


Abstract
CaV2.3 subunits are expressed in neuronal and neuroendocrine cells where they are believed to form native R-type Ca2+ channels. Although R-type currents are involved in triggering neurotransmitter and hormone secretion, little is known about their modulation. Previous studies have shown that muscarinic acetylcholine receptors evoke both inhibition and stimulation of CaV2.3. Muscarinic inhibition of CaV2.3 is mediated by Gβγ subunits, whereas stimulation is mediated by pertussis toxin-insensitive Gα subunits. In the present study, we compared modulation of CaV2.3 by the three Gαq/11-coupled muscarinic receptors (M1, M3, and M5). Our data indicate that these receptors trigger comparable stimulation of CaV2.3. The signaling pathway that mediates stimulation was meticulously analyzed for M1 receptors. Stimulation is blocked by neutralizing antibodies directed against Gαq/11, coexpression of the regulatory domain of protein kinase Cδ (PKCδ), preactivating PKC with phorbol ester, or pharmacological suppression of PKC with bisindolylmaleimide I. Stimulation of CaV2.3 is Ca2+-independent and insensitive to 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole (Gö 6976), a specific inhibitor of Ca2+-dependent PKC isozymes. These results indicate that muscarinic stimulation of CaV2.3 involves signaling by Gαq/11, diacylglycerol, and a Ca2+-independent PKC. In contrast to stimulation, the magnitude of CaV2.3 inhibition depended on receptor subtype, with M3 and M5 receptors producing much larger CaV2.3 inhibition than M1 receptors. Interestingly, muscarinic inhibition of CaV2.3 was notably enhanced during pharmacological suppression of PKC, suggesting the presence of cross-talk between Gβγ-mediated inhibition and PKC-mediated stimulation of R-type channels similar to that described previously for N-type channels.
Native R-type Ca2+ current is defined by its resistance to specific pharmacological antagonists (e.g., 1,4-dihydropyridines, ω-conotoxin GVIA, ω-conotoxin MVIIC, and ω-agatoxin IVA) of other high voltage-activated Ca2+ currents (Zhang et al., 1993). Considerable evidence now supports the view that native R-type Ca2+ currents are conducted primarily through CaV2.3 channel subunits (Piedras-Rentería and Tsien, 1998; Tottene et al., 2000; Lee et al., 2002; Sochivko et al., 2002; but see Wilson et al., 2000). CaV2.3 is expressed in neurons (Niidome et al., 1992; Soong et al., 1993; Williams et al., 1994; Grabsch et al., 2000) and seems to be localized to somatic and dendritic membranes (Yokoyama et al., 1995; Day et al., 1996), suggesting the possible involvement of CaV2.3 in dendritic excitability (Delmas et al., 2000) or control of gene expression. Native R-type Ca2+ channels are present at certain presynaptic terminals, where they participate in evoked neurotransmitter secretion (Wu et al., 1998; Wang et al., 1999; Gasparini et al., 2001). Significantly, recent work demonstrates that CaV2.3 is essential for certain forms of synaptic plasticity within mammalian hippocampus (Dietrich et al., 2003). CaV2.3 is also expressed by various neuroendocrine cells, and R-type currents seem to be important for hormone secretion (Albillos et al., 2000; Weiergraber et al., 2000; Vajna et al., 2001). In mice, genetic deletion of CaV2.3 ablates pharmacologically defined native R-type currents (Lee et al., 2002; but see Wilson et al., 2000) and produces functional deficits in fear behavior, spatial memory, pain perception, and glucose metabolism (Saegusa et al., 2000; Kubota et al., 2001; Matsuda et al., 2001; Lee et al., 2002). These recent studies emphasize the physiological importance of CaV2.3, yet little is known about this channel's modulation through cellular signaling pathways.
Five distinct subtypes of muscarinic acetylcholine receptor (M1-M5) are known, and recent studies using knockout mice have provided new insights into their physiological functions (Bymaster et al., 2003). In many cases, the tissue distributions of muscarinic receptors overlap with those of CaV2.3 (Niidome et al., 1992; Levey, 1993; Soong et al., 1993; Williams et al., 1994; Yokoyama et al., 1995; Day et al., 1996; Grabsch et al., 1999; Weiergraber et al., 2000). It is reasonable to predict that acetylcholine modulates CaV2.3 in vivo and thereby regulates physiological processes involving this channel. Because of the technical difficulties associated with studying modulation of native R-type channels, which typically conduct a small proportion of whole-cell Ca2+ current, we have taken the approach of analyzing CaV2.3 modulation in a heterologous expression system with the belief that information so gained has potential applicability to native systems. This experimental approach has already shown that CaV2.3 channels can be stimulated and inhibited through muscarinic receptors (Meza et al., 1999; Melliti et al., 2000; Kamatchi et al., 2003). As demonstrated by Melliti et al. (2000), muscarinic stimulation of CaV2.3 proceeds through a pertussis toxin-insensitive pathway that is blocked by regulator of G protein signaling 2 (RGS2) or the carboxyl-terminal region of phospholipase C-β1 (PLCβ1ct), two proteins known to antagonize signaling by Gαq/11. Evidence also indicates that muscarinic stimulation of CaV2.3 involves phosphorylation by protein kinase C (PKC) (Stea et al., 1995; Melliti et al., 2000; Kamatchi et al., 2003). Together, these findings suggest that Gαq/11-coupled receptors, which often activate PKC, may be important in modulating CaV2.3.
In the present study, we sought to further elucidate CaV2.3 modulation by Gαq/11-coupled muscarinic receptors. Our data show that M3 and M5 receptors trigger much stronger inhibition of CaV2.3 than M1 receptors. In contrast, all three receptor subtypes produce equivalent stimulation of CaV2.3. We analyzed the pathway that mediates muscarinic stimulation of CaV2.3 and demonstrated that it involves signaling by Gαq/11, diacylglycerol, and a Ca2+-independent PKC isozyme. Intriguingly, we find that muscarinic inhibition of CaV2.3 is significantly enhanced during pharmacological suppression of PKC, suggesting the presence of cross-talk between Gβγ and PKC similar to that described previously for N-type (CaV2.2) Ca2+ channels (Swartz et al., 1993; Zhu and Ikeda, 1994; Hamid et al., 1999). The new information revealed in our study should help in understanding modulation of native R-type Ca2+ channels.
