


Abstract
The aryl hydrocarbon receptor (AhR) functions as a ligand-activated transcription factor that is responsible for the regulation of several response genes, of which the best characterized is the CYP1A1 gene. The present study was undertaken to elucidate the mechanism of activation of the AhR by omeprazole (OME), 2-mercapto-5-methoxybenzimidazole (MMB), and primaquine (PRQ), compounds that have previously been reported to induce CYP1A1 expression but that are not typical AhR ligands. All compounds caused a significant increase in luciferase activity in rat H4IIE and human HepG2 hepatoma cells transfected with a Gal4-AhR construct and the corresponding Gal4-Luc reporter gene. Furthermore, MMB and PRQ, but not OME, were capable of transforming cytosolic AhR to a DNA-binding form and displacing AhR-bound [3H]TCDD in rat hepatic cytosol in vitro. By performing site-directed mutagenesis of residues in the ligand-binding domain of the Gal4-AhR, a construct containing a Y320F substitution was found to be resistant to activation by OME, MMB, and PRQ, but not by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Comparable affinities of [3H]TCDD-binding to the wild-type and the Y320F mutant Gal4-proteins, expressed in human embryonic kidney 293 cells, were obtained in the ligand-binding assay. In contrast, the competition of receptor-bound [3H]TCDD by PRQ was absent from Gal4-Y320F but not from Gal4-AhR cell extracts. The results of this study confirm that MMB and PRQ are low-affinity ligands for the AhR and suggest that high- and low-affinity ligands interact with different residues of the AhR ligand-binding pocket. In addition, the data presented here indicate that Tyr320 plays an important role in AhR activation.
The aryl hydrocarbon receptor (AhR) is a member of the growing family of basic helix-loop-helix Per-Arnt-Sim (PAS) transcription factors whose members play key roles in development, adaptation to hypoxia, control of circadian rhythmicity, and metabolism of xenobiotic compounds (Gu et al., 2000). Mechanistically, the AhR functions as a ligand-activated transcription factor that is responsible for the transcriptional activation of several AhR-responsive genes (see reviews by Hankinson, 1995; Whitlock, 1999). A variety of environmental pollutants (e.g., polycyclic aromatic hydrocarbons and halogenated aromatic hydrocarbons) are high-affinity ligands for the AhR. These ligands, including the prototype AhR ligand 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD), are characterized by being planar, aromatic, and hydrophobic molecules, fitting into a ligand-binding pocket with a maximal dimension of 14 × 12 × 5 Å. In addition, electronic and thermodynamic properties of the ligand have been shown to be critical for favorable interactions between the ligand and the receptor (Gillner et al., 1993; Kafafi et al., 1993; Waller and McKinney, 1995).
Recent identification and characterization of novel AhR ligands or AhR activators (reviewed by Denison and Nagy, 2003), which have physicochemical and structural properties that deviate significantly from these typical AhR ligands, challenge the currently defined ligand-binding model. The structural diversity of these atypical AhR ligands/activators is clearly evident by comparison of the molecular structures of, for instance, carbaryl, primaquine, brevotoxin, and bilirubin (Ledirac et al., 1997; Sinal and Bend, 1997; Washburn et al., 1997; Fontaine et al., 1999). However, corroboration of several of these compounds as AhR ligands has been difficult, especially because [3H]TCDD at high concentrations has often been used in the ligand-binding assays performed. By using a low nonsaturating concentration of [3H]TCDD that permits competition by low-affinity ligands, Denison's group was able to show specific competition of radiolabeled TCDD by, for example, carbaryl and bilirubin in guinea pig hepatic cytosol, indicating that these agents are weak AhR ligands (Denison and Heath-Pagliuso, 1998; Phelan et al., 1998).
We have previously shown that omeprazole (OME), 2-mercapto-5-methoxybenzimidazole (MMB), which constitutes part of the OME molecule, and primaquine (PRQ) (see Fig. 1A for structures) cause induction of the CYP1A1 gene in rat hepatoma H4IIE cells (Backlund et al., 1997, 1999; Werlinder et al., 2001). In the case of OME, it has been shown that this benzimidazole is not capable of transformation of the AhR, in cytosol from H4IIE cells or mouse Hepa1 cells (Backlund et al., 1997; Dzeletovic et al., 1997). Moreover, OME was not capable of competing with [3H]TCDD for ligand-binding to either human, mouse, or rat AhR (Daujat et al., 1992; Daujat et al., 1996; Dzeletovic et al., 1997; Backlund et al., 1999). OME has been shown to induce CYP1A1 in primary hepatocytes or hepatoma cell lines of human but not mouse origin. (Kikuchi et al., 1996). Although CYP1A1 inducibility by OME was not re-established by expression of human AhR in AhR-deficient mouse Hepa1 cells, both the mouse and human AhRs could be activated in a reconstituted yeast model system (Dzeletovic et al., 1997). Therefore, it has been suggested that cellular factors other than the AhR per se are important for proper activation of the AhR signaling pathway by OME.

Induction of CYP1A1 expression by OME, MMB, PRQ, and TCDD in hepatoma cell lines. A, structure of OME, MMB, PRQ, and TCDD. B, rat H4IIE, mouse Hepa1c1c7, and human HepG2 hepatoma cell lines were treated with 200 μM MMB, 200 μM OME, 30 μM PRQ, or 10 nM TCDD for 24 h, and 30 μg of cellular protein was analyzed for CYP1A1 protein expression by Western blot. One representative blot is shown.
The present investigation was undertaken to further characterize the molecular mechanism of activation of the AhR by OME, MMB, and PRQ using rat, mouse, and human hepatoma cell lines and mutant variants of the AhR. Our results delineate MMB and PRQ as low-affinity ligands for the AhR and identify Tyr320 in the ligand-binding domain of the rat AhR as a critical amino acid for binding of the two low-affinity ligands as well as for activation by OME but not for high-affinity binding by TCDD.
Materials and Methods
Materials. OME and MMB were gifts from AstraZeneca (Mölndal, Sweden). 2,3,7,8-Tetrachloro-dibenzo-p-dioxin was from Larondan Fine Chemicals (Malmö, Sweden) and [3H]TCDD (specific activity, 34.7Ci/mmol) (Chemsyn) was purchased from Campro Scientific (Veenendaal, The Netherlands). Primaquine was obtained from Sigma-Aldrich Corporation (St. Louis, MO). The purity of MMB and PRQ was more than 99% and 96%, respectively, as determined by high-performance liquid chromatography. Anti-rat CYP1A1 antibody was purchased from BD Gentest Corporation (Woburn, MA), anti-AhR antibody was from BIOMOL Research Laboratories Inc. (Plymouth Meeting, PA), anti-Gal4(DBD) (RK5C1) antibody (with or without agarose-conjugation) and anti-hsp90 antibody were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). The anti-arnt serum has been described previously (Backlund et al., 1997). Secondary horseradish peroxidase-conjugated immunoglobulins were purchased from Dako AS (Glostrup, Denmark).
