocaine block without significantly affecting the resting channel block (Ragsdale et al., 1996). The structural model proposed to rationalize these findings was an α-helix with side chains of the amino acid residues at positions 1760, 1764, and 1771 on the same face of the helix forming the hydrophobic subsite lining the inner pore of the channel. Lysine mutations at equivalent positions to F1764, N1769, and Y1771 in the rat skeletal muscle (Nav1.4) Na+ channel altered resting and inactivated channel affinity for cocaine and benzocaine (Wright et al., 1998).
A number of studies have implicated amino acid residues in the S6 segments of other domains in channel gating and LA binding. In I-S6, all substitutions of the native asparagine at position 434 in Nav1.4 increased the resting state block by bupivacaine, but only N434K exhibited decreased inactivated state block (Wang et al., 1998; Nau et al., 1999). In I-S6 of Nav1.2, I409A was associated with decreased inactivated state affinity for etidocaine but not for its tricyclic congener sipatrigine or the anticonvulsant lamotrigine (Yarov-Yarovoy et al., 2002). In II-S6, V787K (Nav1.4) increased the affinity of resting channels to bupivacaine (Wang et al., 2001). However, no alanine mutants in II-S6 of Nav1.2 exhibited altered LA blocking affinity (Yarov-Yarovoy et al., 2002). L1465A and I1469A in III-S6 of Nav1.2 reduced the affinity of inactivated channels to lamotrigine; however, these mutations and N1466A increased the affinity of rested channels to lamotrigine (Yarov-Yarovoy et al., 2001). S1276K and L1280K (equivalent to L1465 of Nav1.2) in III-S6 of Nav1.4 exhibited decreased affinity for bupivacaine and complete resistance to batrachotoxin (Wang et al., 2000). It is notable that mutations in all four S6 segments produced changes in both activation and inactivation gating that can complicate the interpretation of mutation-induced alterations in local anesthetic block (Ragsdale et al., 1994, 1996; Wright et al., 1998; Nau et al., 1999; O'Reilly et al., 2001; Yarov-Yarovoy et al., 2001, 2002).
We used cysteine scanning mutagenesis to investigate the contribution of domain I and II S6 segments of the skeletal muscle Nav1.4 channel to the formation of the inner pore and determine the isoform-specific contribution of these residues to channel gating and LA block. Our data suggest that mutation-induced alterations in inactivation gating played an important role in altering sensitivity to LA block. However, several residues in I-S6 (Ile424, Ile425, and Gly428) and II-S6 (Ile782 and Val786) altered lidocaine block of Nav channels independent of any significant changes in channel gating. A preliminary report of these results has been published in abstract form (Kondratiev et al., 2000).
Materials and Methods
Molecular Biology. The cDNA encoding wild-type Nav1.4 μ1 skeletal muscle Na+ channel α subunit (Trimmer et al., 1989) cloned into a vector driving the bicistronic expression of green fluorescent protein and rNAv1.4α subunit using an internal ribosomal entry site (IRES) and the rat brain β1 subunit was cloned pCMV-5 (Isom et al., 1992). Mutant channels were generated polymerase chain reaction using oligonucleotide primers encoding the mutation of interest using the QuikChange site-directed tagenesis kit (Stratagene, La Jolla, CA). All mutations were firmed by DNA sequencing of the region surrounding the mutation and at least two independent clones of each mutation were studied. HEK293 cells (American Type Culture Collection, Manassas, were cotransfected with an equimolar ratio of the Na+ channel subunit and rat brain β1 subunit cDNA using LipofectAMINE Plus according to the manufacturer's instructions. After transfection cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum and penicillin/streptomycin at 37°C in a 5% CO2-humidified incubator and used for experiments after 24 h.
Electrophysiology and Electropharmacology. Transfected cells expressing Na+ channels were identified by epifluorescence microscopy and voltage-clamped 24 h after transfection using whole-cell configuration of the patch-clamp technique with an Axopatch 200B patch-clamp amplifier (Axon Instruments, Inc., Union City, CA) interfaced with a personal computer. Patch electrodes were pulled from borosilicate glass and had 1- to 2-MΔ tip resistances. Voltage command protocols were delivered, and data were acquired using custom-written software. Cell capacitance was calculated integrating the area under an uncompensated capacity transient elicited by a 10-mV hyperpolarizing test pulse from a holding potential of –70 mV. Series resistance was then compensated typically 80 to 85%. Currents were filtered at 5 kHz and digitized with 12-bit resolution. All experiments were performed 10 min after establishment of the whole-cell configuration to ensure adequate dialysis of the cell and to minimize the initial rapid time-dependent shifts in voltage dependence of activation and steady-state inactivation. Standard voltage protocols were used to generate activation and availability relationships. A two-pulse protocol was used to assess recovery from inactivation after short (50-ms) and long (1-s) test depolarizations to –20 mV; the curves were best fit with two exponentials (with the exception of N434C).