Materials and Methods
Cell Culture and Transfection. Human embryonic kidney (HEK) 293 cells were obtained from the American Type Culture Collection (Manassas, VA) and propagated in culture medium containing 90% Dulbecco's modified Eagle's medium (Invitrogen, Carlsbad, CA), 10% defined fetal bovine serum (Hyclone Laboratories, Logan, UT), and 50 μg/ml gentamycin. HEK293 cells of low passage number (<20) were trypsinized weekly and replated onto 60-mm culture dishes at ∼20% confluence. CaPO4 precipitation was used to transfect these cells within 3 to 5 days of plating. The transfection mixture contained expression plasmids encoding rabbit CaV2.3 (formerly known as α1E), rat α2δb, and rabbit β3 channel subunits at 1.25 μg of each cDNA per dish, plus an expression plasmid encoding the M1 muscarinic acetylcholine receptor at 0.5 μg of cDNA per dish. In some experiments, the M3 receptor (0.5 μg per dish) or M5 receptor (0.25 μg per dish) replaced the M1 receptor. These quantities of receptor plasmid were empirically determined to support saturating, agonist-induced modulation of CaV2.3 currents. In selected experiments, plasmids encoding amino-terminal regions of PKCδ (residues 1–121 or 1–298) were transfected at 1.25 μg per dish. These constructs are hereafter denoted PKCδ1–121 and PKCδ1–298, respectively. For all transfections, a plasmid encoding enhanced green fluorescent protein was included at 0.125 μg per dish. The day after transfection, cells were briefly trypsinized, replated onto 12-mm round-glass coverslips, and incubated at 37°C overnight. Electrophysiological experiments were performed 24 to 32 h later. Patch-clamp recordings were made exclusively from green fluorescent cells.
Expression Plasmids. cDNA encoding rabbit CaV2.3 (GenBank accession number X67856) , human M3 muscarinic acetylcholine receptor (GenBank accession number X15266) , and human M5 muscarinic acetylcholine receptor (GenBank accession number M80333) were in pcDNA3.1+ (Invitrogen, Carlsbad, CA). Rat brain α2δb (GenBank accession number M86621) was in pMT2 (Genetics Institute, Cambridge, MA), rabbit β3 (GenBank accession number X64300) was in pcDNA3 (Invitrogen), human M1 muscarinic acetylcholine receptor (GenBank accession number X52068) was in pCD, and jellyfish enhanced green fluorescent protein (GenBank accession number U55763) was in pEGFP (BD Biosciences Clontech, Palo Alto, CA). PKCδ1–121 and PKCδ1–298 (derived from rat PKCδ) (GenBank accession number NM031525) were in pEFLINK (Schuringa et al., 2001).
Patch-Clamp Recordings. Large-bore patch pipettes were pulled from 100-μl borosilicate glass micropipettes (VWR, West Chester, PA) and filled with a solution containing 155 mM CsCl, 10 mM Cs2-EGTA, 4 mM Mg-ATP, 0.32 mM Li-GTP, and 10 mM HEPES (adjusted to pH 7.4 with CsOH). Aliquots of the pipette solutions were stored at –80°C, kept on ice after thawing, and filtered at 0.22 μm immediately before use. In experiments that required intracellular dialysis with antibodies or peptides, anti-Gαq/11, preimmune rabbit IgG, PKC19–31, PKA6–22, or PKCϵ translocation inhibitor peptide (PKCϵTIP) was added to prefiltered pipette solution at the indicated concentrations and not filtered thereafter. The bath solution contained 145 mM NaCl, 40 mM CaCl2 (or 40 mM BaCl2, as indicated), 2 mM KCl, and 10 mM HEPES (adjusted to pH 7.4 with NaOH). CCh (0.5–1.0 mM) was dissolved directly in the bath solution and applied by whole-bath exchange. Phorbol 12 myristate-13-acetate (PMA), 4α-phorbol, bisindolylmaleimide I (Bis I), Bis V, and Gö 6976 were dissolved in DMSO to make stock solutions of 1 to 10 mM. The final DMSO bath solution concentration was between 0.05 and 0.2%. At these concentrations, DMSO alone did not affect channel modulation.
Pipette tips were coated with paraffin to reduce capacitance. Pipettes had d.c. resistances of 1.0 to 1.5 MΩ. Ca2+ currents were recorded in the whole-cell, ruptured-patch configuration. After forming a gigaohm seal in the cell-attached configuration, residual pipette capacitance was compensated using the negative capacitance circuit of the amplifier. After establishing the whole-cell configuration, the d.c. resistance was routinely >1 GΩ. The steady holding potential was –90 mV. No corrections were made for liquid junction potentials. Depolarizations to +30 mV (near the peak of the current-voltage relationship) were delivered at 0.2 Hz unless otherwise noted. Currents were filtered at 2 to 10 kHz using the built-in Bessel filter (four-pole low-pass) of an Axopatch 200B amplifier (Axon Instruments Inc., Union City, CA) and sampled at 10 to 50 kHz using a Digidata 1200 analog-to-digital board (Axon Instruments Inc.) installed in a Gateway Pentium computer. The pCLAMP 8.0 software programs Clampex and Clampfit (Axon Instruments Inc.) were used for data acquisition and analysis, respectively. Figures were drawn using the software program Origin (version 6.0; OriginLab Corp., Northampton, MA).
Linear cell capacitance (C) was determined by integrating the area under the whole-cell capacity transient, which was evoked by a voltage-clamp step from –90 to –80 mV. The average value of C was 21 ± 1 pF (mean ± S.E.M.; n = 187 cells). To minimize voltage errors, the analog series resistance compensation circuit of the amplifier was used to reduce the time constant for decay of the whole-cell capacity transient (τ) as much as possible. Series access resistance (RS) was calculated as τ × (1/C). The average values of τ and RS, measured before electronic compensation, were 85 ± 4 μs and 4.4 ± 0.2 MΩ, respectively. Maximal current amplitude was –1881 ± 109 pA (test potential + 30 mV). After electronic compensation, the average values of τ and RS were 62 ± 3 μs and 3.5 ± 0.3 MΩ, respectively, and the average maximum voltage error was 5.0 ± 0.3 mV.
