


Abstract
Pharmacologically important drugs were examined as potential inhibitors or permeants of human concentrative nucleoside transporters 1 (hCNT1)- and 2 (hCNT2)-producing stable transfectants by assessing their abilities to inhibit uridine transport. hCNT1 exhibited high affinities for uridine analogs (5-fluorouridine, 2′-deoxyuridine, 5-fluoro-2′-deoxyuridine, and 5-fluoro-5′-deoxyuridine) with Ki values of 22 to 33 μM, whereas hCNT2 exhibited moderate affinities for 5-fluoro-2′-deoxyuridine, high affinities for 2′-deoxyuridine and 5-fluorouridine, and low affinity for 5-fluoro-5′-deoxyuridine. The uridine analogs were transported at 2-fold higher rates (at 10 μM) by hCNT1 than by hCNT2. Enantiomeric configuration and the 3′-hydroxyl group of the ribose ring were important determinants for interaction with hCNTs, whereas the 2′-hydroxyl group was less important. Both transporters bound N6-(p-aminobenzyl)adenosine with affinities similar to those of adenosine (Ki = 28-39 μM). Other adenosine receptor ligands, including caffeine, bound better to hCNT1 than to hCNT2 (Ki = 46 versus 103 μM, respectively), whereas 2-chloroadenosine bound better to hCNT2 than to hCNT1 (Ki = 37 and 101 μM, respectively). There was a greater than 3-fold difference in binding affinities between hCNT1 and hCNT2 for nicotine (Ki = 63 versus 227 μM). However, direct measurements of nicotine and caffeine uptake rates (10 μM) failed to demonstrate mediated uptake by either transporter. Although hCNT1 bound several adenosine analogs relatively well, it did not transport 2-chloro-2′-deoxyadenosine (cladribine) or 2-fluoro-9-β-d-arabinofuranosyladenine (fludarabine), whereas hCNT2 transported both, albeit with low activities. The results indicated that although hCNT1 and hCNT2 possess some overlap in transport of several uridine and adenosine analogs, they also exhibit distinct differences in capacity to interact with some adenosine receptor ligands, adenosine-based drugs, and nicotine.
Uridine is a permeant of all mammalian nucleoside transporters identified to date (Cass et al., 1998), and, like adenosine, it seems to act as a signaling molecule (Anderson and Parkinson, 1997). Analogs of uridine, such as 5-fluorouridine, 5-fluoro-2′-deoxyuridine, and 5-fluoro-5′-deoxyuridine, are important in the treatment of disseminated human cancers, especially of the gastrointestinal tract, breast, and ovary (Sadahiro et al., 2001). 5-Fluoro-5′-deoxyuridine is an active metabolite of the orally administered drug capecitabine (Budman, 2000), which has significant activity against solid tumors.
Adenosine also seems to be a universal permeant of mammalian nucleoside transporters.2 However, it is a high-affinity, low-activity permeant of CNT1-mediated processes (Ritzel et al., 1997, 1998; Cass et al., 1998), whereas it is a high-affinity, high-activity permeant of the hCNT2-mediated process (Ritzel et al., 1998; Lang et al., 2001). The role of adenosine in mammalian tissues is diverse, particularly in excitable tissues such as the heart and brain, in which it exhibits protective effects during pathological conditions (Abd-Elfattah et al., 1998). The rapid creation and depletion of extracellular pools of adenosine involved in stimulation and inactivation of adenosine receptors, respectively, are attributed, in part, to fluxes mediated by nucleoside transporters. Adenosine analogs such as N6-(p-aminobenzyl)adenosine and caffeine are known to bind to adenosine receptors (Linden et al., 1985), although nothing is known of whether they interact with hCNTs. Other analogs, such as 2-chloro-2′-deoxyadenosine (cladribine) and fludarabine, are used for the treatment of different hematological malignancies.
The general permeant selectivities of hCNT1 and hCNT2 have been defined for key natural nucleosides in studies with recombinant transporters produced in Xenopus laevis oocytes (Ritzel et al., 1997, 1998). These studies have classified hCNT1 and hCNT2 as mediating, respectively, the cit or cif processes in human cells and are generally referred to as pyrimidine-nucleoside-selective and purine-nucleoside-selective transporters, respectively (Ritzel et al., 1997, 1998; Cass et al., 1999).
Analysis of the functional characteristics of the concentrative nucleoside transporters of human cells was for many years hampered by low activity and the presence of multiple transporter types. The first human stable transfectant (named ARAC/D2) of a member of the CNT family was produced by introducing cDNAs encoding for hCNT2 in a human null background of transport-deficient CEM-ARAC human leukemia cells (Lang et al., 2001). These model cell lines provided the ideal cellular reagent to assess the permeant selectivities of hCNT2 in the absence of other nucleoside-transport activities. Although the purine-nucleoside selectivity of hCNT2 was well known (Ritzel et al., 2001), this transporter exhibited an unexpected capacity to transport several uridine analogs, including 5-fluorouridine and 5-fluoro-2′-deoxyuridine (Lang et al., 2001).
Whereas studies of the permeant-binding domains of hCNT1 and hCNT2 have been undertaken (Loewen et al., 1999), a comparative study of their permeant selectivities has not been performed. In addition, potent inhibitors with similar characteristics to the classic hENT1 inhibitor nitrobenzylthioinosine (NBMPR) have not yet been found for members of the hCNT family. The present work explored uridine and adenosine analogs and some pharmacologically important drugs for their potential inhibitory effects and interactions with hCNT1 and hCNT2. A stable transfectant containing hCNT1 (named TLCT1) was produced in the same transport-deficient cell line that was used to create the ARAC/D2 stable transfectant (Lang et al., 2001). Because hCNT1 is pyrimidine-nucleoside-selective but also accepts adenosine (Ritzel et al., 1997), we wanted to assess the ability of this transporter to interact with other adenosine analogs, notably adenosine receptor ligands such as 2-chloroadenosine, N6-(p-aminobenzyl)adenosine, and caffeine (Varma et al., 2002), as well as some anticancer drugs (e.g., cladribine and fludarabine) (Galmarini et al., 2001). The extent to which hCNT1 interacted with these analogs, as well as several uridine analogs, was compared against that of the purine-nucleoside-selective hCNT2. The comparison included 5-fluorouridine, 5-fluoro-2′-deoxyuridine, and 5-fluoro-5′-deoxyuridine and nucleosides with 1) d- and l-enantiomer configurations, 2) substitution or elimination of the hydroxyl group at the 2′ and 3′ position of the sugar, and 3) the addition of bulky, hydrophobic substituents or halogens at, respectively, the N6 and C5 positions of the base to determine whether modifications of natural nucleosides affect interaction with the transporters and, in some cases, transportability.