Cell Culture Conditions and Treatment. The rat H4IIE, human HepG2, and mouse Hepa1c1c7 hepatoma cell lines and the human embryonic kidney 293 cell line, were obtained from American Type Culture Collection (Manassa, VA). The Hepa1-C12 cell line was a kind gift from Dr Oliver Hankinson (UCLA, Los Angeles, CA) (Hankinson, 1979). All the cells were cultured in 5% CO2 at 37°C in minimum essential medium containing 10% fetal bovine serum and 100 μg/ml streptomycin, 100 IU/ml penicillin, nonessential amino acids, and sodium pyruvate. All cell culture media were obtained from Invitrogen (Carlsbad, CA). For stimulation of cells, the media were replaced with fresh media containing 200 μM OME, 200 μM MMB, 30 μM PRQ, or 10 nM TCDD dissolved in dimethyl sulfoxide (DMSO) and further incubated for the indicated time. Control cells were treated with DMSO alone, at a final concentration that never exceeded 0.2%.
Western Blot Analysis. Western blot analysis of CYP1A1 expression in the 10,000g supernatant prepared from hepatoma cells was carried out as described previously (Backlund et al., 1997). The primary antibody was visualized by using the enhanced chemiluminescence method (Pierce, Rockford, IL) and analyzed on a Fuji Las-1000 luminescent analyzing system (Fujifilm, Stamford, CT).
Plasmid Constructs. The Gal4-AhR hybrid protein was created by cloning of rat AhR into the mammalian expression vector pFA-CMV (Stratagene, La Jolla, CA) containing the Gal4-DNA-binding domain as described previously (Werlinder et al., 2001). The mutations of specific amino acids in the AhR ligand-binding domain was generated by PCR using Pfu DNA polymerase and the pCMV4-AhR vector as a template. The following primer pairs were used (restriction enzyme sites are underlined): for pFA-S274A, S274A-F (GACGGAGCGCTACTCCCTCCACAGTTGGCTTTGTTTGCAATAGCTACTCCACTTCAGCCACCGGCCATCC) and LBD-R (GTCGTCTAGACCCAGTCTTTCCTACTAGTG); for pFA-Y412F, LBD-F (CGACTGCAGCATGTCTGGTTTCCTGGCAATGA) and Y412F-R (CTTTCCTACTAGTGTTGCTTTTGGTGCGTATTGGCAAGGGATCCATTATGGGAGAGAAAGGGCTGGAGATCTCGAACAGTAC); for pFA-S429A, LBD-F and S419A-R (CTTTCCTACTAGTGTTGCTTTTGGTGCGTATTGGCAAGGGATCCATTATGGGAGCGAAAGGGCTG). To generate the remaining amino acid mutations a method, described by Bi and Stambrook (1997), combining a ligase chain reaction and PCR was applied. Briefly, this method uses two external primers (LBD-F and LBD-R) and one internal mutagenic primer in a PCR reaction containing a thermostabile ligase in addition to the DNA polymerase. The internal mutagenic primers containing a 5′-phosphorylation were: Y320F-F (P-CAACAAAGGATCGGGATTTCAGTTTATCCAC); Y376F-F (P-GGAAGACCAGATTTCATCATCGCAACTCAG); T380V-F (P-TACATCATCGCAGTTCAGAGACCGCTAACG); and T385V-R (P-CGTCCTTCTTCATCCACTAGCGGTCTCTG). The reactions were carried out in 20 mM Tris-HCl, pH 7.5, 20 mM KCl, 10 mM MgCl2, 0.1% Igepal, 10 μM rATP, 1 mM DTT, 0.2 mM concentrations of each dNTP, 0.4 mg/ml acetylated bovine serum albumin, 16 U of thermostable Pfu ligase (Stratagene), and 2.5 U of Pfu DNA polymerase, using 0.2 μM concentrations of each of the primers. The PCR was carried out with an initial denaturation for 45 s at 94°C; 30 cycles each of denaturation at 94°C for 30 s, annealing at 60°C for 30 s, and extension at 65°C for 4 min; and finally a 10-min extension time at 65°C before cooling to 4°C. All the PCR products were cut with Eco47III and SpeI and subcloned into the same unique cloning sites in the pFA-AhR plasmid. The correct sequence of the plasmid constructs was confirmed by DNA sequencing using the BigDye terminator cycle sequencing ready reaction kit and analyzed on an ABI PRISM 377 DNA sequencer (both from Applied Biosystems, Foster City, CA).
Transient Transfection. Transient transfections were carried out in 24-well plates, using 250 ng of pFR-Luc reporter plasmid (Stratagene) and 5 pg of pRL-CMV or 5 ng of pRL-SV40 control plasmid (Promega, Madison, WI) per well. The pFA-plasmids, wild type or mutants, were used at 250, 50, and 150 ng in H4IIE, Hepa1-C12, and HepG2 cells, respectively. The Tfx-20 transfection reagent (Promega) was used according to the manufacturer's protocol, and the cells were maintained in OptiMEM (Invitrogen) media during transfection. Approximately 24 h after transfection, the culture medium was exchanged with fresh media containing the inducers as described above. After 20 h, the cells were harvested and luciferase activity was analyzed according to the protocol for the Dual Luciferase Assay System (Promega) on a Turner Designs TD-20/20 luminometer. The results are expressed as the ratio between the firefly luciferase activity of the reporter gene and the Renilla reniformis luciferase activity of the control plasmid, constituting the control for the transfection efficiency.
Cytosol Preparation and Electrophoretic Mobility Shift Assay. Confluent cells were harvested by scraping into PBS, and cytosol was prepared as described previously (Backlund et al., 1997). For activation of cytosolic AhR to a DNA-binding form, cytosolic protein was incubated in the presence of 200 μM MMB, 200 μM OME, 30 μM PRQ, or 10 nM TCDD at 28°C for 3 h followed by electrophoretic mobility shift assay using a 32P-labeled double-stranded XRE oligonucleotide, carried out as described in Backlund et al. (1997). Competition experiments were performed using 40-fold molar excess of unlabeled double-stranded XRE oligonucleotide.