All currents were corrected for linear capacitance and leakage currents using –P/4 subtraction. Ca2+ current amplitudes were measured at the time of peak inward current. Statistical comparisons were drawn by ANOVA or unpaired, two-tailed t test, as appropriate, with p < 0.05 considered significant. Temperature (20–24°C) was continuously monitored using a miniature thermocouple placed in the outflow of the recording chamber.
Reagents. Preimmune rabbit IgG and affinity-purified rabbit polyclonal antibodies directed against 19 amino acids within the extreme carboxyl terminus of Gαq/11 were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). These preparations were supplied in sterile saline without preservatives and used well before the expiration date. Carbachol, Bis I, Bis V, and other standard reagents were purchased from Sigma-Aldrich (St. Louis, MO). PMA, 4α-phorbol, PKC19–31, PKA6–22, Gö 6976, and PKCϵTIP were purchased from CalBiochem (San Diego, CA). Expression plasmids were generously provided by Drs. Y. Mori (National Institute of Physiological Sciences, Okazaki Aichi, Japan) (Cav2.3); T. Snutch (University of British Columbia, Vancouver, BC, Canada) (α2δb); K. Campbell (University of Iowa, Iowa City, IA) (B3); R. Fisher (University of Iowa, Iowa City, IA) (M1 receptor); and L. Dekker (University College London, United Kingdom) (PKCδ1–121 and PKCδ1–298) or obtained from the Guthrie Institute, Sayre, PA (M3 and M5 receptors).
Results
Differential Inhibition of CaV2.3 by M1 versus M3 and M5 Receptors.Figure 1A shows whole-cell Ca2+ currents recorded from an HEK293 cell coexpressing CaV2.3 and M1 receptors. As described previously by Melliti et al. (2000), a saturating concentration of CCh (1 mM) evoked a small and rapid inhibition that was followed by a larger and slower stimulation of CaV2.3 current amplitudes. In the present study, we found that the two other Gαq/11-coupled muscarinic receptor subtypes, M3 and M5, also trigger dual modulation of CaV2.3 (Fig. 1, B and C). However, M3 and M5 receptors produced much stronger inhibition of CaV2.3 than M1 receptors (Fig. 1D). On average, currents were inhibited by 39 ± 3% (n = 12) through M3 receptors and by 24 ± 4% (n = 9) through M5 receptors, compared with only 7 ± 2% (n = 23) inhibition through M1 receptors (p < 0.0001; ANOVA). In contrast, all three receptor subtypes generated similar stimulation of CaV2.3. Thus, CaV2.3 currents were stimulated by 32 ± 3% (n = 23) through M1 receptors, 21 ± 5% (n = 10) through M3 receptors, and 25 ± 4% (n = 8) through M5 receptors (Fig. 1D). These magnitudes of stimulation are indistinguishable (p > 0.05; ANOVA). The onset of stimulation was quite variable, with stimulation reaching a peak within 30 to 90 s of commencing CCh application among cells expressing the same receptor subtype. No consistent differences in the kinetics of stimulation were noted among receptor subtypes.
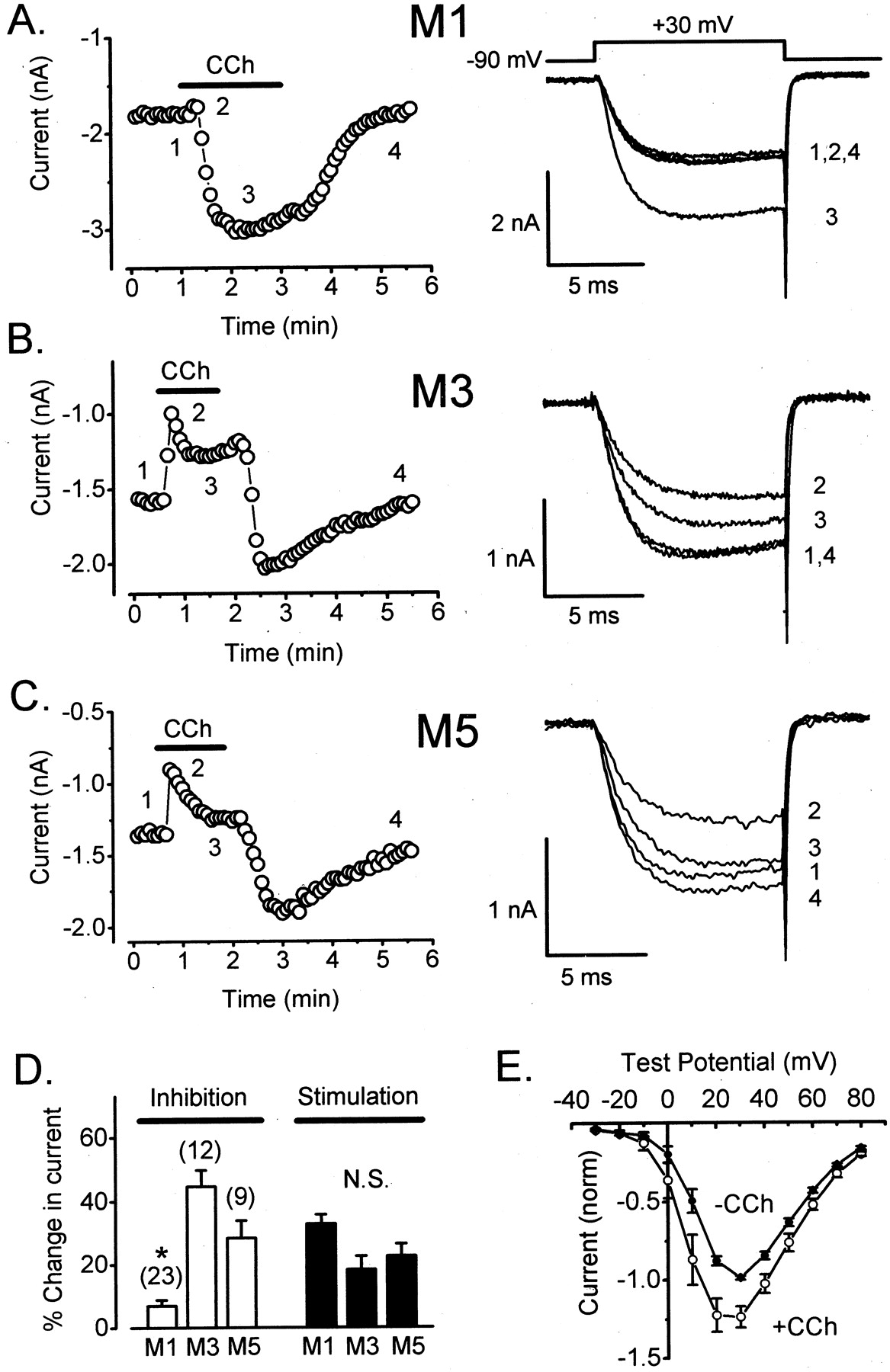
Differential modulation of CaV2.3 by M1, M3, and M5 muscarinic acetylcholine receptors. A, left, maximal Ca2+ current amplitudes recorded from a cell coexpressing CaV2.3 channels and M1 receptors are plotted as a function of time during a representative experiment. Whole-cell Ca2+ currents were evoked every5sby step depolarizations from –90 mV (the steady holding potential) to +30 mV. The standard pipette solution containing 10 mM EGTA was employed. Bath application of CCh (1 mM) is indicated by a heavy horizontal line. Right, currents recorded at times indicated at left. C = 9 pF, RS = 2.0 MΩ, and the maximal voltage error (VE) was 6.0 mV. B, left, modulation of CaV2.3 in a cell coexpressing the M3 