Mutant channels were screened for changes in local anesthetic sensitivity by comparing the first-pulse block and use-dependent block at a stimulation frequency of 10 Hz (50-ms pulse duration) from a holding potential of –120 mV in the presence of 200 μM lidocaine to the wild-type channel. The magnitude of use-dependent block was the ratio of the steady-state current after 30 pulses in the presence of lidocaine to the current in the absence of drug. Dose-response curves were generated by random application of lidocaine to the bath at concentrations ranging from 1 μM to 20 mM. At least three determinations were made at each lidocaine concentration. All lidocaine block was fully reversible with washout of the drug. The IC50 value was determined by fitting the dose-response curves with a binding isotherm of the form I/I0 = 1/(1 + ([lidocaine]/IC50)), where I and I0 are the peak Na+ currents elicited by a 50-ms voltage step from –120 to –20 mV in the presence and absence of lidocaine, respectively. The reduction in the peak current elicited by the first pulse after a long rest (10 s) at –120 mV was used to determine the tonic block IC50, and the current after 30 pulses at 10 Hz was used to determine the use-dependent IC50.
Solutions. Whole-cell Na+ currents were recorded in an extracellular solution containing 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, pH 7.4. The pipette solution contained 35 mM NaCl, 105 mM CsF, 1 mM MgCl2, 10 mM HEPES, 10 mM EGTA, pH 7.2. Experiments were performed at room temperature (18–22°C). Lidocaine HCl (2-diethylamino-N-[2,6-dimethylphenyl]acetamide; Sigma-Aldrich, St. Louis, MO) was dissolved in the extracellular solution and diluted to the appropriate concentration.
Data Analysis. Pooled data were expressed as mean ± S.E.M. Statistical comparisons were performed using an unpaired Student's t test with p values of <0.05 considered statistically significant. Data were fitted to the appropriate functions by nonlinear least-squares methods (Levenburg-Marquardt algorithm; OriginLab Corp, Northampton, MA).
Results
Effect of Mutations on Baseline Na+ Channel Properties. Cysteine was substituted for the native amino acid residues in the S6 segments of domain I from residues Ile424 through Ile436 and domain II from residues Val779 through Phe791 in Nav1.4 (Fig. 1). All cysteine mutants expressed Na+-selective currents (3–5 nA at –20 mV) when cotransfected with the β1 subunit. The mutations had no significant effect on the reversal potential. Figure 2, A and B, shows raw current records and normalized current-voltage (I-V) relations of mutant Na+ channels that were significantly shifted compared with wild-type, grouped by domain. I425C, I436C, L785C, and V787C produced significant depolarizing shifts and V779C a hyperpolarizing shift of the negative slope region of I-V relations compared with wild type. The voltage at half-maximal activation (V0.5) of the conductance-voltage (gNa-V) relations for I425C, I436C, L785C, and V787C were shifted by +5 to +8 mV compared with wild type (Fig. 2, C and D; Table 1). I425C, I432C, and V787C significantly increased the slopes of gNa-V curves (Table 1).

Topology of the first two domains of the Na+ channel α subunit. The residues (13 in each) that were mutated in I-S6 and II-S6 segments are shown below the topology cartoon.

Normalized I-V relations for wild-type and I-S6 (A) and II-S6 (B) mutant channels. Shown are the mutants with significantly shifted I-V relations. The representative families of Na+ currents and are shown above the graphs. The I-V relations were obtained by plotting the peak INa elicited by 50-ms test pulses to various voltages ranging from –70 to +60 mV. The holding potential was –120 mV. Symbols indicate peak INa (mean ± S.E.M.) at different voltages normalized to the maximum INa. The voltage of half-maximal activation (V0.5) and the slope factor (s) were obtained from a fit of the mean normalized data with the Boltzmann function: g/gmax = 1/(1 + exp((V0.5 – V)/s)), where g = INa/(V – VNa), gmax is the maximum Na+ conductance, and VNa is the reversal potential. Solid lines are the nonlinear least-squares fits to the mean data. C and D, bar plots show the shifts of half-maximal activation voltages compared with wild type. Voltages were normalized by subtracting the corresponding values for wild type. For wild type, V0.5 of activation was –33 ± 1 mV (n = 30). Significant differences are indicated by asterisks (p < 0.05).
Basic parameters of Na+ channel gating
Values are presented as mean ± S.E.M.
Figure 3, A and B, shows the steady-state availability curves for mutant Na+ channels that were significantly shifted compared with wild type. In contrast to the relative paucity of effects on activation, mutations at a number of positions in I-S6 and II-S6 altered steady-state availability. The steady-state availability of three mutants in I-S6 (F426C, F430C, and N434C) and one in II-S6 (V779C) were significantly shifted in the hyperpolarizing direction compared with wild type. Two mutant channels in I-S6 (Y431C and I436C) and three in II-S6 (I782C, V786C, and L790C) exhibited significant depolarizing shifts in availability (Fig. 3, C and D; Table 1). Of the I-S6 mutants that exhibited a shift in the voltages of half-inactivation (V0.5), only N434C had a significantly greater slope of the steady-state availability relation (Table 1). Two mutations in II-S6 (M780C and G783C) significantly increased the slope of availability curves without altering the V0.5, whereas V786C significantly reduced the slope compared with wild type (Table 1).