Ligand Binding Assay. Rat liver cytosol was prepared essentially as described previously (Backlund et al., 1997), except that a HEPES/EDTA/DTT/glycerol buffer [25 mM HEPES pH 7.4, 1.5 mM EDTA, 1 mM DTT, and 10% (v/v) glycerol] was used. Measurement of AhR ligand binding was carried out using sucrose gradient centrifugation (Poland et al., 1976) with modifications described by Denison et al. (1998) to allow for detection of low-affinity AhR ligands. Rat liver cytosol, 5 mg/ml, was incubated for 2 h at 4°C with 5 nM [3H]TCDD in the presence or absence of 1 μM TCDD, OME (300 μM and 3 mM), PRQ (200 μM and 1 mM), and MMB (1 and 5 mM). The samples were treated with dextran-coated charcoal for 5 min to remove unbound ligand. After a brief centrifugation to pellet the dextran-charcoal, the resulting supernatant was layered onto a 5.1-ml 5 to 30% linear sucrose gradient prepared in HEPES/EDTA/DTT/glycerol buffer and centrifuged for 160 min at 4°C in a Beckman VTi65 rotor at 60,000 rpm. After centrifugation, fractions (∼200 μl) were collected from the bottom of the gradient and radioactivity was measured by liquid scintillation counting. Specific binding of [3H]TCDD to the AhR was calculated by subtracting the amount of [3H]TCDD in the peak fractions in the presence and absence of 200-fold TCDD or other competitor.
Immunoprecipitation. For immunoprecipitations with anti-Gal4 antibody, H4IIE cells were transfected in 60-mm dishes with 2.5 μg of pFA-plasmids; 44 h after transfection, total cell extracts was prepared in 700 μl of cell lysis buffer, consisting of 20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 10% glycerol, 1% Triton X-100, 2 mM EDTA, 5 mM NaF, 1 mM Na3VO4, 10 mM Na2MoO4, 20 μM leupeptin, 10 μg/ml aprotinin, and 1 mM PMSF. The cells were lysed by incubation at 4°C for 15 min under gentle agitation, and the cell extract was passed through a 20-gauge needle to shear the DNA and was thereafter centrifuged at 10,000g for 10 min. The total cell extracts were precleared by addition of 2 μl of normal mouse IgG (Santa Cruz Biotechnology) and 20 μl of protein A/G Plus agarose and incubated for 30 min at 4°C under gentle agitation. To immunoprecipitate the wild type or mutant Gal4-AhR proteins, 20 μlofthe Gal4(DBD) agarose-conjugated antibody was added to the precleared total cell lysate and incubated for 1 h to over-night at 4°C under gentle agitation. The bound complexes were washed twice with immunoprecipitation buffer containing, 20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 1% glycerol, 0.1% Triton X-100, 2 mM EDTA, 5 mM NaF, 1 mM Na3VO4, 10 mM Na2MoO4, 20 μM leupeptin, 10 μg/ml aprotinin, and 1 mM PMSF and then twice with wash buffer containing 20 mM Tris-HCl, pH 7.4, 137 mM NaCl, 5 mM NaF, 1 mM Na3VO4, 10 mM Na2MoO4, 20 μM leupeptin, 10 μg/ml aprotinin, and 1 mM PMSF. Finally, the agarose beads were resuspended in 25 μl of 1× SDS sample buffer and immunoprecipitated proteins were analyzed by Western blot.
Expression of Gal4-AhR and Gal4-Y320F in HEK293 Cells. HEK293 cells were plated in 150-mm dishes and grown until approximately 90% confluent. The cells were transfected with 40 μg of pFA-AhR or pFA-Y320F plasmid or with the empty pFA-CMV vector, using the LipofectAMINE 2000 reagent (Invitrogen) according to the protocol supplied by the manufacturer. After incubation for 55 to 60 h, the cells were harvested by scraping in PBS. After a brief centrifugation, the cell pellet was suspended in a buffer containing 20 mM potassium phosphate, pH 7.2, 1 mM EDTA, 1 mM DTT, 10% glycerol, 20 μM leupeptin, 10 μg/μl aprotinin, and 1 mM PMSF and homogenized in a Dounce homogenizer with 20 up-and-down strokes using a B-type pestle. The homogenate was centrifuged at 17,000g for 30 min and the resulting supernatant was frozen at –70°C until used. For ligand binding studies, 1.5 mg of protein was used to analyze [3H]TCDD binding essentially as described above.
Results
Induction Pattern of CYP1A1 in Different Hepatoma cell Lines. It has been shown that OME can cause induction of CYP1A1 in primary hepatocytes or hepatoma cells from rat and human, but not from mouse (Kikuchi et al., 1996; Backlund et al., 1997; Dzeletovic et al., 1997). To determine the generality of CYP1A1 induction by compounds of structurally different nature in various hepatoma cell lines, we performed Western blot analysis of CYP1A1 protein levels in rat H4IIE, mouse Hepa1c1c7 and human HepG2 hepatoma cell lines treated with OME, MMB, and PRQ for 24 h. As expected, all tree compounds induced CYP1A1 in rat H4IIE cells, and OME and PRQ caused CYP1A1 induction in HepG2 cells (Fig. 1B). Surprisingly, MMB but neither OME nor PRQ caused induction of CYP1A1 protein in mouse Hepa1c1c7 cell. In contrast, MBB did not cause an induction of CYP1A1 in human HepG2 cells. Overall, these results indicate the importance in the selection of appropriate cell system when novel AhR ligands or activators are being investigated.
Time-Dependent Activation of Gal4-AhR in H4IIE Cells. To address the question of a direct involvement of the AhR in the CYP1A1-induction response caused by OME, MMB, and PRQ, activation of a Gal4-AhR hybrid protein, containing the Gal4 DNA-binding domain, was examined in transfected H4IIE cells. The activity of the Gal4-AhR construct, when transfected together with the appropriate Gal4-Luc reporter gene, seems to be independent of, for example, Arnt, thus avoiding some other factors that could be crucial for proper transcriptional activation of AhR response genes. H4IIE cells were transiently transfected with the Gal4-AhR construct and the Gal4-Luc reporter, and luciferase activity was measured at different time points after stimulation with OME, MMB, PRQ, and TCDD. As shown in Fig. 2A, TCDD caused a rapid increase in luciferase activity observed already at 4 h. Similarly, PRQ also elicited a fast response and, notably, activation of the Gal4-AhR protein by PRQ and TCDD was transient with a peak at 12 h. On the other hand, activation of the Gal4-AhR construct by MMB and OME was much slower, in that increased luciferase activity was observed only at the 24-h time point (Fig. 2A). H4IIE cells stimulated for 20 h with MMB, OME, PRQ, and TCDD showed 5.7-, 8.2-, 17-, and 22-fold increases in luciferase activity, respectively (Fig. 2B). Thus, these results demonstrate the involvement of the AhR in the CYP1A1-induction response exerted by OME, MMB, and PRQ. However, the delayed Gal4-AhR activation response elicited by MMB and OME could indicate a different mechanism of activation of the AhR by these compounds compared with TCDD.