receptor. Application of CCh (500 μM) is indicated by a heavy horizontal line. Right, currents recorded at times indicated at left. C = 19 pF; RS = 2.0 MΩ; VE = 3.1 mV. C, left, modulation of CaV2.3 in a cell coexpressing the M5 receptor. Application of CCh (500 μM) is indicated by a heavy horizontal line. Right, currents recorded at times indicated at left. C = 19 pF; RS = 2.6 MΩ; VE = 3.5 mV. D, summary of results. Inhibition was measured as [1 – (inhibited current/control current)] × 100%. Stimulation was measured as [(stimulated current – inhibited current)/control current] × 100%. Error bars represent ± S.E.M.; means were compared by ANOVA. E, stimulation of CaV2.3 through M1 receptors is voltage-independent. Control I-V data (•) were obtained at 0.2 Hz before CCh (1 mM) application. During the peak of stimulation, I-V data (○) were again obtained at 0.2 Hz. Symbols represent data obtained from the same six cells before and during CCh application. For each cell, current amplitude was normalized to the peak current recorded before CCh application. Statistical significance (★, p < 0.05; ANOVA) is indicated.
Muscarinic Stimulation of CaV2.3 Is Voltage-Independent. Muscarinic inhibition of CaV2.3 is fast and mediated by Gβγ subunits, whereas stimulation is slow and mediated by PTX-insensitive Gα subunits (Meza et al., 1999; Melliti et al., 2000). Previous studies of N- and P/Q-type Ca2+ channels have indicated that Gβγ subunits typically cause a voltage-dependent form of channel inhibition, whereas channel modulation by Gα and its associated downstream signaling pathways is typically voltage-independent (Dunlap and Ikeda, 1998). To determine the voltage dependence (or independence) of muscarinic stimulation, we measured CaV2.3 current-voltage (I-V) relationships before and during maximal stimulation. As illustrated in Fig. 1E, the I-V relationship for CaV2.3 currents did not change appreciably during exposure to CCh. This result suggests that muscarinic stimulation of CaV2.3 is voltage-independent.
Gαq/11 Mediates Muscarinic Stimulation of CaV2.3. Previous experiments have shown that muscarinic stimulation of CaV2.3 is PTX-insensitive and blocked by coexpression of either RGS2 or PLCβ1ct (Melliti et al., 2000). Although RGS2 and PLCβ1ct are thought to interact preferentially with Gαq/11, the Gα-specificities of these two signaling proteins have not been exhaustively examined, and HEK293 cells may express PTX-insensitive Gα subunits in addition to Gαq/11. To determine whether Gαq/11 mediates muscarinic stimulation of CaV2.3, we dialyzed cells with neutralizing antibodies directed against the carboxyl terminus of Gαq/11. Such antibodies have been previously demonstrated to block slow muscarinic inhibition of L-type Ca2+ channels reconstituted in HEK293 cells (Bannister et al., 2002). Figure 2B illustrates a representative experiment in which anti-Gαq/11 in the pipette solution (10 ng/μl) prevented muscarinic stimulation of CaV2.3. Anti-Gαq/11 reduced stimulation to very low levels (6 ± 2%) in 12 of 16 cells. In the four remaining cells, CCh evoked normal magnitudes of stimulation presumably reflecting poor movement of anti-Gαq/11 into the cell. Data from these latter four cells were excluded from further analysis. Inhibition of CaV2.3 through M1 receptors was unaffected (6 ± 2%; n = 12) by anti-Gαq/11. Control experiments showed that both inhibition (10 ± 2%; n = 7) and stimulation (28 ± 5%; n = 7) of CaV2.3 were unaltered in cells dialyzed for equal periods with rabbit preimmune IgG (10 ng/μl). These data (summarized in Fig. 2C) support the conclusion that muscarinic stimulation of CaV2.3 is mediated by Gαq/11.