Materials and Methods
Materials. Radioisotopes were purchased from Moravek Biochemicals Inc. (Brea, CA). 3H-Labeled nucleosides were purified by high-performance liquid chromatography using water-methanol gradients on a C18 reverse-phase column. All chemicals were of analytical grade and were obtained from either Sigma-Aldrich (Oakville, ON, Canada), Fisher Scientific Co. (Nepean, ON, Canada), or the Cross Cancer Institute Pharmacy (Edmonton, AB, Canada), unless otherwise stated. Other chemicals were of analytical grade and commercially available. Cell-culture supplies were from Invitrogen (Carlsbad, CA).
Plasmid Construction. The open reading frame of hCNT1 was polymerase chain reaction-amplified using hCNT1 (Ritzel et al., 1997) cDNA in the original cloning vector pGem (BD Biosciences Clontech, Palo Alto, CA) as a template and the following 5′EcoRI- and 3′XbaI-containing primers (restriction sites underlined): 5′-GGGAATTCATGGAGGAACCTGCAGCCAGCCCTG-3′ and 5′-CCTCTAGATCACTGTGCGCAGATCGTGTG-3′. The EcoRI-hCNT1-XbaI fragment was sub-cloned into the multiple cloning site downstream of the enhancer/promoter sequences of the immediate early gene of human cytomegalovirus of the mammalian expression vector pcDNA3 (Invitrogen). The structure of pcDNA3/hCNT1 was verified by restriction endonuclease mapping and DNA sequencing. DNA sequences were determined by Taq DyeDeoxy terminator cycle sequencing with an automated model 310 DNA sequencer (Applied Biosystems, Foster City, CA). Sequence analysis used MacVector DNA analysis software (Oxford Molecular Limited, Bethesda, MD).
Production of Stable Transfectants. The production of hCNT2 stable transfectants (named ARAC/D2) in CEM-ARAC cells has been described previously (Lang et al., 2001). hCNT1 stable transfectants (named TLCT1) were produced by adaptations of the procedures described previously (Lang et al., 2001). pcDNA3/hCNT1 was transformed into Top10F′ Escherichia coli by the heat-shock method and plasmid DNA was prepared using the Maxi Plasmid Purification Kit (QIAGEN, Valencia, CA) according to the manufacturer's instructions. The pcDNA3/hCNT1 was linearized with BlgII restriction enzyme and diluted to 0.25 μg/μl with RPMI 1640 medium that contained no glutamine (Gln-free RPMI).
Recipient cells were prepared for electroporation from actively proliferating CEM-ARAC cultures by 1) centrifugation (800g for 10 min), 2) resuspension at 5 × 104 cells/ml in RPMI 1640 medium with 10% HIHS, and 3) incubation at 37°C in a humidified (95%) atmosphere of 5% CO2 in air for 72 h to a concentration of approximately 4 × 105 cells/ml, after which they were 4) collected by centrifugation (800g for 10 min), 5) washed twice with Gln-free RPMI, and 6) concentrated in Gln-free RPMI to 3 × 108 cells/ml. Electroporation was performed using a gene pulser equipped with a capacitance extender (Bio-Rad, Mississauga, ON, Canada) and a 4-mm gap cuvette (Bio-Rad) in which was placed 170 μl of cell suspension and 30 μl of the linearized plasmid mixture. After electroporation (190 V, 960 μF for 65-75 ms), cells were 1) resuspended in RPMI 1640 supplemented with 15% HIHS, 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) and 2) incubated for 24 h at 37°C in a humidified (95%) atmosphere of 5% CO2 and air, after which they were 3) triturated, 4) diluted to 2 × 105 cells/ml with RPMI 1640 supplemented with 10% HIHS, 5) incubated at 37°C for an additional 24 h, 6) collected by centrifugation (800g for 10 min), 7) resuspended at 1 × 105 cells/ml in RPMI 1640 supplemented with 10% HIHS and 0.2 μg/μl geneticin (G418; Invitrogen), and 8) incubated at 37°C in a humidified (95%) atmosphere of 5% CO2 with medium changes at 3- to 4-day intervals for approximately 1 month.
Geneticin-resistant cells were identified by cloning of cells in soft agarose. Geneticin-resistant cells were grown in soft-agarose cloning medium that consisted of equal volumes of fresh RPMI 1640 supplemented with 0.2 μg/μl G418, 20% HIHS, 1 mM α-ketoglutarate (Sigma-Aldrich), and 6 mM glutamine and the same medium that had been “conditioned” by exposure to actively proliferating CEMARAC cells for 24 h. Geneticin-selection cultures were suspended in cloning medium with 1% Seaplaque agarose (Mandel, Guelph, ON, Canada) to yield 5 × 103 to 1 × 104 cells/100-mm plate (Fisher Scientific) and incubated for 3 weeks at 37°C in a humidified (95%) atmosphere of 5% CO2 in air. Surviving colonies were selected, expanded, and screened for nucleoside transport activity in transport assays.
Growth and Maintenance of Cell Lines. CEM-ARAC is a human T-lymphoblastic leukemia cell line that is nucleoside transport-defective because of the lack of hENT1 (Lang et al., 2001). CEMARAC and its derivatives were subcultured every 3 to 4 days in growth medium, RPMI 1640 (Invitrogen) supplemented with 10% fetal bovine serum (v/v), to which was added 0.25 μM 7-deazaadenosine and 0.5 μM 1-β-d-arabinofuranosylcytosine for CEM-ARAC cells and 0.25 μM 1-β-d-arabinofuranosylcytosine and 0.2 μg/μl G418 for the TLCT1 and ARAC/D2 transfectants. Cells were incubated at 37°C in a humidified (95%) atmosphere of 5% CO2 in air, and cell numbers were determined using a Coulter Z2 electronic particle counter equipped with a size analyzer (Beckman Coulter Inc., Burlington, ON). All cell lines were routinely assessed and shown to be free of Mycoplasma species by direct culture in agar/cell-free medium (Medical Microbiology Laboratory, Edmonton, AB, Canada).