Steady-state availability curves for wild-type and mutant Na+ channels in I-S6 (A) and II-S6 (B). Data are shown for mutant channels that significantly shifted the availability curves compared to wild type. The steady-state availability curves were generated by plotting the peak current elicited by a test pulse of 50 ms to –20 mV after a 500-ms prepulse to a range of voltages from –160 to 0 mV in steps of 5 mV (the protocol is shown in the inset). The holding potential was –120 mV. Currents obtained during the test pulse were normalized to the maximum current (Imax) and plotted as a function of the prepulse potential. Half-maximal inactivation voltage (V0.5) and the slope factor (s) were obtained from a fit of the mean normalized data with the Boltzmann function: I/Imax = 1/(1 + exp((V0.5 – V)/s)). Symbols indicate peak INa (mean ± S.E.M.) at different voltages normalized to the maximum INa (Imax). Solid lines are the Boltzmann fits to the data. C and D, summary plots show the shifts of half-maximal steady-state inactivation voltages compared with wild type. For wild type, V0.5 of steady-state availability was –76.8 ± 0.7 mV (n = 30). The asterisks indicate statistically significant differences at p < 0.05.
Effect of Mutations on Kinetics of Inactivation. Mutations in I-S6 and II-S6 altered the kinetics of inactivation. Figure 4, A to D, shows plots of the fractional recoveries of peak INa as a function of time at –120 mV after 50-ms (Fig. 4, A and C) and 1-s (Fig. 4, B and D) prepulses to –20 mV; the wild-type and mutant channels that exhibited significantly different kinetics are shown. The recoveries from inactivation after the 50-ms prepulse (Fig. 4, A and C) were best fit with two exponentials. The dominant fast time constant was 2.1 ± 0.2 ms comprising 96 ± 11% of the recovery; the remaining slow component had a time constant of 13.5 ± 2.8 ms for wild-type channels. L427C recovered faster than wild type with a fast time constant of 1.2 ± 0.1 ms (Fig. 4A; Table 2). Two other mutants, N434C (I-S6) and V779C (II-S6), recovered more slowly than wild type. N434C exhibited a slowed τfast (3.3 ± 0.1 ms) of reduced amplitude (62% of total) compared with wild type. The slow component of recovery of N434C was enhanced (37% of total) and was more than an order of magnitude slower (τslow = 263 ± 9 ms) than the slow component of wild type; however, channels still recovered fully in 1 s (Fig. 4A; Table 2). V779C recovered with a dominant fast time constant of 3.2 ± 0.1 ms after 50-ms prepulses (Fig. 4C; Table 2).

Kinetics of inactivation. A and B, kinetics of recovery from inactivation of wild type and significantly different I-S6 (A) and II-S6 (B) mutant channels at –120 mV after a 50-ms depolarization to –20 mV (P1). Symbols indicate peak INa (mean ± S.E.M.) obtained during the P2 pulse relative to the peak INa obtained during the P1 pulse. The solid lines are biexponential function fits to the recovery data. C and D, kinetics of recovery from inactivation of wild-type and significantly different I-S6 mutant channels (C) and II-S6 mutant channels (D) at –120 mV after a 1-s depolarization to –20 mV (P1). E and F, kinetics of entry into slow inactivated states. Wild type and mutant channels in I-S6 (E) and II-S6 (F) that significantly altered kinetics of entry into slow inactivated states are shown. The first pulse (P1) of varied duration to –20 mV was followed by a 10-ms recovery period at –120 mV to permit recovery from fast inactivation before a second 50-ms pulse (P2) to –20 mV was applied. Current amplitudes obtained during the second pulse were normalized by the current amplitude obtained during the first pulse and plotted as a function of the duration of P1. The solid lines are best fits to the data using a second-order exponential function. The cells were held at –120 mV between pulses. Voltage protocols for the recovery from inactivation and entry into the slow inactivated states are shown in the insets.
Kinetics of recovery from and entry into the slow inactivated states Values are presented as mean ± S.E.M.
To study slow inactivated states, we examined recoveries of the channels after longer prepulses. A number of mutations slowed the kinetics of recovery from inactivation after 1-s prepulses with N434C producing the most dramatic effect (Fig. 4, B and D; Table 2). Wild-type recovery after long prepulses was best fit with two exponentials: the fast component (45% of total amplitude) with the time constant of 3.0 ± 0.3 ms and the slow component (51% of total amplitude) with the time constant of 155 ± 8 ms (Table 2). N434C (Fig. 4B; Table 2) recovered with a single-exponential time course that was 3-fold slower than the slow component of recovery of the wild-type channel (τ = 429 ± 78). In addition to N434C, eight other mutations, six in I-S6 (I424C, L427C, G428C, L432C, I433C, and I436C) and two in II-S6 (I782C and V786C) slowed the fast time component of recovery from inactivation without a significant change in the slow component compared with wild type (Fig. 4, B and D; Table 2). Entry into slow inactivated states was assessed using a standard two-pulse protocol. The time constant for entry into slow inactivated states for wild type was 1,200 ± 30 ms. A number of mutant Na+ channels in I-S6 and fewer in II-S6 significantly altered the kinetics of entry into slow inactivated states (Fig. 4, E and F; Table 2). Several mutant channels with slowed recovery kinetics after long prepulses exhibited faster entry into slow inactivated states (I-S6: I424C, G428C, L432C, I433C, N434C, and I436C; II-S6: I782C and V786C). Two mutants in I-S6 (S429C and F430C) and one in II-S6 (V781C) delayed the entry into slow inactivated states (Fig. 4, E and F; Table 2). The residual current after a 10-s depolarizing pulse is the component that resisted entry into slowly recovering inactivated states. The wild-type channel exhibited a residual current that was 6.1% of the peak; F430C, M780C, V781C, V787C, L790C, and F791C had significantly greater residual currents and I424C, F426C, L427C, N434C, and V786C had significantly smaller residual currents compared with wild type (Table 2).