Muscarinic stimulation of CaV2.3 is mediated by Gαq/11. Data from cells coexpressing M1 receptors. A, stimulation of CaV2.3 is blocked by intracellular dialysis for >8 min with a neutralizing antibody (10 ng/μl) directed against Gαq/11. C = 20 pF; RS = 3.4 MΩ; VE = 4.2 mV. B, stimulation of CaV2.3 is unaffected by intracellular dialysis for >8 min with preimmune rabbit IgG (10 ng/μl). C = 21 pF; RS = 2.2 MΩ; VE = 2.7 mV. C, summary of results. Error bars represent ± S.E.M. Statistical significance (★, p < 0.05; t test) is indicated.
Muscarinic Stimulation of CaV2.3 Involves Signaling by 1,2-Diacylglycerol. A major downstream effector of Gαq/11 is phospholipase C-β1, which cleaves phosphoinositol 4,5-bisphosphate to produce 1,4,5-inositol trisphosphate and 1,2-diacylglycerol (DAG). DAG activates numerous isozymes of PKC (Mellor and Parker, 1998). To investigate the potential importance of DAG in muscarinic stimulation of CaV2.3, we expressed the regulatory region (residues 1–298) of PKCδ. This construct (PKCδ1–298) lacks the catalytic domain of PKCδ but contains the DAG-binding C1 region, which is conserved among all DAG-dependent PKC isozymes (Mellor and Parker, 1998). PKCδ1–298 is predicted to sequester DAG and thereby interfere with the activation of endogenous PKCs. Additionally, PKCδ1–298 may act as a dominant-negative to more specifically block signaling by endogenous PKCδ (Schuringa et al., 2001). As seen in Fig. 3A, expression of PKCδ1–298 prevented stimulation of CaV2.3 through M1 receptors. On average, stimulation was decreased to very low levels (4 ± 2%; n = 8) in cells transfected with this construct. Importantly, Gβγ-mediated inhibition of CaV2.3 was not reduced in these cells. On the contrary, inhibition was actually larger (17 ± 3%) in PKCδ1–298–transfected cells (Fig. 3C). We expressed a shorter region of PKCδ (residues 1–121) that lacks the DAG-binding C1 region as a control for these experiments. As seen in Fig. 3B, this shorter construct (PKCδ1–121) failed to reduce muscarinic stimulation of CaV2.3 (32 ± 8%; n = 11). These results show that PKCδ1–298 selectively blocks stimulation of CaV2.3 through M1 receptors and support the idea that muscarinic stimulation of CaV2.3 requires signaling by DAG.

Stimulation of CaV2.3 requires signaling by diacylglycerol (DAG). Data from cells coexpressing M1 receptors. A, stimulation of CaV2.3 is blocked by coexpression of the regulatory region of PKCδ (amino acids 1–298; PKCδ1–298). C = 26 pF; RS = 2.4 MΩ; VE = 4.2 mV. B, stimulation of CaV2.3 is unaffected by coexpression of a shorter construct (PKCδ1– 121) that lacks the DAG-binding C1 domain. C = 25 pF; RS = 4.3 MΩ; VE = 6.9 mV. C, summary of results with PKCδ1–121 and PKCδ1–298. Error bars represent ± S.E.M. ★, p < 0.05; t test. D, PMA occludes stimulation of CaV2.3 through M1 receptors. Application of PMA (500 nM) is indicated by the hatched horizontal bar. Application of CCh (1 mM) is indicated by a heavy horizontal line. E, stimulation of CaV2.3 is unaffected by 4α-phorbol (500 nM). C = 36 pF; RS = 1.3 MΩ; VE = 2.7 mV. F, summary of results with PMA and 4α-phorbol. Error bars represent ± S.E.M. ★, p < 0.05; t test.
PMA Occludes Stimulation of CaV2.3. Previous studies have shown that CaV2.3 currents are robustly enhanced by phorbol esters such as PMA (Stea et al., 1995; Meza et al., 1999; Kamatchi et al., 2003). PMA effectively activates many PKC isozymes (Mellor and Parker, 1998). As expected, PMA (500 nM) triggered a significant increase (61 ± 7%; n = 8) in the amplitude of CaV2.3 currents (Fig. 3D). To determine whether currents enhanced by PMA could be stimulated further through M1 receptors, we applied CCh after the PMA-induced current increase had stabilized (Fig. 3D). All together, muscarinic stimulation of CaV2.3 was greatly reduced (6 ± 3%; n = 8) in cells pre-exposed to PMA. In control experiments, application of inactive 4α-phorbol (500 nM) failed to enhance CaV2.3 currents (4 ± 3%; n = 10) and failed to reduce agonist-dependent stimulation of CaV2.3 through M1 receptors (33 ± 8%; Fig. 3, E and F). Inhibition and stimulation of CaV2.3 were also unaltered by DMSO (p > 0.05), the vehicle used to solubilize PMA and 4α-phorbol (Fig. 3F). These results support the conclusion that muscarinic stimulation of CaV2.3 requires signaling by DAG.