Nucleoside Transport Assays. Initial rates of nucleoside uptake were measured at room temperature using the oil-stop method (Lang et al., 2001). Transport assays involving adenosine deaminase-sensitive nucleosides were conducted in the presence of 2 μM 2′-deoxycoformycin to prevent deamination. Time courses of uptake of 3H-nucleosides were determined using rapid sampling procedures in which the transport process was initiated by the addition of cells to the 3H-nucleoside solution (1:1) and terminated by rapid addition of excess nonradioactive nucleoside solution followed by immediate centrifugation (16,000g for 30 s) through transport oil [mixture of paraffin oil (Fisher Scientific) and silicone 550 oil (Dow Corning, Mississauga, ON, Canada) with final specific gravity of 1.03 g/ml] to separate the cells from the permeant solution. Replicate assay mixtures were exposed to either [14C]polyethylene glycol or 3H2O to determine trapped extracellular and intracellular water volumes, respectively. The cell pellets were solubilized in 0.5 ml of 5% Triton X-100, and cell-associated radioactivity was determined by liquid scintillation counting. Transport rates were derived from regression analysis of the linear sodium-dependent component of uptake, and kinetic parameters (apparent Km and Vmax values) were calculated using Prism software (GraphPad Software Inc., San Diego, CA). IC50 values were determined from nonlinear regression analyses of transport rates plotted as a function of the logarithmic concentration of compound tested. Ki values were derived from IC50 using the mean Km values for uridine transport in TLCT1 and ARAC/D2 cells according to the equation used by Cheng and Prusoff (1973).
Results
Kinetics of Uridine Transport by hCNT1 in TLCT1 Stable Transfectants. It has been shown that the purine-nucleoside-selective hCNT2 possesses the capacity to transport several uridine analogs with high affinity (Lang et al., 2001). These uridine analogs were explored further to assess the extent of interaction with the pyrimidine-nucleoside-selective hCNT1. A similar procedure used in the production and characterization of hCNT2-producing stable transfectants (ARAC/D2) (Lang et al., 2001) was used in this study to generate hCNT1-producing stable transfectants (TLCT1, results not shown).
The kinetic properties of hCNT1-mediated uridine transport into TLCT1 stable transfectants were determined by calculating initial rates of uridine uptake, derived from uptake time courses as a function of graded uridine concentrations as presented in Fig. 1. Uridine influx into TLCT1 cells was saturable, and the Michaelis-Menten kinetic parameters (Km and Vmax) obtained from three independent experiments (mean ± S.D.) were 34 ± 2 μM and 1.01 ± 0.17 pmol/μl cell water/s, respectively.

Kinetic analysis of uridine transport mediated by hCNT1 in TLCT1 stable transfectants. Transport assays with TLCT1 transfectants were performed with graded concentrations of [3H]uridine in sodium-containing (▪) and sodium-free (□) transport buffer as described under Materials and Methods. Inset, a representative uptake time course with 10 μM[3H]uridine is shown. Initial rates of uptake were determined from the slopes of linear portions of uptake time courses over 180-s time periods similar to those shown in the inset. The sodium-dependent component of [3H]uridine uptake rates are plotted as a function of the uridine concentrations tested (0-1000 μM). Results are the means ± S.D. of triplicate determinations, and error bars are not shown where S.D. values were smaller than the size of the data symbols. The data were analyzed by nonlinear regression using GraphPad Prism software. Shown is one of three independent experiments that yielded similar results. The calculated kinetic parameters Km and Vmax are 35 μM and 1.12 pmol/μl cell water/s, respectively.
Inhibitory Activities of 5-Fluoro-5′-Deoxyuridine and Other Uridine Analogs on Uridine Uptake by hCNT1 and hCNT2 in Stable Transfectants. Capecitabine) is metabolized to 5-fluoro-5′-deoxyuridine, which is believed to be the form that enters target tumor cells. A comparison of the inhibitory activities of 5-fluoro-5′-deoxyuridine on [3H]uridine transport by hCNT1 and hCNT2 was assessed in the concentration-effect relationships shown in Fig. 2. Although both cell types exhibited reductions in uridine transport rates with increasing concentrations of 5-fluoro-5′-deoxyuridine, hCNT1-mediated uridine transport was more sensitive than hCNT2-mediated transport, with IC50 values of 31 and 498 μM, respectively.

Concentration-dependent inhibition of uridine transport by 5-fluoro-5′-deoxyuridine in TLCT1 and ARAC/D2 stable transfectants. [3H]uridine uptake was measured as described under Materials and Methods in sodium-containing or sodium-free transport buffer alone or with graded concentrations of 5-fluoro-5′-deoxyuridine in TLCT1 (hCNT1-containing) (▪) and ARAC/D2 (hCNT2-containing) (□) transfectants. Sodium-dependent transport rates were calculated by subtracting uptake measurements performed in sodium-free transport buffer from uptake measurements performed in sodium-containing transport buffer and are presented as the percentage of sodium-dependent uridine transport remaining in the absence of 5-fluoro-5′-deoxyuridine. Values are means ± S.D. obtained from triplicate determinations. Results were analyzed by nonlinear regression using GraphPad Prism analysis software. IC50 and Ki values are given in Table 1.
The inhibitory effects of several other structurally related uridine analogs were also examined in TLCT1 (hCNT1-containing) or ARAC/D2 (hCNT2-containing) stable transfectants in experiments similar to those shown in Fig. 2. The computed Ki values are presented in Table 1. Ki values in the range of 22 to 33 μM were obtained in hCNT1-containing TLCT1 transfectants for 5-fluorouridine, 2′-deoxyuridine, 5-fluoro-2′-deoxyuridine, and 5-fluoro-5′-deoxyuridine. Although similarly low Ki values were obtained in hCNT2-containing ARAC/D2 transfectants for 5-fluorouridine and 2′-deoxyuridine (34 and 39 μM, respectively), the Ki values for 5-fluoro-2′-deoxyuridine and 5-fluoro-5′-deoxyuridine were, respectively, slightly (82 μM) and much (411 μM) higher. Capecitabine itself did not inhibit [3H]uridine uptake into either TLCT1 or ARAC/D2 cells when tested at 1 mM.