Activation of rat Gal4-AhR hybrid protein in transiently transfected hepatoma cell lines. Cells were cotransfected with the pFA-AhR plasmid, the pFR-Luc reporter, and the pRL-CMV control plasmid. The next day, the medium was exchanged with medium containing DMSO, 200 μM MMB, 200 μM OME, 30 μM PRQ, and 10 nM TCDD in quadruplicate wells, and further incubated for the time indicated. The cells were harvested by passive lysis, and cell extracts were analyzed for firefly and R. reniformis luciferase activity. The values in the diagrams represent mean ± S.E. of several independent experiments as indicated in A. Time-dependent activation of the Gal4-AhR hybrid in H4IIE cells (n = 4). B–D, activation of Gal4-AhR after 20 h of stimulation in H4IIE (n = 5), Hepa1-C12 (n = 3), and HepG2 (n = 3) cells, respectively.
Activation of Rat Gal4-AhR Hybrid Protein in Mouse and Human Hepatoma Cell Lines. The lack of CYP1A1 induction response by OME and PRQ in Hepa1c1c7 cells and MMB in HepG2 cells, despite the fact that they elicited a response in H4IIE cells, could be caused by structural or functional differences between the rat, mouse, and human AhR proteins. To investigate this possibility, the Gal4-AhR hybrid construct containing the rat receptor was transiently transfected into human HepG2 cells or mouse Hepa1-C12 cells. The Hepa1-C12 cell line, which is almost completely deficient in AhR protein expression (Hankinson, 1994), turned out to be more suitable for these transfection studies than the wild-type Hepa1c1c7 cells. As shown in Fig. 2C, TCDD caused a 3.1-fold increase in Gal4-AhR–dependent luciferase activity in Hepa1-C12 cells. In contrast, an attenuation of the response was seen by OME treatment. On the other hand, PRQ-dependent activation (1.7-fold) of Gal4-AhR was obtained in Hepa1-C12 cells, indicating that the lack of PRQ-mediated CYP1a1 induction in mouse cells could be caused by an incapability of this compound to interact with the murine AhR. In HepG2 cells, a 2.8-, 4.4-, 4.6-, and 15-fold activation of the Gal4-AhR construct was obtained by OME, MMB, PRQ, and TCDD, respectively (Fig. 2D). Unexpectedly MMB caused an activation of the Gal4-AhR construct in transfected HepG2 cells but not in Hepa1-C12 cells, which is the opposite of what was seen on CYP1A1 protein expression (compare Fig. 1B). As in the case of PRQ in mouse cells, these results could indicate an incapability of MMB to interact with the human AhR; however, the reason for the lack of activation of the rat Gal4-AhR in transfected Hepa1-C12 cells is currently not known.
Activation of Cytosolic AhR to a DNA-Binding Form. To assess the ability of the compounds investigated to interact directly with the AhR, ligand-dependent transformation (i.e., activation of the cytosolic AhR to a DNA-binding form in vitro) was carried out. Thus, cytosols from H4IIE and Hepa1c1c7 cells were incubated with OME, MMB, or PRQ for 2 h and DNA binding was analyzed by electrophoretic mobility shift assay. As shown in Fig. 3, incubation of either rat H4IIE or mouse Hepa1c1c7 cytosol with MMB or PRQ in vitro resulted in an increased intensity of the band corresponding to the DNA-binding form of the AhR. In contrast, stimulation of H4IIE or Hepa1c1c7 cytosol with OME caused an attenuation of the band of the AhR-complex compared with the DMSO control. These data suggest that both MMB and PRQ could directly interact with the murine and rat AhR.
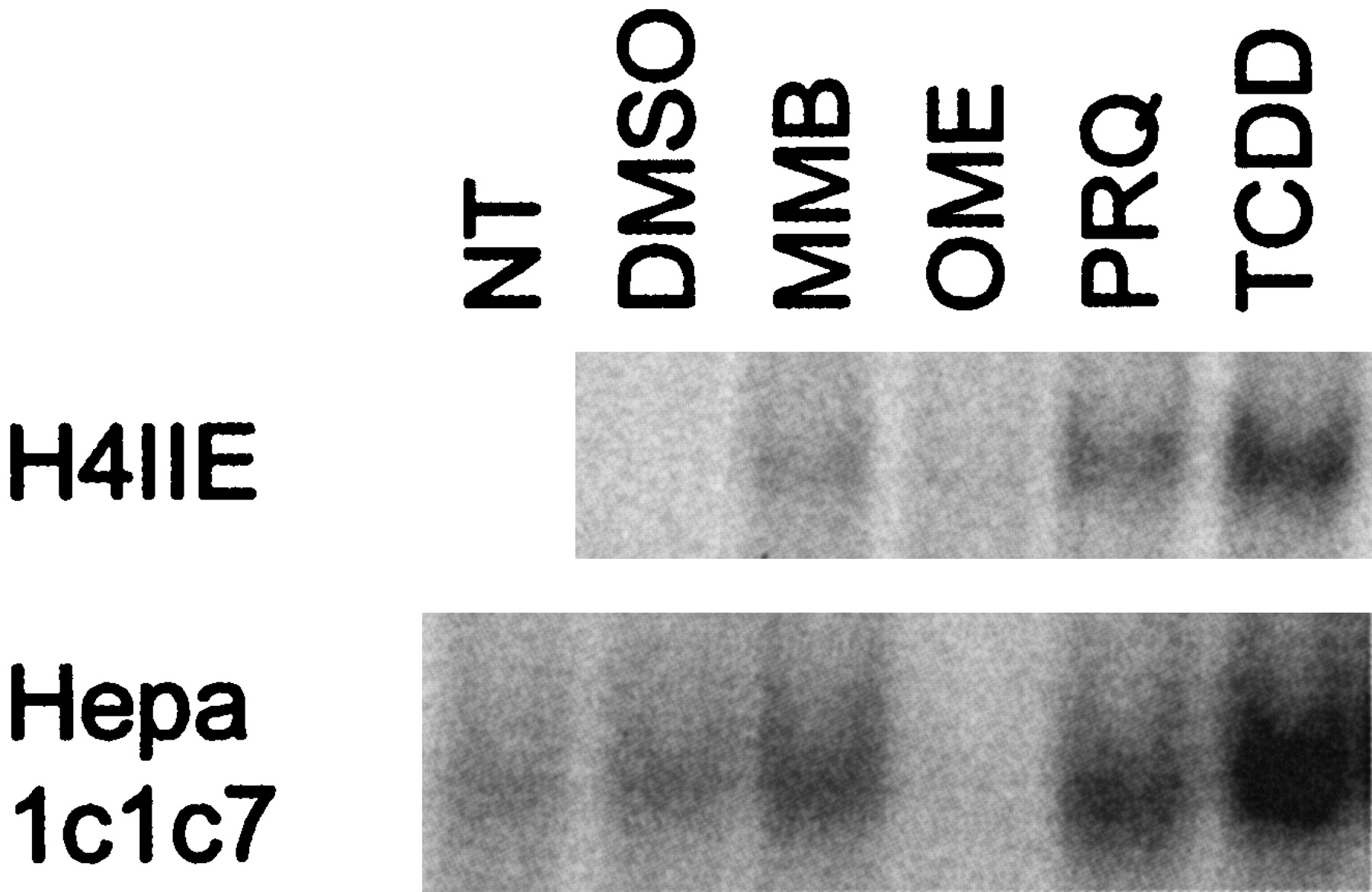
Transformation and DNA-binding of the AhR-complex in vitro. Cytosolic extracts were prepared from H4IIE (3.5 mg/ml) or Hepa1c1c7 (1.5 mg/ml) cells and 45 or 20 μg of protein, respectively, was incubated with DMSO, 200 μM MMB, 200 μM OME, 30 μM PRQ, and 10 nM TCDD or received no treatment (NT) for 2 h at 28°C. Aliquots were incubated with a 32P-labeled XRE-probe for 20 min and then analyzed by electrophoretic mobility shift assay. Only the bands corresponding to the AhR complex are shown.