Lidocaine Block of Na+ Channels. The S6 membrane repeats, particularly in domains III and IV, are important components of local anesthetic binding sites in the Na+ channel. All mutant channels were assessed for their sensitivity to tonic (rested) and use-dependent block by lidocaine. Lidocaine was continuously added to the bath for 10 min before beginning of recordings. With infrequent stimulation (0.1 Hz) lidocaine (200 μM) reduced maximum Na+ conductance of the wild-type channels by 19 ± 2% (Fig. 5A; Table 3). G428C (I-S6) exhibited significantly reduced block of gNa,max by lidocaine compared with wild type, whereas N434C, I436C, L785C, and V787C displayed enhanced rested state block (Fig. 5A).

State-dependent block of wild-type and mutant channels with significantly different sensitivity to lidocaine. A, plots of the fractional first-pulse rested block of maximum Na+ conductance (gmax) and use-dependent of INa at –20 mV by 200 μM lidocaine. Cells were held at –120 mV for 10 min during the application of lidocaine. Each bar represents mean ± S.E.M. The statistically significant differences at p < 0.05 are indicated for rested (#) and use-dependent (*) block. B, plot of the IC50 values for first-pulse rested (squares) and use-dependent (circles) block of the mutant channels with altered gating and/or sensitivity to 200 μM lidocaine. Mutants with significantly different IC50 values compared with wild type are indicated by solid symbols.
IC50 concentrations for tonic and use-dependent block Values are presented as mean ± S.E.M.
Local anesthetic block is conformation-sensitive with the drug preferentially binding to the open and inactivated states (Hille, 1977; Hondghem and Katzung, 1977; Balser et al., 1996,Balser et al., 1996). At 10-Hz stimulation frequency (50-ms pulse durations and 50-ms recovery periods) from a holding potential of –120 mV, 200 μM lidocaine reduced the peak wild-type current by 68 ± 2% (Fig. 5A). F430C, I782C, and V786C exhibited significantly attenuated block at rapid rates of stimulation compared with wild type (Fig. 5A).
Dose-response curves were determined for mutant channels that exhibited significantly altered basic biophysical properties or sensitivity to 200 μM lidocaine compared with wild type. The IC50 for tonic block of G428C by lidocaine was 2-fold greater than the IC50 for wild type, whereas the IC50 values for N434C, I436C, and L785C mutant channels were 40 to 60% lower (Fig. 5B; Table 3). The IC50 values for use-dependent block of I424C, I425C, F430C, I782C, and V786C were more than 2-fold greater than the IC50 value for wild type (Fig. 5B; Table 3).
Effect of Lidocaine on Kinetics and Voltage Dependence of Gating. Apparent differences in lidocaine affinity may result from changes in the voltage dependence or kinetics of channel gating (Nuss et al., 2000). A number of cysteine substitutions in I-S6 and II-S6 produced changes in the voltage dependence and kinetics of gating (Figs. 2, 3, 4; Tables 1 and 2). Lidocaine shifted all of the steady-state availability curves at least 10 mV in the hyperpolarizing direction compared with drug-free controls (Fig. 6; Table 4) but had more modest and inconsistent effects on the activation curves (Table 4). The V0.5 of activation and steady-state inactivation for both I-S6 and II-S6 mutant channels were not significantly different from wild-type in the presence of lidocaine with few exceptions. I425C, I436C, L785C, and N789C had significantly depolarized activation V0.5 values (Table 4). F426C and N434C had significantly hyperpolarized availability V0.5s in the presence of lidocaine compared with wild type, whereas Y431C, I782C, and V786C significantly had depolarized availability V0.5s in the presence of lidocaine compared with wild type (Table 4). In general, lidocaine produced shifts of similar magnitude in the voltage dependence of the steady-state availability relations in mutant and wild-type channels. The only exception was N434C that exhibited an exaggerated hyperpolarizing shift in the availability curve (Fig. 6A).

Plots of V0.5 of steady-state availability for wild-type and mutant channels. The V0.5 values of availability under control conditions (open circles) and in the presence of 200 μM lidocaine (filled circles) for I-S6 (A) and II-S6 (B) mutant channels. Each symbol represents mean ± S.E.M. The statistically significant V0.5 differences at p < 0.05 are indicated for control (*) and in the presence of 200 μM lidocaine (#).
Basic parameters of Na+ channel gating in 200 μM lidocaine
Values are presented as mean ± S.E.M.