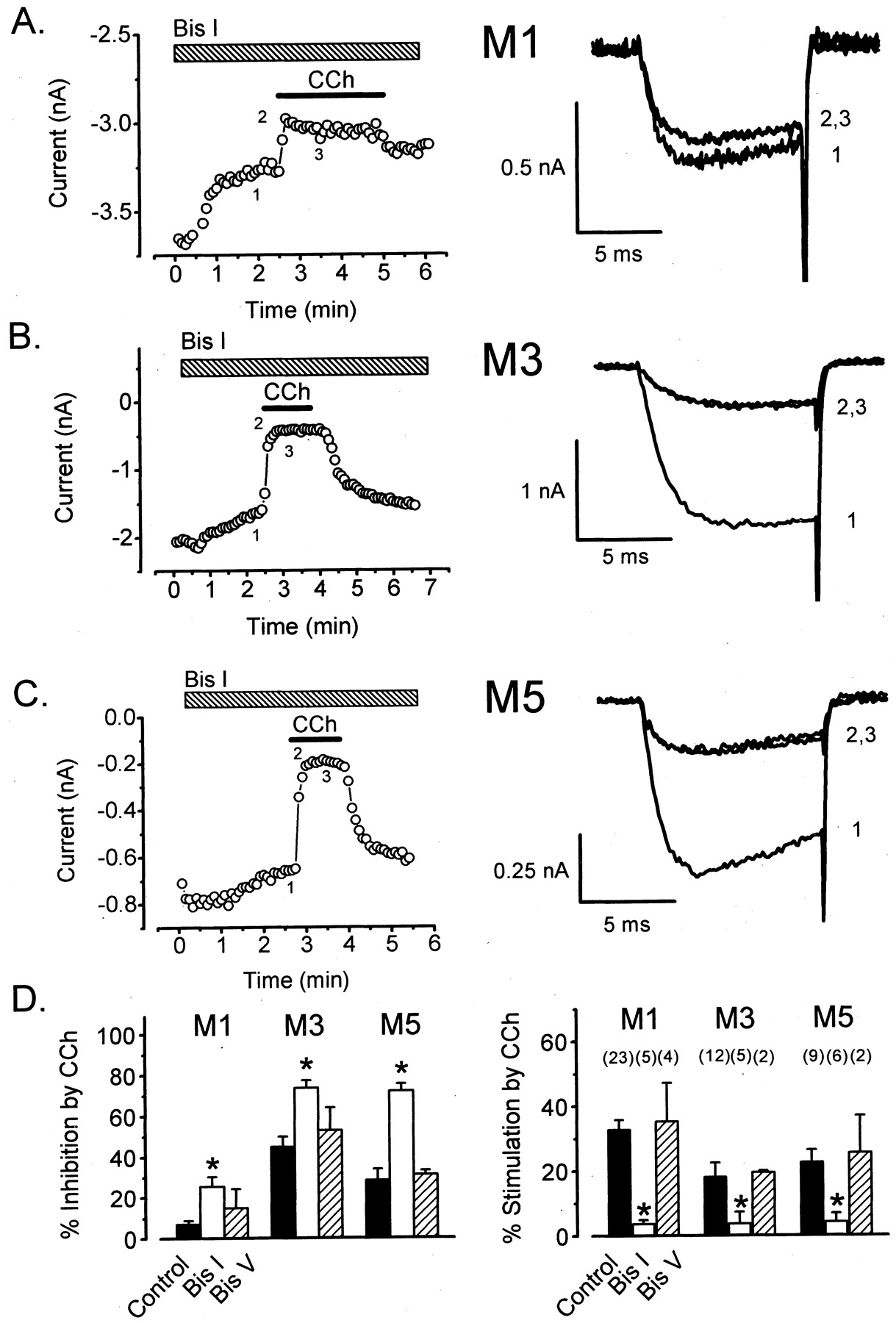
A DAG-Dependent but Ca2+-Independent PKC Isozyme Mediates Stimulation of CaV2.3. The requirement for DAG suggests that muscarinic stimulation of CaV2.3 involves PKC (Mellor and Parker, 1998). To further test this idea, we exposed cells to the specific PKC inhibitor Bis I. As seen in Fig. 4A, application of Bis I (500 nM) greatly reduced stimulation of CaV2.3 through M1 receptors (4 ± 1%; n = 5). In contrast, stimulation was unaffected by the same concentration of Bis V (Fig. 4D), the inactive control compound for Bis I. As seen in Fig. 4, A and C, the application of Bis I often produced a substantial decline in the baseline current, whereas Bis V did not (data not shown), suggesting that CaV2.3 is basally phosphorylated by PKC. We also found that stimulation of CaV2.3 through M1 receptors was significantly reduced (11 ± 4%; n = 6) after intracellular dialysis with PKC19–31 (100 μM), a pseudosubstrate peptide inhibitor of PKC (data not shown). However, stimulation was unaffected (25 ± 4%; n = 4) by intracellular dialysis with PKA6–22 (100 μM), a pseudosubstrate peptide inhibitor of PKA (data not shown).

PKC mediates stimulation of CaV2.3 through M1, M3, and M5 muscarinic receptors. A, left, stimulation of CaV2.3 through M1 receptors is blocked by bath application of Bis I (hatched bar). Right, currents recorded at times indicated at left. C = 11 pF; RS = 2.1 MΩ; VE = 1.5 mV. B, left, stimulation of CaV2.3 through M3 receptors is blocked by Bis I. Right, currents recorded at times indicated at left. C = 19 pF; RS = 2.7 MΩ; VE = 5.7 mV. C, left, stimulation of CaV2.3 through M5 receptors is blocked by Bis I. Right, currents recorded at times indicated at left. C = 13 pF; RS = 3.3 MΩ; VE = 2.7 mV. D, summary of results. Left, inhibition; right, stimulation. Statistical significance (★, p < 0.05; t test) between control and Bis I-exposed cells is indicated. Error bars represent ± S.E.M. The concentrations of Bis I and Bis V were 500 nM; CCh was 0.5–1.0 mM.
As shown in Fig. 4, B and C, Bis I also effectively blocked stimulation of CaV2.3 through M3 receptors (4 ± 4%; n = 5) and M5 receptors (4 ± 3%; n = 6). In control experiments (Fig. 4D, right), Bis V failed to reduce stimulation through either M3 receptors (20 ± 1%; n = 2) or M5 receptors (26 ± 12%; n = 2). Together, these pharmacological results indicate that muscarinic stimulation of CaV2.3 involves signaling by PKC.
Interestingly, each muscarinic receptor subtype triggered stronger inhibition of CaV2.3 in the presence of Bis I (Fig. 4D). This finding may partly reflect a somewhat more accurate measurement of inhibition in the absence of stimulation. However, because inhibition is considerably faster than stimulation (Meza et al., 1999), this explanation is unlikely to completely account for the effect of Bis I. A more likely possibility is that PKC-dependent phosphorylation antagonizes the inhibition of CaV2.3 by Gβγ, as has been previously shown for N-type (CaV2.2) and P/Q-type (CaV2.1) Ca2+ channels (Hamid et al., 1999).