Competition of [3H]TCDD Ligand Binding. The results presented above indicate that MMB and PRQ could be weak ligands for the AhR. Based on the dose dependence of TCDD and PRQ in the transformation assay in vitro (Werlinder et al., 2001; M. Backlund, unpublished observations), the relative potency of PRQ was estimated to be about 50,000-fold lower than TCDD. To address the question of weak ligands, rat liver cytosol, which is a rich source of AhR protein, was incubated with a low concentration of [3H]TCDD (5 nM) but concentrations of the competitors were kept as high as possible to retain their solubility. Upon sucrose density gradient centrifugation, the [3H]TCDD-labeled AhR was recovered approximately in the 9S region of the gradient, consistent with the sedimentation properties of the AhR-hsp90 complex. As shown in Fig. 4A, PRQ at a 40,000-fold molar excess (200 μM) displaced approximately 50% of the radiolabeled TCDD in the 9S-fractions, whereas complete competition of [3H]TCDD was obtained at a 200,000-fold molar excess of PRQ. By using a 200,000-fold molar excess of MMB, displacement of approximately 65% of [3H]TCDD was obtained (Fig. 4B). In contrast to the results obtained by PRQ and MMB, OME was not capable of competing with [3H]TCDD for binding to the AhR in hepatic rat cytosol, even at concentrations as high as 3 mM (Fig. 4C). Although the potencies of OME and MMB in activation of the Gal4-AhR construct or induction of CYP1A1 gene expression was approximately the same (see Fig. 2 and Backlund et al., 1999), a 600,000-fold molar excess of OME was not enough to displace [3H]TCDD, whereas the same fold molar excess of MMB was sufficient. Taken together, these results indicate that MMB and PRQ constitute low-affinity ligands for the rat AhR; nevertheless, in agreement with previous reports, OME apparently does not bind directly to the AhR protein.

Competitive [3H]TCDD ligand binding assay. Cytosolic extract was prepared from rat liver, and aliquots of 400 μl (5 mg/ml protein) were incubated at 4°C for 2 h with 5 nM [3H]TCDD in the presence or absence of competitors at the concentrations indicated. Ligand binding activity of the receptor was analyzed on a linear 5 to 30% (w/v) sucrose gradient by density centrifugation, and bound [3H]TCDD was measured by scintillation counting of fractions collected from the bottom of the gradient.
Effect of Point Mutations of the Ligand-Binding Domain of Rat Gal4-Ahr on the Activation Response. A polymorphism causing an amino acid exchange in the ligand-binding domain (LBD) of the AhR has been reported to be responsible for the difference in responsiveness to toxicity between C57BL/6 and DBA/2J mice, and substitution of this amino acid (A375V) results in a receptor with decreased TCDD-binding capacity (Ema et al., 1994). By creating the same mutation of the amino acid in question in the rat Gal4-AhR construct (A379V), we found that activation of the Gal4-A379V by TCDD was maintained, although the response by OME, MMB, and PRQ was almost abolished (results not shown). These findings indicated that the LBD is important for activation of the AhR by these compounds and encouraged us to carry out a more extensive structure-function analysis of the LBD. Seven different single point mutations (based on our interest in potential phosphorylation sites of the AhR) were created in the Gal4-AhR construct as schematically depicted in Fig. 5A. The wild-type and mutant Gal4-AhR hybrids were expressed in rat H4IIE cells; subsequently, the cells were stimulated with MMB, PRQ, OME, and TCDD for 20 h. Five of the seven constructs, Gal4-S274A, Gal4-Y376F, Gal4-T380, Gal4-Y412F, and Gal4-S419A, responded to the treatment in a manner very similar to that of the wild-type Gal4-AhR (Fig. 5B). On the other hand, activation of the Gal4-T385V construct was moderately reduced by PRQ and TCDD and almost completely lost by MMB and OME. Most striking, however, was the selective and total abrogation of activation of the Gal4-Y320F hybrid caused by OME, MMB, and PRQ, whereas the response by TCDD was still maintained (Fig. 5B). Similar results were obtained when the Gal4-Y320F or Gal4-T385V constructs were transfected into human HepG2 cells (results not shown).