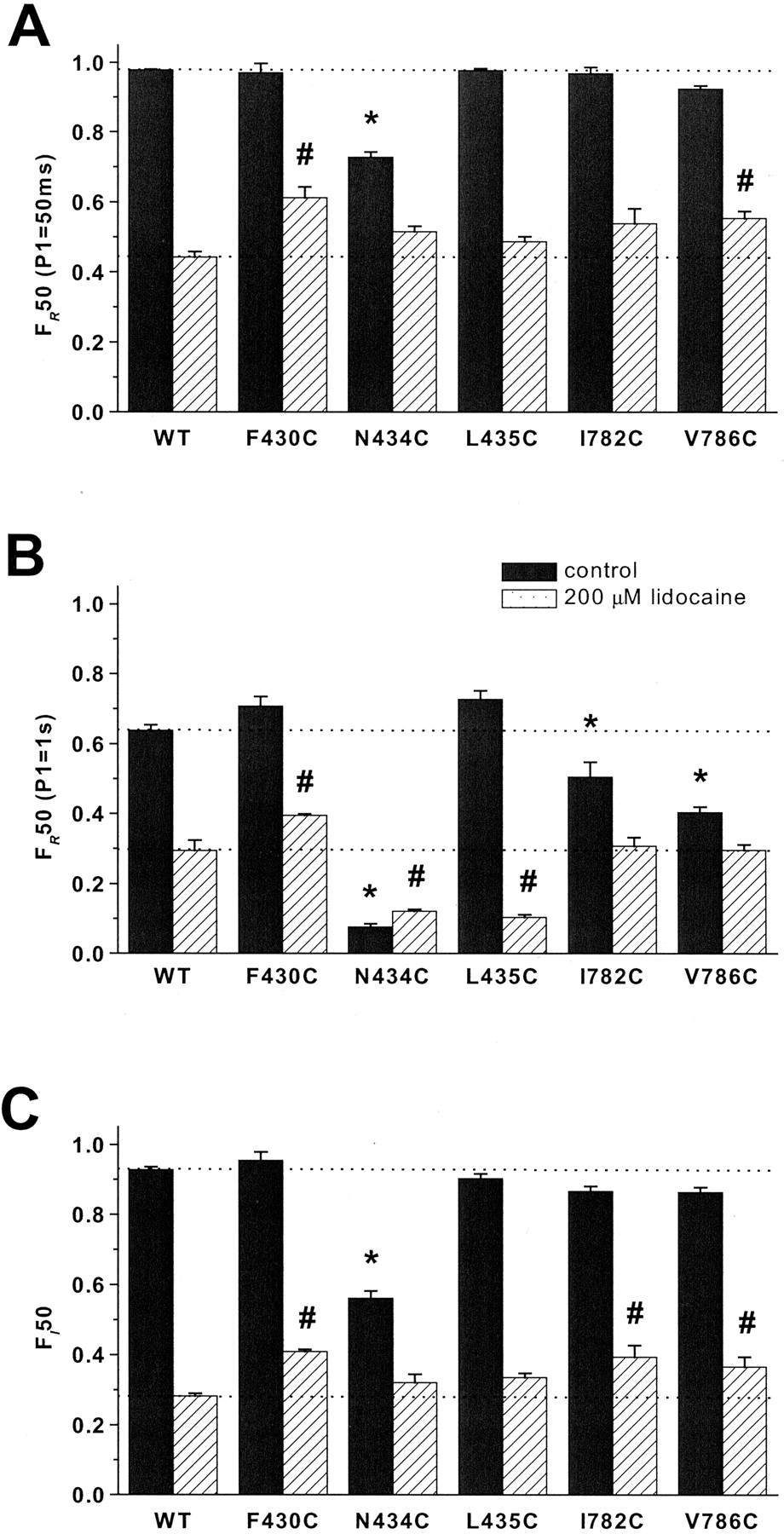
Lidocaine slowed the recovery from inactivation and hastened the entry into the inactivated states increasing the kinetic complexity of both processes. We used the fractional current at 50 ms (F50) as a model-independent index of the rate of recovery from (FR50) and entry into (FI50) inactivated states in the absence and presence of 200 μM lidocaine. In the presence of drug, F430C exhibited hastened recovery from inactivation and slowed the entry into inactivated states (Fig. 7, A and C). Similar to F430C, two mutants in II-S6, I782C and V786C, exhibited hastened recovery after 50-ms depolarizations (V786C) and slowed entry into inactivated states (I782C and V786C; Fig. 7, A and C). Despite dramatic enhancement of the rate of entry and slowed recovery from inactivation in the absence of drug (Fig. 4; Table 2), the entry rate (Fig. 7C) and recovery after short pulses (Fig. 7A) for N434C were not significantly different from wild type in the presence of lidocaine. However, recoveries after 1-s prepulses were significantly slower for N434C and L435C compared with wild type (Fig. 7B).

Gating kinetics of wild-type and I-S6 and II-S6 mutants with significantly different inactivation gating in the absence and presence of 200 μM lidocaine. A and B, fractional recovery at 50 ms (FR50) is plotted after 50-ms (A) and 1-s (B) depolarizing pulses to –20 mV. C, plots of the fractional entry into slow inactivated states after 50-ms (FI50) depolarizing pulses to –20 mV from a holding potential of –120 mV. Each column represents mean ± S.E.M. The statistically significant differences at p < 0.05 are indicated for control (*) and lidocaine (#).
Discussion
Effects of Cysteine Substitutions in I-S6 and II-S6 on Na+ Channel Properties. In contrast to serial lysine substitution (Wang et al., 2000, 2001), replacement of residues in I-S6 and II-S6 segments with cysteine was well tolerated; all mutant channels were functional when the α subunits were coexpressed with the β1 subunits in HEK293 cells. Our data reveal that a distinct pattern of mutant channels that display significant changes in the voltage dependence and kinetics of gating compared with wild-type Nav1.4.