HEK293 cells express the α, βI, βII, and γ isozymes of PKC, which are activated by DAG and Ca2+, and the δ and ϵ isozymes of PKC, which are activated by DAG but not by Ca2+ (Mellor and Parker, 1998; Leaney et al., 2001). Robust muscarinic stimulation of CaV2.3 observed in recordings made with 10 mM intracellular EGTA (Fig. 1) argues against the involvement of Ca2+-dependent PKC isozymes. However, such isozymes might mediate stimulation if they operated within a Ca2+ microdomain not buffered by EGTA. To examine this possibility, we substituted BAPTA (20 mM) for EGTA in the pipette solution. Surprisingly, BAPTA completely eliminated both inhibition and stimulation of CaV2.3 (Fig. 5A). In complementary experiments, we substituted Ba2+ for Ca2+ in the bath solution under the assumption that Ba2+ cannot activate Ca2+-dependent PKC isozymes. As shown in Fig. 5B, Ba2+ currents exhibited normal muscarinic stimulation (28 ± 6%; n = 7). Next, we exposed cells to Gö 6976, a selective inhibitor of Ca2+-dependent PKCs (Martiny-Baron et al., 1993). Cells were exposed to a fairly high concentration (500 nM) of Gö 6976 for at least 15 min before CCh application. As indicated in Fig. 5C, Gö 6976 had no effect on muscarinic stimulation of CaV2.3 (26 ± 3%; n = 7). These results (summarized in Fig. 5D) indicate that muscarinic stimulation of CaV2.3 does not involve a Ca2+-dependent PKC isozyme.

Muscarinic stimulation of CaV2.3 is Ca2+-independent. Data from cells coexpressing M1 receptors. A, muscarinic modulation of CaV2.3 is blocked by high intracellular BAPTA. Cells were dialyzed for >5 min with a pipette solution containing BAPTA (20 mM) in place of EGTA (10 mM). C = 15 pF; RS = 2.6 MΩ; VE = 1.1 mV. B, Ba2+ currents also exhibit muscarinic stimulation. The bath solution contained 40 mM Ba2+in place of Ca2+. For these experiments, the standard (10 mM EGTA) pipette solution was used. C = 10 pF; RS = 6.1 MΩ; VE = 7.6 mV. C, muscarinic stimulation of CaV2.3 is unaffected by Gö 6976, an inhibitor of Ca2+-dependent PKC isozymes. Cells were exposed to Gö 6976 (500 nM) for >15 min before applying CCh. The standard (10 mM EGTA) pipette solution was employed. C = 16 pF; RS = 4.6 MΩ; VE = 5.7 mV. D, summary of results. Error bars represent ± S.E.M. Before CCh application, cells were dialyzed for >3 min with PKCϵ translocation inhibitor peptide (PKCϵTIP; 40 μM) dissolved in the standard pipette solution. Bis I data from Fig. 4 are shown for comparison. Statistical significance (★, p < 0.05; ANOVA) between control and Bis I-treated or BAPTA-dialyzed cells is indicated.
The effectiveness of PKCδ1–298 (Fig. 3, A and C) suggests that stimulation might be mediated by PKCδ. However, because PKCδ1–298 is expected to sequester DAG, it may block activation of other Ca2+-independent isozymes such as PKCϵ. To examine this possibility, we dialyzed cells with a relatively high concentration (40 μM) of PKCϵTIP, a non-apeptide that specifically blocks translocation and activation of PKCϵ (Johnson et al., 1996). As summarized in Fig. 5D, muscarinic stimulation of CaV2.3 was unaffected (30 ± 6%; n = 7) by PKCϵTIP, arguing against involvement of this isozyme. We conclude that muscarinic stimulation of CaV2.3 is mediated by a DAG-dependent but Ca2+-independent PKC isozyme, most likely PKCδ.
Discussion
In this study, we compared modulation of neuronal/neuroendocrine CaV2.3 Ca2+ channels by the three Gαq/11-coupled muscarinic acetylcholine receptors (M1, M3 and M5). We found that M3 and M5 receptors produce much stronger inhibition of CaV2.3 than M1 receptors (Fig. 1D). This differential inhibition of CaV2.3 is evident under relatively physiological conditions (i.e., in the presence of stimulation) and observed when stimulation has been blocked through pharmacological suppression of PKC (Fig. 4). In contrast to inhibition, we found that all three receptor subtypes trigger comparable stimulation of CaV2.3 (Fig. 1D). Using M1 receptors, we analyzed the pathway underlying stimulation of CaV2.3 and demonstrated that it involves signaling by Gαq/11, diacylglycerol, and a Ca2+-independent PKC isozyme, most likely PKCδ. Because M1, M3, and M5 receptors all couple primarily to Gαq/11, and because stimulation involves signaling by Gαq/11, we assume that each muscarinic receptor subtype activates the same stimulatory pathway.
The similar stimulation of CaV2.3 by M1, M3, and M5 receptors may indicate that each receptor subtype activates PKC with similar efficacy or that PKC activation is saturating in each case. The differential inhibition of CaV2.3 by M1 versus M3/M5 receptors is interesting and potentially significant. Previously, we showed that M1 receptors inhibit CaV2.3 by liberating Gβγ subunits (Melliti et al., 2000). Based on this result, it is reasonable to assume that Gβγ also mediates inhibition of CaV2.3 through M3 and M5 receptors. In rat sympathetic neurons, native N-type Ca2+ channels (CaV2.2) are differentially inhibited by different Gβγ subunits (Ruiz-Velasco and Ikeda, 2000). These previous findings suggest that M1 receptors may couple to Gβγ subunits that have a lower affinity for CaV2.3 than Gβγ subunits that couple to M3 and M5 receptors. Another possibility is that M3 and M5 receptors are positioned closer to CaV2.3 than are M1 receptors, such that the channels experience different concentrations of Gβγ during receptor activation. Regardless of the mechanism, our results suggest that the effects of acetylcholine on CaV2.3 may depend on which muscarinic receptor subtypes are present.
