


Abstract
Cells selected for resistance to cisplatin are often cross-resistant to copper and vice versa, and the major copper influx transporter copper transport protein 1 (CTR1) has been shown to regulate the uptake of cisplatin, carboplatin, and oxaliplatin in yeast. To further define the role of hCTR1 in human tumor cells, the ovarian carcinoma cell line A2780 was molecularly engineered to increase expression of hCTR1 by a factor of 20-fold. Enhanced expression of hCTR1 in the A2780/hCTR1 cells was associated with a 6.5-fold increase in basal steady-state copper content and a 13.7-fold increase in initial copper influx, demonstrating that the exogenously expressed hCTR1 was functional in altering copper homeostasis. The A2780/hCTR1 cells accumulated 46% more platinum after a 1-h exposure to 2 μM cisplatin, and 55% more after a 24 h exposure, than the control A2780/empty vector cells. The initial uptake of cisplatin was 81% higher in the A2780/hCTR1 cells when measured at 5 min. Thus, increased expression of hCTR1 had a substantially larger effect on the cellular pharmacology of copper than cisplatin. Interestingly, the increased uptake of copper and cisplatin was accompanied by only a marginal increase in sensitivity to the cytotoxic effect of copper and cisplatin, and there was no increase in the extent of cisplatin-DNA adduct formation. Thus, although increased expression of hCTR1 mediates greater cellular accumulation of copper and cisplatin, hCTR1 delivers these compounds into intracellular compartments from which they do not have ready access to their key cytotoxic targets.
Although copper is an essential trace element necessary for the activity of many critical enzymes, free copper is dangerous to the cell because of its ability to undergo redox reactions that generate oxygen free radicals. The dual challenge of acquiring copper from the environment and protecting it from oxidation is achieved by a highly conserved system of copper transporters and chaperones that transport copper(I) into the cell and then guard it against oxidation while distributing it to copper-requiring enzymes (Linder and Hazegh-Azam, 1996). Current evidence indicates that copper(I) moves through the cell via direct protein-protein interactions between transporters and intracellular chaperones that ensure that copper is virtually never free in the cell (Pufahl et al., 1997; Rae et al., 1999).
The protein responsible for the transport of copper across the plasma membrane is human copper transporter 1 (hCTR1). The function of hCTR1 is essential for development, because deletion of both CTR1 alleles in the mouse, which shares 92% sequence identity with hCTR1 (Lee et al., 2001), causes embryonic death in utero (Zhou and Gitschier, 1997; Kuo et al., 2001). The extracellular region of hCTR1 possesses two metal-binding domains that play a role in scavenging copper under conditions of copper starvation. Under basal conditions, two methionines in the extracellular domain (40Met and 45Met) and two methionines in the second transmembrane region are necessary for copper transport (Eisses and Kaplan, 2002). Copper transport by hCTR1 is sensitive to temperature, copper concentration, extracellular pH, and extracellular potassium ion concentration (Lee et al., 2002). Like copper, the cellular uptake of cisplatin is also influenced by temperature, pH, and potassium ion concentration (Atema et al., 1993; Gately and Howell, 1993; Endo et al., 2000; Amtmann et al., 2001).
Although much is known about the conditions necessary for cisplatin uptake, the mechanism by which cisplatin enters and exits from tumor cells has remained poorly defined for many years. Early studies suggested that one component of cisplatin uptake is mediated by a transport mechanism or channel (Andrews et al., 1988; Andrews and Albright, 1991; Mann et al., 1991). Cells selected for acquired resistance to cisplatin commonly exhibit impaired uptake (Andrews and Howell, 1990; Gately and Howell, 1993). Recent studies from this laboratory have demonstrated that cells selected for resistance to cisplatin are cross-resistant to copper and vice versa and that parallel changes in the cellular pharmacokinetics of both cisplatin and copper are observed in cisplatin-resistant cells (Katano et al., 2002). Strong evidence implicating CTR1 as the major cisplatin influx transporter was provided by studies of yeast in which a knockout of yCTR1 was shown to markedly reduce the uptake of cisplatin and to render the cells resistant to its cytotoxic effects (Ishida et al., 2002; Lin et al., 2002). Embryo fibroblasts established from mice in which both CTR1 alleles were disabled also exhibited reduced cisplatin uptake and cell kill (Ishida et al., 2002). Human CTR1 differs substantially from its yeast and murine homologs in amino acid sequence and copper binding domains, and no information is available on whether hCTR1 can mediate the influx of cisplatin in human cells. We report here studies of the cellular pharmacology of cisplatin in human ovarian carcinoma cells molecularly engineered to express increased levels of hCTR1. In this system, the overexpression of hCTR1 lead to an increase in platinum accumulation and a decreased growth rate, yet it had limited effect on the sensitivity to cisplatin and to the amount of platinum binding to DNA.