Ki values for inhibition of uridine transport into TLCT1 and ARAC/D2 stable transfectants by analogs of uridine
The concentrations of test compounds that reduced uridine uptake rates by 50% (IC50 values) were determined from concentration-effect relationships obtained as shown in Figure 2. Ki values were derived from IC50 values according to the equation of Cheng and Prusoff (1973) using mean (n = 3) Km values of 34 ± 2 and 46 ± 4 μM, respectively, for uridine transport in TLCT1 and ARAC/D2 transfectants.
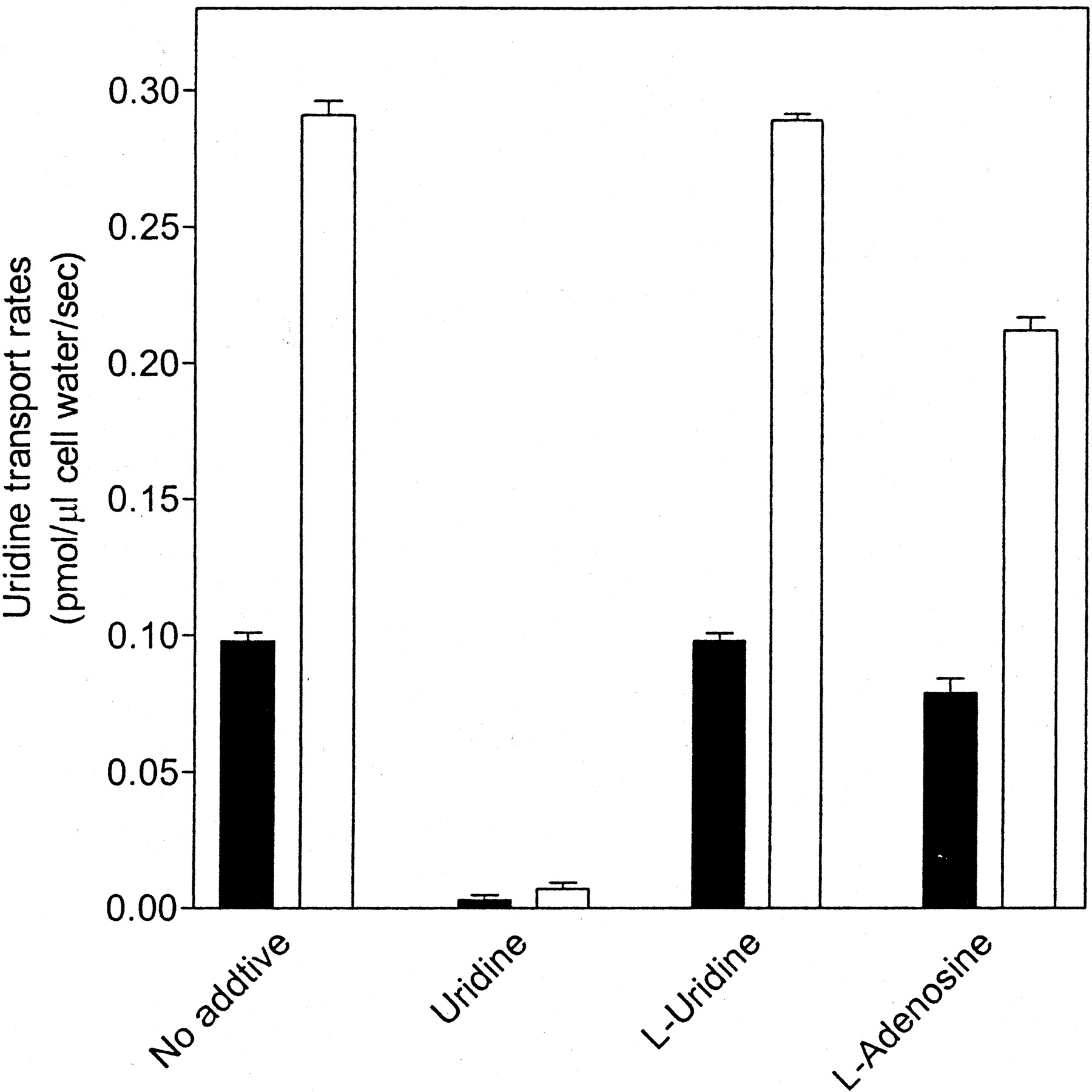
Inhibition of [3H]Uridine Transport into TLCT1 and ARAC/D2 Cells by l-Adenosine but Not by l-Uridine. Earlier studies with murine cells have demonstrated that the equilibrative NBMPR-sensitive transport process exhibits stereoselectivity for the d-enantiomer of various nucleosides (Gati et al., 1989). The extent to which l-enantiomers of uridine and adenosine inhibited the uptake of 3H-labeled d-uridine by TLCT1 or ARAC/D2 cells producing, respectively, hCNT1 and hCNT2 was assessed in the experiments shown in Fig. 3. Transport rates of d-uridine in the presence and absence of high concentrations of either l-uridine or l-adenosine were derived from linear regression analysis of uptake time courses. Uridine transport rates, which were higher for TLCT1 cells (0.291 ± 0.009 pmol/μl cell water) than for ARAC/2 cells (0.098 ± 0.006 pmol/μl cell water), were reduced to negligible levels in the presence of nonradiolabeled d-uridine. There was no apparent change in the uridine transport rates observed for either cell line in the presence of 1 mM l-uridine, whereas there were small reductions in uridine transport rates in the presence of l-adenosine to 0.212 ± 0.008 and 0.079 ± 0.009 pmol/μl cell water, respectively, in hCNT1-containing TLCT1 and hCNT2-containing ARAC/D2 cells. The inhibition by l-adenosine of hCNT1-mediated uridine influx was greater than that of hCNT2-mediated influx, although both inhibitions were much less than those observed with d-uridine. These results indicated weak interaction of l-adenosine with hCNT1 and hCNT2.

Effects of l-uridine and l-adenosine on uridine influx into TLCT1 and ARAC/D2 stable transfectants. Uptake of 10 μM [3H]uridine into TLCT1 (hCNT1-containing, □) and ARAC/D2 (hCNT2-containing, ▪) transfectants was measured in sodium-containing transport buffer in the absence or presence of 1 mM uridine, l-uridine or l-adenosine as described under Materials and Methods. Initial rates of uptake were obtained from 180-s time courses similar to those shown in Fig. 1. Each value represents the mean ± S.D. of three determinations.