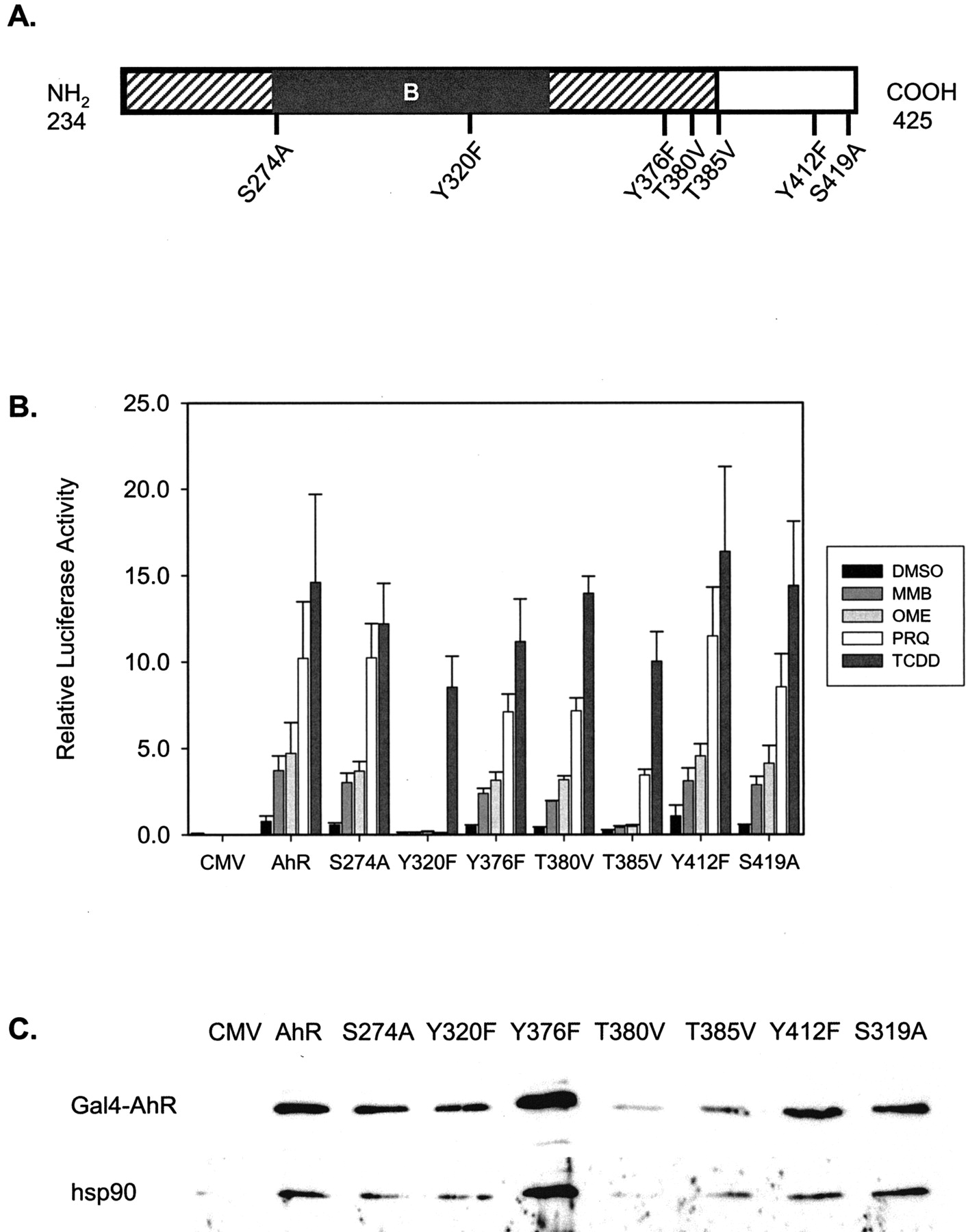
Effect of point mutations within the LBD of the AhR. A, schematic representation of the amino acid sequence of the LBD of rat AhR showing the position of single point mutations created in the Gal4-AhR construct. The diagonal and black boxes represent the C-terminal parts of the PAS domain and the PAS-B domain, respectively. B, activation pattern of the Gal4-mutant constructs by MMB, OME, PRQ, and TCDD. H4IIE cells were transfected with the pFA plasmids containing the point mutations as indicated; after stimulation, the samples were analyzed as described in the legend to Fig. 2. The diagram represents mean ± S.E. values of four independent experiments. C, expression levels of the Gal4-AhR mutant proteins and their interaction with hsp90. H4IIE cells were transfected with the wild-type and mutant pFA-plasmids as described above. After incubation for 24 h, total cell extracts was prepared, and immunoprecipitation was carried out using an anti-Gal4 antibody. Expression of Gal4AhR protein level was analyzed by Western blot on the immunoprecipitates by using the same anti-Gal4 antibody. Interaction of Gal4-AhR wild-type and mutant protein with hsp90 was detected using an antihsp90 antibody. One representative blot of six experiments is shown; the variations in Gal4-protein expression level seen are not consistent between the different experiments.
To rule out the possibility that the incapability of activation of the Gal4-Y320F or Gal4-T385V was caused by a lower expression level of the mutant proteins, the level of Gal4-protein expression was analyzed by immunoprecipitation with an anti-Gal4 antibody, followed by immunoblotting with the same antibody. As shown in Fig. 5C, the mutant Gal4-hybrid proteins were expressed at levels similar to those of the wild-type Gal4-AhR, indicating that the lack of activation was not caused by insufficient protein expression. In addition, the ability of the Gal4-hybrid proteins to interact with the hsp90 chaperone and Arnt was analyzed, given that AhR LBD has been shown to be important for these interactions. As shown by coimmunoprecipitation with the anti-Gal4 antibody, the wild-type and mutant Gal4-AhR hybrid proteins displayed comparable capacity to interact with hsp90 (Fig. 5C). In contrast, coimmunoprecipitation analysis failed to detect any interaction between the Gal4-proteins and Arnt, regardless of whether total cell extracts from untreated or treated cells were analyzed (results not shown). Taken together, these results show a selective loss of the AhR activation response when Tyr320 was mutated to Phe by the low-affinity ligands MMB and PRQ, as well as by OME, even though the response caused by the high-affinity ligand TCDD was unaffected.
Binding of [3H]TCDD to Gal4-AhR and Gal4-Y320F. To examine whether the Gal4-Y320F construct shows an impaired ligand-binding capacity compared with the wild-type Gal4-AhR, the mutant and wild-type Gal4-proteins were expressed in HEK293 cells, and [3H]TCDD ligand-binding assay was carried out. The HEK293 cells express very low levels of endogenous AhR protein (Fig. 6B), and the expression levels of the Gal4-AhR and Gal4-Y320F proteins were approximately 5- to 10-fold higher. Incubation of HEK293 cell extracts with [3H]TCDD and subsequent sucrose density gradient centrifugation revealed sedimentation of a ∼9S [3H]TCDD-binding complex in cell extracts prepared from Gal4-AhR– and Gal4-Y320F–expressing HEK293 cells, but not in extracts prepared from cells transfected with the empty CMV vector (Fig. 6A). Thus, binding of [3H]TCDD to the low levels of endogenously expressed AhR in HEK293 cells was not detected. Comparison of [3H]TCDD binding to full-length AhR and Gal4-AhR hybrid indicated that the heterologous Gal4 DNA-binding domain did not interfere with ligand binding (results not shown). When saturation binding analysis was carried out with increasing concentrations of [3H]TCDD, maximal binding was obtained already at approximately 10 nM [3H]TCDD (Fig. 6C). Regression analysis verified a similar binding capacity of [3H]TCDD for Gal4-AhR and Gal4-Y320F proteins, because Bmax values were very similar (9976 and 10088 cpm, respectively). Furthermore, the calculated Kd values for Gal4-AhR and Gal4-Y320F were 1.6 and 3.7 nM, respectively, indicating that the binding affinity of [3H]TCDD was not significantly changed. Thus, these results indicate that the Tyr320 to Phe substitution did not affect high-affinity ligand binding by TCDD. However, the complete lack of activation of the Gal4-Y320F construct by MMB and PRQ could indicate a failure of low-affinity ligands to bind to the mutant receptor. To investigate this possibility, competitive ligand-binding analysis was carried out using PRQ as well. Incubation of extracts prepared from Gal4-AhR–expressing HEK293 cells with 5 nM [3H]TCDD in the absence or presence of 1 μM TCDD or 1 mM PRQ revealed a complete competition by TCDD and approximately 60% competition by PRQ (Fig. 6D). Similarly 200-fold molar excess of TCDD was adequate to completely displace the binding of [3H]TCDD to the Gal4-Y320F protein. In contrast, incubation of the cell extracts containing the Gal4-Y320F protein with a 200,000-fold molar excess of PRQ showed no displacement at all of the bound radiolabeled TCDD. Evidently, binding of the low-affinity ligand PRQ was abrogated by substitution of Tyr320 with a Phe in the LBD of the AhR, whereas binding of the high-affinity ligand TCDD was not significantly affected.