Rotational movement of the M2 (analogous to S6) segment accompanies opening of the KcsA channel (Perozo et al., 1999). Similarly, activation of voltage-dependent channels has been associated with movements of the S6 segments; indeed these regions likely underlie the activation gate (Liu et al., 1997; Holmgren et al., 1998; del Camino et al., 2000; Li-Smerin et al., 2000). Mutations of Nav1.4 in I-S6 and II-S6 that altered activation gating (I425C, I436C, L785C, and V787C) shift the curves in the depolarizing direction as if substituting these hydrophobic residues with cysteine eliminated or generated new intramolecular contacts that stabilized the closed state (Fig. 2). A similar depolarizing shift and reduction in the slope of V787C has been described (O'Reilly et al., 2001). In contrast to N434C, the alanine substitution at position 434 in Nav1.4 produced a depolarizing shift in the activation curve (Wang and Wang, 1997; Nau et al., 1999). This pattern of alterations in the voltage dependence of activation is distinct from that of alanine scanning mutagenesis of analogous residues of I-S6 and II-S6 in Nav1.2. In Nav1.2, only the alanine substitutions at positions 418 and 975 (analogous to Asn434 and Leu788 in Nav1.4) exhibited significant depolarizing shifts in the activation curve (Yarov-Yarovoy et al., 2002).
The discordance of the effects of some of the mutations on activation and inactivation gating suggests that the S6 segments undergo conformational changes during inactivation gating that are not simply linked to activation even though opening and inactivation are coupled processes in Na+ channels (Armstrong and Bezanilla, 1977; Chahine et al., 1994; Yang and Horn, 1995; Yang et al., 1996). I436C on the cytoplasmic side of I-S6 exhibited activation and steady-state availability curves that were shifted in the depolarizing direction (Figs. 2 and 3; Table 1), consistent with resistance to channel opening and subsequent inactivation. I425C on the extracellular side of I-S6 had a similar shift in the activation curve and a smaller shift in the availability curve in the same direction. V779C on the extracellular side of II-S6 exhibited hyperpolarizing shifts in the activation and availability curves suggesting that this mutation destabilized the closed state and promoted closed state inactivation (Figs. 2 and 3; Table 1). Mutations separated by four residues in I-S6 (F426C, F430C, and N434C) and II-S6 (I782C, V786C, and L790C) exhibited significant hyperpolarizing and depolarizing shifts in the voltage dependence of steady-state availability, respectively (Fig. 3). This pattern suggests that a face of the α-helices in I-S6 and II-S6 stabilizes and destabilizes inactivation, respectively. This would be consistent with rotation and/or translation of the S6 helices during inactivation. Our results differ from that of alanine mutants in the analogous region of Nav1.2 expressed in Xenopus laevis oocytes where small shifts of the half-inactivation voltages, mostly in the depolarizing direction were observed (Yarov-Yarovoy et al., 2002). The substituting amino acid has profound effects on the magnitude and direction of the gating shift observed in S6 mutant channels (Nau et al., 1999), consistent with the notion that a number of interactions are altered during movement of the S6 during inactivation gating.
A number of kinetically distinct inactivated states have been described in Na+ channels (Bezanilla and Armstrong, 1977; Balser et al., 1996,Balser et al., 1996; Featherstone et al., 1996; Hayward et al., 1997; Benitah et al., 1999). Mutations in I-S6 and II-S6 significantly altered the rate at which the channels enter or recover from inactivated states (Fig. 4). Longer depolarizations favored the recruitment of slower forms of inactivation. The precise mechanism of slow inactivation in Na+ channels is not known but residues in I-S6 and II-S6 altered the kinetics of these processes. The effects were particularly prominent in I-S6 where six of the 13 mutants delayed recovery from inactivation after long pulses and hastened entry into inactivated states (Figs. 4 and 7) seemingly favoring occupancy of slowly recovering inactivated states. Arguably, the most dramatic inactivation gating changes were observed with mutation of a highly conserved asparagine in I-S6 (N434C). Conserved asparagines are present at sequence-aligned positions in the S6 segments of all four domains: Asn434 (I), Asn789 (II), N1281 (III), and N1584 (IV). N434C uniquely modulated the voltage dependence and kinetics of channel gating stabilizing inactivated states, whereas N789C produced no significant changes in the voltage dependence or kinetics of gating. Our data are consistent with previous reports that have highlighted the importance of Asn434 in channel gating (Wang and Wang, 1997, 1998; Nau et al., 1999). Substitutions at position 434 in Nav1.4 with aromatic amino acids and cysteine produced large hyperpolarizing shifts in the availability curves, whereas substitution with polar and charged residues tended to produce small depolarizing shifts (Nau et al., 1999). Hydrophobic interactions involving the residue at position 434 seem to stabilize inactivation. Replacement of the asparagines in III-S6 and IV-S6 of Nav1.2 also produced significant effects on the voltage dependence and kinetics of gating that resulted in altered sensitivity to LAs (Ragsdale et al., 1994; Wright et al., 1998; Yarov-Yarovoy et al., 2001). Alanine substitutions in III-S6 and IV-S6 caused significant hyperpolarizing shifts of availability curves and increased rested state sensitivity to etidocaine (Ragsdale et al., 1994) and lamotrigine (Yarov-Yarovoy et al., 2001), whereas a large depolarizing shift of the availability curve of N1584K was associated with less rested and inactivated state LA block (Wright et al., 1998).