Inhibitory Activities of Adenosine Analogs and Nicotine on Uridine Uptake by hCNT1 and hCNT2 in Stable Transfectants. Several derivatives of adenine and adenosine were examined for their ability to inhibit uridine transport into either TLCT1 or ARAC/D2 cells. Initial rates of uptake of 10 μM [3H]uridine were measured in the presence of 1 mM test compounds, and the results are presented in Table 2. High concentrations (1 mM) of nonradiolabeled uridine reduced [3H]uridine transport to negligible levels in both TLCT1 and ARAC/D2 cells (0.005 ± 0.002 and 0.002 ± 0.003 pmol/μl cell water/s, respectively). Transport of [3H]uridine into TLCT1 cells was inhibited completely by 1 mM concentrations of N6-(p-aminobenzyl)adenosine, nicotine, caffeine, adenosine, and 2′-deoxyadenosine, almost completely by 5′-deoxyadenosine, 2-chloroadenosine, partially by 8-chloroadenosine, and not at all by erythro-9-(2-hydroxy-3-nonyl)adenine, 2′-3′-dideoxyadenosine, and adenine. Transport of [3H]uridine into ARAC/D2 cells was inhibited completely by 1 mM concentrations of N6-(p-aminobenzyl)adenosine, caffeine, adenosine, and 2′-deoxyadenosine, partially by nicotine, 5′-deoxyadenosine, and 2-chloroadenosine and poorly, or not all, by 8-chloroadenosine, erythro-9-(2-hydroxy-3-nonyl)adenine, 2′-3′-dideoxyadenosine, and adenine.
Inhibition by analogs of adenine and adenosine on uridine transport by TLCT1 and ARAC/D2 stable transfectants
Transport of 10 μM [3H]uridine into TLCT1 and ARAC/D2 transfectants was measured in sodium-containing transport buffer in the absence or presence of 1 mM test compound as described under Materials and Methods. Initial rates of uptake were obtained from 180-s uptake time courses. The results are the means ± S.D. of triplicate determinations. The ratios of transport rates were determined by dividing rates in the presence of test compound by rates obtained in their absence.
Concentration-Effect Relationships of Adenosine Analogs and Nicotine for Inhibition of [3H]uridine Transport into TLCT1 and ARAC/D2 Stable Transfectants. It has been shown that N6-(p-aminobenzyl)adenosine is a high-affinity ligand of A1 and A3-type adenosine receptors as well as a potent inhibitor of es (i.e., ENT1-mediated) transporters (Durand and Green, 2001). Because both N6-(p-aminobenzyl)adenosine and 5′-deoxyadenosine were strong inhibitors of [3H]uridine transport into TLCT1 and ARAC/D2 cells, their relative inhibitory activities were determined in the experiments shown in Fig. 4. The IC50 values for N6-(p-aminobenzyl)adenosine inhibition of [3H]uridine transport into TLCT1 and ARAC/D2 cells were 36 and 43 μM, respectively, indicating that N6-(p-aminobenzyl)adenosine was a moderately potent inhibitor of hCNT1- and hCNT2-mediated uridine influx. The inhibitory activity of 5′-deoxyadenosine was examined in the experiment shown in Fig. 4. 5′-Deoxyadenosine exhibited more pronounced inhibition against TLCT1 cells (IC50 = 88 μM) than against ARAC/D2 cells (IC50 = 287 μM).

Concentration-dependent inhibition of uridine transport by N6-(p-aminobenzyl)adenosine and 5′-deoxyadenosine in TLCT1 and ARAC/D2 stable transfectants. Initial rates of sodium-dependent [3H]uridine uptake were measured in the presence or absence of graded concentrations of either N6-(p-aminobenzyl)adenosine (v, θ) (0.1 μM to 1 mM) or 5′-deoxyadenosine (λ, μ) (0.1 μM to 1.5 mM) in TLCT1 (hCNT1-containing, v, λ), or ARAC/D2 (hCNT2-containing, θ, μ) transfectants as described in Fig. 2. IC50 and Ki values are given in Table 3.
Both caffeine and nicotine exert important physiological effects on the central nervous system in association with adenosine (von Borstel et al., 1986). The molecular structure of caffeine is similar to that of adenosine, and it can act as a ligand of adenosine receptors (Evoniuk et al., 1987). To determine whether either caffeine or nicotine interacts well with hCNT1 or hCNT2, the IC50 values for inhibition of uridine transport were determined in the experiments shown in Fig. 5. Both caffeine and nicotine produced concentration-dependent reductions in uridine transport rates, yielding IC50 values of 59 and 82 μM, respectively, in TLCT1 cells, and 125 and 276 μM, respectively, in ARAC/D2 cells. These values were 2- to 3-fold higher for ARAC/D2 cells than for TLCT1 cells, suggesting differences in the ability of these compounds to interact with hCNT1 or hCNT2.