[3H]TCDD binding to Gal4-AhR and Gal4-Y320F expressed in HEK293 cells. HEK293 cells were transfected with pFA-CMV, pFA-AhR, and pFA-Y320F plasmids, and cell extracts (∼2.0 mg/ml protein) were prepared 55 h after transfection. A, [3H]TCDD ligand binding assay. Cell extracts (1.5 mg, total protein) prepared from the transfected HEK293 cells were incubated with 15 nM [3H]TCDD for 2 h in the presence or absence of 200-fold TCDD. Ligand binding assay was carried out by centrifugation on a 10 to 40% sucrose gradient, and bound [3H]TCDD was measured by scintillation counting of the fractions collected from the gradient. B, expression level of Gal4-AhR and Gal4-Y320F. The level of expression of endogenous AhR (arrowhead) and Gal4-hybrid proteins (arrow) in 20-μg cell extracts was analyzed by Western blot using anti-AhR (top) and anti-Gal4(DBD) (bottom) antibodies. C, saturation analysis of [3H]TCDD binding to Gal4-AhR and Gal4-Y320F. HEK293 extracts (1.5 mg, total protein) from Gal4-AhR– or Gal4-Y320F–expressing cells were incubated with increasing concentration of [3H]TCDD and analyzed by sucrose density centrifugation as described above. Bound [3H]TCDD was calculated as the amount of [3H]TCDD in the peak fractions (six fractions) of the ∼9S complex. Nonlinear regression analysis was calculated in SigmaPlot by using the formula for saturation binding, y = Bmax × x/(Kd + x). D, competitive ligand-binding assay with PRQ. HEK293 extracts from Gal4-AhR– or Gal4-Y320F–expressing cells were incubated with 5 nM [3H]TCDD in the presence of 1 μM TCDD or 1 mM PRQ, and bound [3H]TCDD was analyzed as above.
Discussion
The results of this study demonstrate that OME, MMB, and PRQ were capable of activation of the AhR and subsequently could cause transcriptional activation of an AhR-responsive gene. However, the mode of interaction of each of these compounds with the receptor, and with the high-affinity ligand TCDD, differs. First, a larger variation of the CYP1A1-induction response between species or cell lines was apparent by stimulation of different hepatoma cell lines with these atypical CYP1A1 inducers compared with what is observed for polycyclic aromatic hydrocarbons or halogenated aromatic hydrocarbons. Second, a Tyr-residue identified in the LBD of the AhR was shown to be crucial for activation of the receptor by OME, MMB, and PRQ, but not by TCDD. Third, a direct, albeit weak, interaction by MMB or PRQ with the AhR was observed; however, no such interaction could be detected by OME. Thus, based on these results, it is apparent that one can distinguish three different mechanisms of AhR activation by OME, MMB/PRQ, and TCDD, respectively, and that these pathways exhibit special structural and cellular requirements.
It has previously been shown that rat, mouse, and human hepatoma cell lines respond differently to, for example, OME (Kikuchi et al., 1996; Backlund et al., 1997; Dzeletovic et al., 1997), and in this study, we found that MMB and PRQ also showed a cell-specific CYP1A1 expression pattern. Similarly to OME, PRQ was capable of causing increased expression of CYP1A1 in rat H4IIE and human HepG2 cells but not in mouse Hepa1c1c7 cells. When the pFA-AhR expression vector, which is based on rat AhR, was transfected in mouse Hepa1c1c7 cells, activation of the Gal4-AhR by PRQ, but not by OME, was restored. These results are in agreement with previous findings by Kiuchi et al. (1996) indicating that the lack of induction of Cyp1a1 by OME in mouse cell lines is dependent on some other cellular component apart from the AhR protein itself. Concerning PRQ, the results of the transfection experiments indicate that this compound could interact with the human and rat AhR but not with murine AhR. However, PRQ was capable of transforming both rat and mouse AhR in cytosolic extracts in vitro, implying that the lack of induction of Cyp1a1 in Hepa1c1c7 cells also was most probably caused by cellular factors other than the AhR per se. Overall, these results indicate that the structural differences of the AhR protein itself is not the major determinant in defining the responsiveness of a cell line. In contrast, other cellular factors seem to be responsible for the observed variability in AhR activation exerted by these novel CYP1A1 inducers.
By using a nonsaturable concentration of [3H]TCDD in the ligand-binding assay, it was possible to show competition of radiolabeled TCDD by MMB and PRQ, thus demonstrating for the first time a direct interaction of these compounds with the AhR. This was further supported by the finding that MMB and PRQ caused transformation of cytosolic AhR prepared from either rat H4IIE or mouse Hepa1c1c7 cells, to a DNA-binding form in vitro. PRQ was initially shown by Fontaine et al. (1999) to increase both the ethoxyresorufin-O-deethylase activity and CYP1A1 mRNA in human hepatocytes as well as in HepG2 cells, but no displacement of [3H]TCDD from AhR in competitive binding studies using 9S-enriched fractions of human hepatic cytosol was seen. However, the conditions used by this group would result in a molar difference of only 8000-fold between [3H]TCDD and PRQ. By applying these conditions in our modified ligand-binding assay, no displacement of AhR-bound [3H]TCDD was observed in rat liver cytosol (results not shown). It is likely, therefore, that the apparent discrepancy between their study and ours is not a consequence of species differences (human versus rat) but is a result of the modified ligand-binding assay used in this study. As demonstrated by the ligand-binding assay, the relative potencies of PRQ and MMB are in the range of 5 to 6 orders of magnitude lower than TCDD, indicating that the interaction with the receptor is very weak. Thus, the results of this study, as well as those of Fontaine et al. (1999), designate MMB and PRQ as low-affinity AhR ligands and represent two novel groups of chemicals as agonists for the receptor.