Lidocaine Block of Cysteine Mutants in I-S6 and II-S6. The cytoplasmic portion of S6 is presumed to line in the inner mouth of the channel if the structure of voltage-gated ion channels is analogous to that of KcsA (Doyle et al., 1998; Lipkind and Fozzard, 2000). Mutations of S6 residues in all four domains alter LA block of the Na+ channel (Ragsdale et al., 1994; Wright et al., 1998; Nau et al., 1999; Wang et al., 2001; Yarov-Yarovoy et al., 2001, 2002). Prominent destabilization of LA block occurs with mutations in IV-S6 (Ragsdale et al., 1994, 1996) and to a lesser extent III-S6 (Yarov-Yarovoy et al., 2001). The role that I-S6 and II-S6 play in LA binding is less certain. However, recent alanine scanning mutagenesis of I-S6 and II-S6 in the Nav1.2 channel reveals a single mutation (I409A) that selectively reduced the affinity for etidocaine (Yarov-Yarovoy et al., 2002). Other studies in the Nav1.4 channel background have demonstrated the importance of residues in I-S6 in LA block and have suggested that the quaternary ammonium interacts with Asn434 (Wang et al., 1998; Nau et al., 1999).
Our data implicate a number of other residues in I-S6 and II-S6 of Nav1.4 in LA block. The interpretation of mutation-induced alterations in LA block is often complicated by concomitant changes in channel gating. In fact, given the extent of changes in channel gating demonstrated by I-S6 and II-S6 cysteine mutants in the absence of drug, it is notable that larger changes in the sensitivity to block by lidocaine were not observed. It is likely that mutations in this region of the channel concomitantly alter both gating and drug binding (Wang et al., 1998; Nau et al., 1999; Yarov-Yarovoy et al., 2002). The effects of the mutations that significantly altered lidocaine sensitivity were modest with the IC50 values for tonic and use-dependent block of mutants differing by less than a factor of 3 (Fig. 5B; Table 3; Wang et al., 1998). Several mutant channels reduced the sensitivity to use-dependent lidocaine block and none significantly enhanced sensitivity (Table 3). The reduced use-dependent block was associated with slower entry and faster recovery from inactivated states in the presence of lidocaine (F430C). However, other mutants with increased IC50 values for use-dependent block (I424C, I425C, I782C, and V786C) did not exhibit gating changes that could explain the reduced sensitivity to lidocaine, suggesting that LA binding is altered in these mutants. G428C was uniquely resistant to first-pulse block (Fig. 5), yet underwent inactivation more rapidly and recovery more slowly than the wild-type channel (Fig. 4, B and E). Mutants with enhanced first-pulse block can be divided into those with gating changes that served to explain the enhanced sensitivity (N434C and I436C) and those that exhibited no significant differences in gating (L785C and V787C) accounting for the increased rested state affinity for lidocaine, although enhanced drug access in the rested state and other mutation-induced structural changes cannot be excluded by our data (Figs. 5, 6, 7; Table 4).
Orthodoxy in LA block of Na+ channels predicts that stabilization of inactivation should enhance block, particularly use-dependent block. Thus, the enhanced lidocaine block of mutant channels such as N434C is likely the result of mutation-induced changes in inactivation gating. However, other mutants that stabilized steady-state inactivation (F426C, F430C, and V779C) failed to enhance or even reduced rested state or use-dependent sensitivity to lidocaine (Figs. 3, 5, and 7). Lidocaine block may also rely on the occupancy of slowly recovering inactivated states (Balser et al., 1996,Balser et al., 1996). F430C resisted the entry into slow inactivated states, recovered more rapidly from inactivation (Fig. 4) in the presence of lidocaine, and exhibited reduced use-dependent block (Fig. 5). Similarly, enhanced rested block seen for N434C likely resulted from enhanced slow inactivation. This is similar to the previously reported effects of bupivacaine on N434C (Nau et al., 1999). Counterexamples include I782C and V786C, which exhibited reduced sensitivity to use-dependent block despite stabilization of slowly recovering inactivated states manifested by hastened recovery from and entry into these states (Fig. 4).
Our data are consistent with movement of the S6 α-helices during activation and inactivation gating. Changes in the position of the side chains of S6 residues during gating may alter channel-drug interactions and thereby modify sensitivity to block by LAs. Indeed, fundamentally different physicochemical mechanisms may characterize the molecular interactions between LAs and varying states of the Na+ channel (Li et al., 1999). Alternatively, mutations in I-S6 and II-S6 may produce allosteric changes in the drug receptor conformation and channel gating machinery. In the context of previously published site-specific mutagenesis data (Ragsdale et al., 1994, 1996; Nau et al., 1999; Wang et al., 1998; Yarov-Yarovoy et al., 2001, 2002) and our own, we suggest the topological relationship of the S6 segments of the Na+ channel shown in Fig. 8. This model is largely in agreement with that of Yarov-Yarovoy et al. (2002) but differs from previously published models. In particular, the changes in LA affinity of N434C does not mandate a position of this side chain in the pore of the channel. This model suggests that binding is highly asymmetric with I-S6 and II-S6 being quantitatively less important than IV-S6 and III-S6, where potent π-stacking and π-quaternary ammonium interactions are likely to occur.