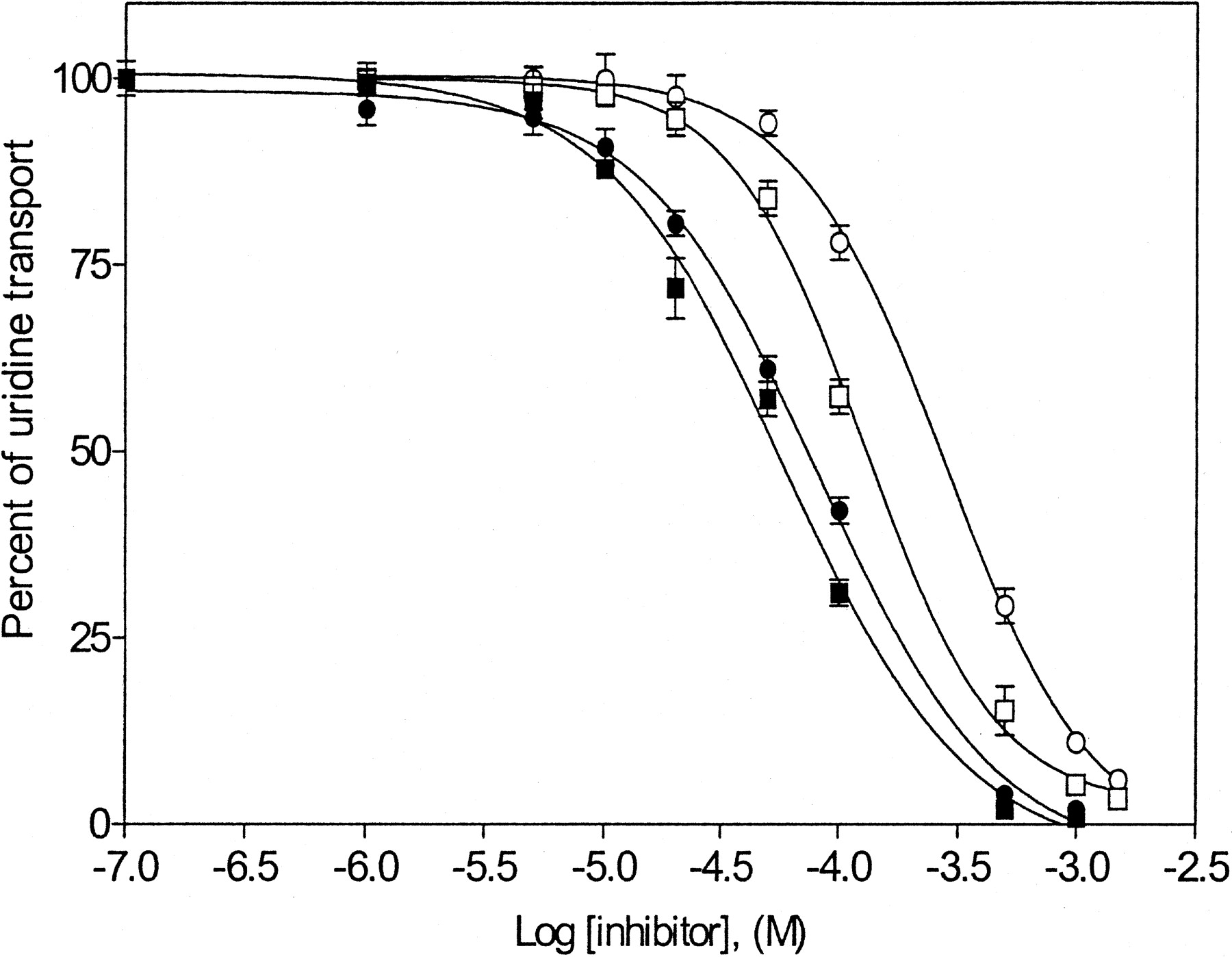
Concentration-dependent inhibition of uridine transport by caffeine and nicotine in TLCT1 and ARAC/D2 stable transfectants. Initial rates of sodium-dependent [3H]uridine uptake were measured in the presence or absence of graded concentrations of either caffeine (▪, □) (0.1 μM to 1 mM) or nicotine (•, ○) (0.1 μM to 1.5 mM) in TLCT1 (hCNT1-containing, ▪, •) or ARAC/D2 (hCNT2-containing, □, ○) transfectants as described in Fig. 2. IC50 and Ki values are given in Table 3.
Comparison of the Inhibitory Constants of Adenosine Analogs and Nicotine in TLCT1 and ARAC/D2 Cells. Because several of the adenosine analogs and nicotine inhibited uridine transport, other adenosine analogs were also examined in similar inhibition experiments. The IC50 values from concentration-effect relationships were used to calculate Ki values, and the results are presented in Table 3. For both TLCT1 and ARAC/D2 cells, Ki values for N6-(p-aminobenzyl)adenosine, adenosine, and 2′-deoxyadenosine were in the range of 28 to 44 μM. The Ki values for TLCT1 cells were lower for caffeine, nicotine, and 5′-deoxyadenosine (46, 63, and 68 μM, respectively) than for ARAC/D2 cells (103, 227, and 237 μM, respectively). The Ki values for 2-chloroadenosine were 37 μM in ARAC/D2 cells and 101 μM in TLCT1 cells. The other analogs of adenosine (8-chloroadenosine, l-adenosine, and 2′-3′-dideoxyadenosine) exhibited little, if any, inhibitory effects on uridine transport in either cell line.
Assessment of Transportability of Natural and Therapeutic Nucleosides by hCNT1 and hCNT2 in TLCT1 and ARAC/D2 Transfectants. The inhibition experiments presented above revealed differences in the inhibitory activities of uridine and adenosine analogs on [3H]uridine transport into the TLCT1 and ARAC/D2 stable transfectants, suggesting differences in interactions of these compounds with hCNT1 and hCNT2. The nature of this interaction was addressed further by comparison of the rates of uptake of 3H-labeled uridine analogs, adenosine analogs, and nicotine at a single concentration (10 μM). Initial rates were determined from time courses of uptake of 3H-labeled compounds and the results are summarized in Table 4. TLCT1 cells, which possessed hCNT1, exhibited approximately 3-fold higher sodium-dependent transport of 2′-deoxyuridine and uridine than ARAC/D2 cells, which possessed hCNT2. Thymidine was not transported by ARAC/D2 cells but was transported by TLCT1 cells, albeit at relatively low levels (net rate, 0.067 pmol/μl cell water/s) compared with uridine (net rate, 0.293 pmol/μl cell water/s).
Comparison of initial rates of uptake of nucleoside, caffeine, and nicotine by TLCT1 and ARAC/D2 stable transfectants
Uptake of 10 μM 3H-labeled compound into TLCT1 and ARAC/D2 cells was measured as described under Materials and Methods. Initial rates of uptake were determined from linear regression analysis of 3-min time courses of uptake in sodium-containing or sodium-free transport buffer. Net transport rates (i.e., sodium-dependent transport) were determined by subtracting uptake rates in the absence of sodium from uptake rates in the presence of sodium. For all compounds tested, transport rates (pmol/μl cell water/s) in the absence of sodium ranged from 0.003 ± 0.002 to 0.018 ± 0.014 for TLCT1 transfectants and from 0.001 ± 0.002 to 0.012 ± 0.008 for ARAC/D2 transfectants.
Adenosine was the only purine nucleoside among the compounds examined that was transported by hCNT1-containing TLCT1 cells, although the net rate (0.040 pmol/μl cell water/s) was only 16% of that observed with hCNT2-containing ARAC/D2 cells (net rate, 0.248 pmol/μl cell water/s). ARAC/D2 cells exhibited approximately 2-fold higher transport rates of adenosine analogs than uridine analogs, and the purine 2′-deoxynucleosides were transported with rates that were almost similar to those of the purine ribonucleosides. Thus, hCNT2 exhibited a greater capacity to transport adenosine analogs than uridine analogs, whereas hCNT1 exhibited a greater capacity to transport uridine analogs than adenosine analogs.