Given the relatively high concentration needed of MMB and PRQ in the ligand-binding assay, it could be argued that a contaminant with extremely high affinity for the AhR could be present in these preparations. Although this possibility cannot be conclusively ruled out, several lines of evidence support an argument against this possibility: First, the difference in characteristics of AhR activation, such as lack of activation of the Gal4-Y320F mutant, by these compounds compared with TCDD. A high affinity contaminant would most probably share the characteristics of TCDD. Second, PRQ and MMB dissolved in water can activate the AhR. High-affinity AhR agonists are generally very hydrophobic and would have limited solubility in water. Finally, the difference in CYP1A1-induction response seen in the various hepatoma cell lines would normally not be observed for high-affinity AhR ligands. Therefore, our conclusion is that the parent compounds, not minor contaminants, are responsible for the ligand-binding properties of PRQ and MMB.
Although we and others previously have shown that OME cannot compete with AhR-bound [3H]TCDD (Daujat et al., 1992, 1996; Backlund et al., 1997; Dzeletovic et al., 1997) we reexamined OME with the use of the modified ligand-binding assay. However, no displacement of [3H]TCDD in hepatic rat cytosol was seen, even with a 600,000-fold molar excess of OME. Furthermore, OME was incapable of transforming cytosolic AhR to a DNA-binding form in vitro. Thus, the results of this study fully confirm previous findings and provide the basis for a strong argument against a direct binding of OME to the AhR. We have therefore proposed that OME mediates AhR activation via a ligand-independent mechanism, presumably through a tyrosine kinase-dependent signal transduction pathway, leading to conformational changes of the AhR similar to those caused by ligand binding (Backlund et al., 1997). On the other hand, we cannot fully rule out the possibility that a metabolite of OME is the active principle, as suggested by Dzeletovic et al. (1997), although our attempts to identify, detect, or, for instance, trap such an agent in conditioned media have not been successful.
The LBD of the AhR has also been shown to be important for, in addition to ligand-binding, association with the molecular chaperone hsp90 and for dimerization with Arnt (Coumailleau et al., 1995; Fukunaga et al., 1995; Lindebro et al., 1995). The minimal LBD spans the PAS-B domain, a domain structure that has been identified as an important signaling module that senses, transduces, and regulates adaptation to environmental changes (Taylor and Zhulin, 1999; Gu et al., 2000). Recently, a three-dimensional model for the LDB of the mouse AhR was published, using the known crystal structure of the O2-sensing PAS domain of the bacterial FixL protein as a template (Procopio et al., 2002). According to this model, the ligand-binding pocket is situated between a β-sheet on one side and a long α-helix, the so-called Fα-helical connector, on the other side. The main difference between all known PAS structures is attributed to the positioning of the Fα-helical connector, which is placed closer to the β-sheet in the PAS proteins that do not bind ligand compared with those that bind a heme (e.g., FixL) (Gong et al., 1998). Notwithstanding the known important role of the PAS-containing LBD of the AhR, amazingly few studies have dealt with detailed structural-functional aspects of this region. One exception is the recognition of the responsibility of Ala375 in the AhR LBD of C57BL/6 mice for the higher ligand-binding capacity compared with the Val375 variant found in the AhR of DBA/2J mouse as well as in the human AhR (Ema et al., 1994; Dzeletovic et al., 1997). Because substitution of the corresponding amino acid (Ala379) of the rat AhR resulted in an attenuation of Gal4-AhR activation by all compounds tested (not shown), we thought that the Gal4-AhR construct could be used successfully for structural-functional analysis.
Among seven single point mutations created in the Gal4-context, the Gal4-Y320F mutant revealed a selective and complete loss of activation by PRQ and MMB as well as by OME. However, the activation of Gal4-Y320F by TCDD was unaltered, and the affinity for [3H]TCDD was similar to that of the wild type receptor, as analyzed by saturation ligand-binding assay. Furthermore, coimmunoprecipitation analysis showed that the interaction of the Gal4-Y320F variant with hsp90 was preserved. These results indicate that the Gal4-Y320F variant was correctly folded and maintained the properties of TCDD ligand binding and transactivation. On the other hand, low-affinity ligand binding was apparently impaired, because no competition of [3H]TCDD-bound Gal4-Y320F receptor by PRQ was obtained. Based on our findings, it is unlikely that TCDD would directly be recognized by the Tyr320 residue. However, one interpretation of the results obtained by PRQ could be that this compound interacts with the Tyr320-site and that this interaction would be crucial both for correct positioning of the ligand and for the functional outcome of the AhR. In support of different positioning of various ligands of the AhR LBD, the localization of cofactors in several PAS proteins has been shown to occur in the same general hydrophobic region of the PAS domain; however, the actual positioning and ligand binding site interactions differ considerably (Gong et al., 1998; Pellequer et al., 1998; Crosson and Moffat, 2001). It is conceivable that, to encounter the environmental stress, the AhR has evolved a rather promiscuous ligand-binding pocket, in which structurally diverse compounds can be positioned. The data of the present study would fully support such an idea and might explain how chemicals as diverse as MBB and bilirubin, for instance, could acquire the necessary ligand binding interactions required to attain AhR activation. Further investigations combining site-directed mutagenesis, generation of pharmacophores of high- and low-affinity AhR ligands, and molecular modeling will be necessary to get a more detailed picture of the accommodation of ligands in the ligand-binding cavity of the receptor.
In conclusion, the results of this study define MMB and PRQ as low-affinity ligands for the AhR; furthermore, a conserved Tyr residue in the LBD was identified as a critical amino acid for binding of the two low-affinity ligands, as well as for AhR-activation by OME, but not for high-affinity TCDD binding and activation. Our results support the characterization of the AhR as a promiscuous receptor that can bind and be activated by multiple exogenous and endogenous ligands and functions as a sensor eliciting biological responses with varying efficacies depending upon the incoming signal.
Acknowledgments
We appreciate the kind gift of the Hepa1-C12 cell line from Dr Oliver Hankinson. We thank Dr. Lars Weidolf (AstraZeneca) for all the kind help and for providing us with OME and MMB. We also thank Susanne Ahlberg and Susanne Andersson for skillful technical assistance.
Footnotes
-
This work was supported by grants from Karolinska Institutet and The Swedish Research Council.
-
ABBREVIATIONS: AhR, aryl hydrocarbon receptor; Arnt, aryl hydrocarbon receptor nuclear translocator; PAS, Per-Arnt-Sim; TCDD, 2,3,7,8-tetrachlorodibenzo-p-dioxin; OME, omeprazole; MMB, 2-mercapto-5-methoxybenzimidazole; PRQ, primaquine; HEK, human embryonic kidney; DMSO, dimethyl sulfoxide; CMV, cytomegalovirus; PCR, polymerase chain reaction; DTT, dithiothreitol; XRE, xenobiotic response element; PMSF, phenylmethylsulfonyl fluoride; hsp90, 90-kDa heat shock protein; LBD, ligand-binding domain.
- Received June 26, 2003.
- Accepted November 7, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}