In an earlier study, Stea et al. (1995) expressed CaV2.3 in Xenopus oocytes and studied its modulation by Gαq/11-coupled metabotropic glutamate 1a receptors and phorbol esters. Stea et al. (1995) showed that CaV2.3 currents were enhanced by phorbol esters, and that current enhancement was blocked by staurosporine, a broad spectrum inhibitor of serine/threonine kinases. Our present results show that muscarinic stimulation of CaV2.3 is blocked by Bis I (Fig. 4) and PKC19–31, which are more specific inhibitors of PKC than staurosporine. Thus, our results clearly indicate that PKC mediates muscarinic stimulation of CaV2.3. We also found that Bis I enhances muscarinic inhibition of CaV2.3 (Fig. 4). This secondary effect of Bis I suggests that PKC-dependent phosphorylation antagonizes inhibition of CaV2.3 by Gβγ. Previously, PKC was shown to antagonize inhibition of native N-type Ca2+ channels through either Gαi- or Gαs-coupled neurotransmitter receptors (Swartz et al., 1993; Zhu and Ikeda, 1994). It has been proposed that PKC-dependent phosphorylation of a specific threonine residue within the I-II linker of CaV2.2 (N-type Ca2+ channels) reduces binding of Gβγ to this channel region and thereby antagonizes Gβγ-mediated channel inhibition (Hamid et al., 1999). Our present results suggest that a similar cross-talk between PKC and Gβγ may apply to CaV2.3.
Inhibition and stimulation of CaV2.3 were both completely blocked by intracellular dialysis with 20 mM BAPTA (Fig. 4, A and D). This finding recalls our previous demonstration that BAPTA eliminates slow muscarinic inhibition of CaV1.2c (L-type) Ca2+ channels (Bannister et al., 2002). In that study, we found that 5, 5′-dinitro BAPTA, which binds Ca2+ with very low affinity, blocks inhibition as effectively as BAPTA. The effectiveness of 5, 5′-dinitro BAPTA indicates that the effect of BAPTA is unrelated to Ca2+ chelation. Together, our previous results (Bannister et al., 2002) and present findings (Fig. 5) support the conclusion that BAPTA uncouples signaling through M1 receptors independently of Ca2+ buffering.
In the present study, we showed that CaV2.3 Ba2+ currents also exhibit muscarinic stimulation (Fig. 5), indicating that Ca2+ influx is not required. Ba2+ currents were recorded with 10 mM EGTA in the pipette solution, which should have effectively buffered Ca2+ released from intracellular stores. Thus, our data indicate that muscarinic stimulation of CaV2.3 is Ca2+-independent. We also found that muscarinic stimulation of CaV2.3 was insensitive to Gö 6976, a specific inhibitor of Ca2+-dependent PKC isozymes. Together, these results suggest that muscarinic stimulation of CaV2.3 is mediated by a Ca2+-independent PKC isozyme. Two Ca2+-independent PKCs, PKCδ and PKCϵ, are expressed in HEK293 cells (Leaney et al., 2001). The failure of PKCϵTIP to reduce stimulation of CaV2.3 (Fig. 5D) argues against the involvement of this isozyme. Thus, it seems that PKCδ is the most likely candidate. Consistent with this possibility, we found that muscarinic stimulation of CaV2.3 was blocked by PKCδ1–298, the regulatory region of PKCδ. This construct has been previously reported to function as a dominant-negative in blocking substrate phosphorylation by wild-type PKCδ (Schuringa et al., 2001). However, because PKCδ1–298 should sequester DAG, it may block activation of any Ca2+-independent PKC isozyme. Thus, although our results suggest that muscarinic stimulation of CaV2.3 is mediated by PKCδ, we cannot exclude the involvement of other Ca2+-independent isozymes.
Potential Physiological Significance. Native R-type Ca2+ currents trigger hormone secretion by certain neuroendocrine cells (Grabsch et al., 2000; Weiergraber et al., 2000; Matsuda et al., 2001; Vajna et al., 2001) and contribute to evoked secretion of neurotransmitters at certain central synapses (Wu et al., 1998; Wang et al., 1999; Albillos et al., 2000; Gasparini et al., 2001; Vajna et al., 2001). Furthermore, recent work indicates that CaV2.3 is essential for certain forms of synaptic plasticity within mammalian hippocampus (Dietrich et al., 2003). The presence of CaV2.3 on neuronal somata and dendrites (Yokoyama et al., 1995; Day et al., 1996; Delmas et al., 2000) raises the possibility that R-type Ca2+ currents are involved in Ca2+-dependent gene expression or in helping to determine the postsynaptic responses of neuronal membranes. For example, CaV2.3 might influence action-potential duration and frequency or the response of neuronal membranes to receptor agonists. Stimulation of CaV2.3 currents by Gαq/11-coupled receptors may produce significant effects on neurosecretion, gene expression, membrane excitability, or synaptic plasticity. Recent studies have shown that CaV2.3 is involved in pain transduction, fear behavior, spatial memory, and glucose metabolism (Saegusa et al., 2000; Kubota et al., 2001; Matsuda et al., 2001; Lee et al., 2002; Dietrich et al., 2003). Thus, in addition to influencing the basic biophysical properties of neurons and neuroendocrine cells, the muscarinic modulation of CaV2.3 could potentially affect higher-order physiological processes. Our present study provides new information about signaling mechanisms that modulate CaV2.3. This new information should contribute to a better understanding of the physiology of native R-type Ca2+ channels.
Footnotes
-
This work was supported by grants from the National Institutes of Health (NS34423), the American Heart Association (AHA) (0040067N), the Utah Agricultural Experimental Station (Project 638) (to B.A.A.), and by Predoctoral Fellowship 9910094Z from the AHA (to R.A.B.)
-
ABBREVIATIONS: RGS, regulator of G protein signaling; PLCβ1ct, the carboxyl-terminal region of phospholipase C-β1; PKC, protein kinase C; HEK, human embryonic kidney; PKA, protein kinase A; TIP, translocation inhibitor peptide; PMA, phorbol 12-myristate 13-acetate; Bis, bisindolylmaleimide; DMSO, dimethyl sulfoxide; C, linear cell capacitance; τ, time constant for decay of the whole-cell capacity transient; RS, series access resistance; ANOVA, analysis of variance; I-V, current-voltage; DAG, 1,2-diacylglycerol; BAPTA, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; Gö 6976, 12-(2-Cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo(2,3-a)pyrrolo(3,4-c)-carbazole.
- Received June 6, 2003.
- Accepted October 31, 2003.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}