Because the hCNT1 and hCNT2 proteins differed in their capacity to transport physiological uridine and adenosine analogs, further experiments were conducted to compare their relative abilities to transport therapeutically active analogs of uridine, adenosine, and nicotine. As shown in Table 4, both TLCT1 and ARAC/D2 cells transported 5-fluorouridine, although the rate in TLCT1 cells was 2.4-fold greater than that in ARAC/D2 cells. TLCT1 cells exhibited no detectable uptake of cladribine or fludarabine, whereas ARAC/D2 cells transported both compounds, albeit at low rates and the rate for cladribine (0.023 pmol/μl cell water/s) was 2.3-fold higher than that for fludarabine (0.010 pmol/μl cell water/s). Despite the general preference of hCNT2 for purine nucleosides, the rate for 5-fluorouridine was 4.8- and 11-fold higher than the rates for cladribine and fludarabine, respectively. Whereas hCNT1 was also capable of transporting adenosine and 2′-deoxyadenosine, it did not transport either cladribine or fludarabine.
Because inhibition experiments revealed that caffeine and nicotine differentially inhibited uridine influx by hCNT1 and hCNT2, the transportability of these substances was assessed by measuring their uptake directly in TLCT1 and ARAC/D2 cells (Table 4). The results indicated little, if any, uptake of either caffeine or nicotine by hCNT1 or hCNT2, although both compounds exhibited inhibitory activities that were comparable with those of compounds that are permeants of the transporters.
Discussion
The present work used the stable transfectants TLCT1 and ARAC/D2 producing, respectively, hCNT1 and hCNT2 in a transport-deficient leukemia cell line to compare the two major members of the hCNT family in the same genetic background. This approach overcomes some of the difficulties associated with the multiplicity of nucleoside transporters in cell preparations (Roovers and Meckling-Gill, 1996).
The inhibition and transport studies presented here compared hCNT1 and hCNT2 in their capacity to interact with analogs of uridine and adenosine, including anticancer drugs. The Ki values for uridine analogs (5-fluorouridine, 2′-deoxyuridine, 5-fluoro-2′-deoxyuridine, and 5-fluoro-5′-deoxyuridine) were similarly low for hCNT1 in the range of 22 to 33 μM, suggesting that hCNT1 exhibited high affinities for these compounds. The purine-nucleoside-selective hCNT2 exhibited moderate- to high-affinity interactions with 5-fluorouridine, 2′-deoxyuridine, and 5-fluoro-2′-deoxyuridine but low-affinity interactions with 5-fluoro-5′-deoxyuridine. It has been shown that 5-fluoro-5′-deoxyuridine, a metabolite of capecitabine, is a permeant of recombinant hCNT1 produced in X. laevis oocytes (Mata et al., 2001). Our study demonstrated that 5-fluoro-5′-deoxyuridine interacted well with hCNT1 with a Ki value of 24 μM. We also showed that capecitabine itself is not likely to be transported by either hCNT1 or hCNT2 because it was unable to inhibit [3H]uridine influx into either TLCT1 or ARAC/D2 cells when tested at 1 mM. The high affinity of 5-fluoro-5′-deoxyuridine for hCNT1, as indicated by our result, suggests that this transporter plays an important role in its uptake at low concentrations.
Analyses of the inhibitory activities of adenine and adenosine analogs on uridine uptake demonstrated that erythro-9-(2-hydroxy-3-nonyl)adenine, 2′-3′-dideoxyadenosine, and adenine exhibited negligible inhibitory effects, and therefore they were probably not permeants of either hCNT1 or hCNT2. The inability of 2′-3′-dideoxyadenosine to reduce hCNT1- or hCNT2-mediated [3H]uridine rates was consistent with the inability of 2′-3′-dideoxyuridine to inhibit hCNT2-mediated uridine uptake, suggesting the importance of the 3′-hydroxyl moiety for interaction with hCNTs. The 3′ hydroxyl group is evidently an important structural feature for permeant recognition for most nucleoside transporters because es (i.e., hENT1-mediated) transport in erythrocytes was also poorly inhibited by several 2′,3′-dideoxynucleosides (Domin et al., 1993). Rat CNT1, which shares 83% amino acid identity with hCNT1, exhibits the ability to mediate the uptake of the antiviral nucleoside 2′,3′-dideoxycytidine, albeit with low transport activity, when produced in X. laevis oocytes (Ritzel et al., 1997).
Several nucleoside drugs highly effective against viral and cancer diseases are l-nucleosides (Pastor-Anglada et al., 1998). l-Enantiomers of antiviral drugs are important in that they have been shown to be more potent than their corresponding d-enantiomers, and they are essentially noncytotoxic to human peripheral blood mononuclear cells (Stuyver et al., 2002). Uptake of 3H-labeled d-uridine by hCNT1- and hCNT2-containing cells was not affected by l-uridine. However, l-adenosine inhibited hCNT1-mediated d-uridine uptake, albeit poorly, suggesting that it was either a nonpermeant or, at best, a poor permeant of this transporter. The d-nucleosides inhibited [3H]uridine uptake completely, suggesting that the enantiomeric configuration of the ribofuranosyl moiety was an important determinant for interaction with hCNT1 and hCNT2. This result is in agreement with an earlier study that examined sodium-dependent and -independent nucleoside transport processes in L1210 murine leukemia cells and showed that these processes exhibited distinct preferences for d-adenosine over l-adenosine (Dagnino et al., 1991).
Ligands of adenosine receptors such as N6-(p-aminobenzyl)adenosine, 2-chloroadenosine, and caffeine completely blocked uridine influx by hCNT1 and hCNT2. The Ki values for N6-(p-aminobenzyl)adenosine inhibition of uridine influx were similarly low for hCNT1 (28 μM) and hCNT2 (35 μM) and were in the concentration range that was observed for adenosine (39 and 25 μM, respectively, for hCNT1 and hCNT2). N6-(p-aminobenzyl)adenosine has been shown to inhibit adenosine transport in cultured glial cells from chick embryo brain (Thampy and Barnes, 1983). Although the molecular identity of the nucleoside transporter responsible for the interaction with N6-(p-aminobenzyl)adenosine in cells of brain tissue is not known, it may potentially be a CNT family member. There is evidence that CNT family members are present in human brain, and their distribution was correlated with that of adenosine A1 receptors in different brain areas (Lang et al., unpublished results).