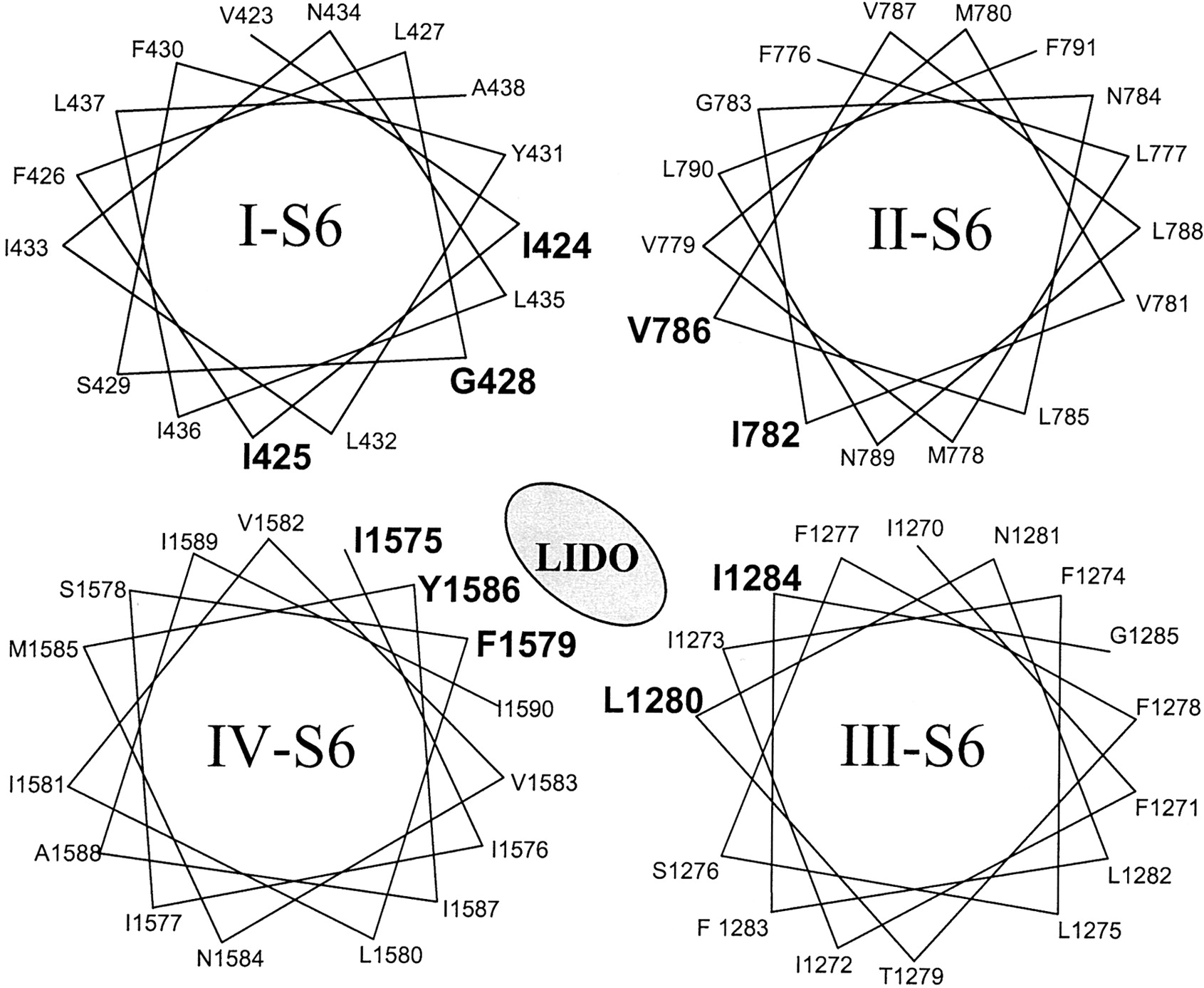
Helical wheel representations of the S6 segments of four domains. The S6 mutations in each of the domains that altered local anesthetic binding are shown in bold.
The seemingly contradictory data regarding the role of individual residues in S6 can be reconciled by consideration of the differing experimental approaches (Ragsdale et al., 1994, 1996; Wang et al., 1998; Wright et al., 1998; Nau et al., 1999; Yarov-Yarovoy et al., 2001, 2002). The detailed structure of the binding sites may vary in different Na+ channel isoforms. Importantly, utilization of mutagenic strategies to determine which residues influence LA binding depends to a significant degree on the substituting amino acid and the structure of the pharmacophore.
Acknowledgments
We thank Ailsa Mendez for technical assistance with site-directed mutagenesis.
Footnotes
-
This work was supported by the Training Grant T32 HL07227-24 (to A.K.) and National Institutes of Health R01 HL 50411 (to G.F.T.).
-
ABBREVIATIONS: HEK, human embryonic kidney; I-V, current-voltage.
- Received January 24, 2003.
- Accepted June 11, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}