The Ki values for inhibition of uridine transport by 2-chloroadenosine were 37 and 101 μM, respectively, in hCNT2-containing ARAC/D2 and hCNT1-containing TLCT1 cells, indicating a greater than 2-fold difference in binding affinities. The Ki value for caffeine inhibition of hCNT2-mediated uridine transport was more than 2-fold greater than the Ki value for its inhibition of hCNT1-mediated transport (103 versus 46 μM). Although hCNT1 exhibited moderate affinity for caffeine that was within the range of Km values observed for many natural nucleosides transported by the CNT family members (Ritzel et al., 2001), direct measurements of rates of uptake of 10 μM caffeine failed to demonstrate mediated uptake in either TLCT1 or ARAC/D2 cells.
The unexpected inhibitory activity of caffeine led to the analysis of nicotine for its interaction with the two hCNTs. There was an approximate 3.5-fold difference in Ki values (63 versus 227 μM) between hCNT1 and hCNT2. However, measurements of uptake rates of 10 μM nicotine demonstrated that it was either a poor permeant or a nonpermeant for hCNT1 and hCNT2. The serum level of caffeine achieved after an average (150-ml) cup of coffee is normally at least 4.9 μM (Nehlig and Debry, 1994). The resulting nicotine concentration in blood after smoking one cigarette is in the range of 0.23 to 0.32 μM (Lawson et al., 1998). The physiological significance of average levels of caffeine and nicotine on hCNTs is uncertain, although it is tempting to speculate, from our study, that an increased consumption above normal levels may potentially affect the ability of the hCNTs to transport other permeants and potentially affect their physiological functions. Permeants such as adenosine may be competitively inhibited from being transported intracellularly, leading to an increase in local adenosine concentrations near adenosine receptors. An increased extracellular adenosine concentration has been shown to be neuroprotective through its interaction with adenosine receptors by preventing the release of neurotransmitters and reducing neuronal excitability (Dunwiddie and Masino, 2001).
A comparison of the rates of transport of uridine analogs at 10 μM by hCNT1 and hCNT2 showed that 2′-deoxyuridine, uridine, and 5-fluorouridine were transported at 2-fold higher rates by hCNT1 than by hCNT2. This suggests that hCNT1 exhibited a greater capacity for mediating uptake of uridine analogs than hCNT2, although all of these compounds seemed to bind well to both transporters. In contrast, the rates of transport of 10 μM guanosine, 2′-deoxyguanosine, adenosine, and 2′-deoxyadenosine by hCNT2 were approximately 4-fold higher than the rates of adenosine transport by hCNT1. These results are consistent with the view that adenosine is a high-affinity, low-capacity permeant of hCNT1 and a high-affinity, high-capacity permeant of hCNT2 because adenosine bound well to both transporters with Ki values of 39 and 25 μM, respectively, for inhibition of uridine transport by hCNT1 and hCNT2. The concentrations of adenosine in the extracellular milieu range from 50 to 200 nM and can increase as much as 46-fold greater than physiological concentrations in pathological conditions such as hypoxia and ischemia (Latini and Pedata, 2001). It is possible that hCNT1 and hCNT2 act together to regulate extracellular adenosine pools, with hCNT2 mediating the majority of adenosine influx at physiological adenosine concentrations and hCNT1 providing an additional route for adenosine uptake at higher adenosine levels.
The adenosine-containing analogs (cladribine and fludarabine) were relatively poor permeants of hCNT2 compared with natural nucleosides and were also not permeants of hCNT1. This finding raises an important issue regarding similarities and differences in permeant recognition by human and rodent CNT family members. It has been shown that cladribine was a relatively good permeant of rCNT2 (also known as SPNT1) (Schaner et al., 1997). This indicates a species difference in interaction of cladribine with rat and human CNT2 proteins. The difference in transportability of cladribine is perhaps not surprising, given that these proteins share less amino acid identity (69%) at the amino-terminal half in which the substrate binding domain of CNTs is believed to reside (Loewen et al., 1999).
The results presented here demonstrated that several important substances which mediate effects on the central nervous system exhibited affinities that were similar to those of the natural nucleosides that are permeants of hCNT1 and hCNT2. Although hCNT1 is pyrimidine-nucleoside-selective, it exhibited higher affinities for interaction with several adenosine analogs as well as caffeine and nicotine, than hCNT2. In contrast, hCNT2, which is perceived to be purinenucleoside-selective, was capable of interacting and transporting several uridine analogs with high affinity. These somewhat surprising findings reveal new complexities with respect to permeant selectivities within the hCNT family. Perhaps more importantly, the differences between hCNT1 and hCNT2 in their abilities to transport nucleoside analogs provide useful new information for the design and use of nucleoside drugs.
Footnotes
- Received August 27, 2003.
- Accepted December 22, 2003.
↵2 The correspondence between human nucleoside transporter proteins (GenBank accession numbers given in parentheses) and their activities is: hENT1 (U81375), es; hCNT1 (U62966), cit; and hCNT2 (AF036109), cif.
C.E.C. is Canada Research Chair of Oncology, and J.D.Y. is Heritage Scientist of the Alberta Heritage Foundation for Medical Research. This work was supported by the Alberta Cancer Board and Canadian Cancer Society and Terry Fox Foundation operating grants from the National Cancer Institute of Canada.
ABBREVIATIONS: CNT, concentrative nucleoside transporter; ENT, equilibrative nucleoside transporter; h, human; r, rat; cladribine, 2-chloro-2′-deoxyadenosine; NBMPR, nitrobenzylmercaptopurine ribonucleoside (6-[(4-nitrobenzyl)thio]-9-β-d-ribofuranosylpurine); HIHS, heat-inactivated horse serum; Gln-free RPMI, RPMI 1640 medium without glutamine; fludarabine, 2-fluoro-9-β-d-arabinofuranosyladenine; abbreviations used in transporter acronyms: c, concentrative; e, equilibrative; s and i, sensitive and insensitive to inhibition by nitrobenzylmercaptopurine ribonucleoside (6-[(4-nitrobenzyl)thio]-9-β-d-ribofuranosylpurine), respectively; f, formycin B (nonmetabolized purine nucleoside); t, thymidine.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}