Materials and Methods
Drugs and Reagents. Platinol-AQ containing cisplatin at a concentration of 3.3 mM was a gift from Bristol-Myers Squibb Co. (Princeton, NJ). It was stored in the dark at room temperature, and a 100 μM stock solution was created by dilution in 0.9% NaCl. Copper in the form of cupric sulfate was obtained from Fisher Scientific Co. (Tustin, CA). Protein concentration was measured using Bradford's reagent from Bio-Rad (Hercules, CA). The generation of the rabbit polyclonal antibodies against amino acids 1 through 67 of hCTR1 used for immunofluorescence is described elsewhere (Klomp et al., 2002). Fluorescein isothiocyanate-conjugated goat anti-rabbit antibody was obtained from Jackson Immunoresearch Laboratories Inc. (West Grove, PA). Hoechst 33342 dye for nuclear staining was purchased from Molecular Probes (Eugene, OR). The monoclonal mouse anti-metallothionein was obtained from U.S. Biologicals (Swampscott, MA). The polyclonal rabbit anti-hCTR1 antibody used for Western blotting was generated by immunizing rabbits with a peptide containing amino acids 2 through 22 of the N terminus of hCTR1 from Biocarta Inc. (San Diego, CA). Horseradish peroxidase-conjugated goat anti-rabbit secondary antibody was purchased from Amersham Biosciences Inc. (Piscataway, NJ). All other chemicals and reagents were obtained from Fisher Scientific.
Cell Lines and Vectors. The A2780 ovarian carcinoma cell line (Hamilton et al., 1985) was grown in RPMI 1640 medium containing 10% fetal bovine serum at 37°C in 5% CO2. A pcDNA3.1 vector containing full-length hCTR1 cDNA and expressing a geneticin resistance marker constructed as described previously (Moller et al., 2000) was generously provided by Dr. Lisbeth Birk Moller (John F. Kennedy Institute, Glostrup, Denmark). Cells were transfected with either hCTR1/pcDNA3.1 or empty vector (EV) using LipofectAMINE (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. Transfected cells were selected with 500 μg/ml geneticin. Surviving clones were combined to create a multiclonal generation.
Western Blot Analysis. Cells from an 80% confluent T75 flask were trypsinized and recovered by centrifugation at 4°C, after which they were frozen at -20°C for 1 h and then thawed on ice. The pellet was resuspended in 100 μl of buffer (250 mM sucrose, 10 mM Tris-HCl, pH 7.4, 1 μg/ml antipain, 1 μg/ml pepstatin, 1 μg/ml leupeptin, and 20 μg/ml phenylmethylsulfonyl fluoride) and homogenized using a dounce homogenizer for 1min. The suspension was centrifuged for 15 min at 500g at 4°C to remove nuclei and large particulate matter (Deutscher, 1990), and the protein concentration of the resulting supernatant was determined. Samples containing 100 μg of protein were boiled before electrophoresis in a 4 to 20% SDS-polyacrylamide gel electrophoresis gel and transfer to nitrocellulose membranes (Bio-Rad) via electrophoresis for 30 min at 200 V using a Transblot SD apparatus (Bio-Rad). Membranes were blocked in 5% milk in Tris-buffered saline for 1 h at room temperature. Blots were incubated at 4°C overnight with anti-hCTR1 antibody diluted 1:1000 or with anti-metallothionein diluted 1:1000 and mouse antitubulin (Sigma-Aldrich, St. Louis, MO) diluted 1:20,000 in 5% milk in Tris-buffered saline. Membranes were then washed three times with the same buffer containing 0.05% Tween 20 and incubated with horseradish peroxidase-conjugated anti-mouse and anti-rabbit antibodies for 1 h at room temperature. Before detection, membranes were again washed three times with Tris-buffered saline containing 0.05% Tween 20, and detection was performed using the enhanced chemiluminescence Western detection system from Amersham Biosciences according to manufacturer's instructions.
Fixation and Staining. Cells were grown in T75 flasks until 80% confluence and then were harvested by trypsinization and pelleted by centrifugation in 10% RPMI 1640 medium with 10% fetal bovine serum. The resulting pellets were resuspended in 5 ml of 10% RPMI 1640 medium with 10% fetal bovine serum, and 100 μl of cells were added to each well of a 24-well plate. Before the addition of cells, a 0.16-mm thick coverslip and 300 μl of 10% RPMI 1640 medium containing 10% fetal bovine serum and 200 μM bathocuprione disulphonic acid were placed in each well. Bathocuprione disulphonic acid was added to chelate any copper in the media, ensuring that copper could not affect cisplatin binding. Once cell growth on the coverslips reached 80% confluence at approximately 48 h after plating, the cells were exposed to 10% RPMI 1640 medium with 10% fetal bovine serum containing copper for 5 min. After the specified duration of exposure, the coverslips were placed at 4°C and washed three times with phosphate-buffered saline and then fixed in 3.7% formaldehyde in the same buffer at room temperature for 30 min. Subsequent staining and washings were performed as described previously (Klomp et al., 2002).
Measurement of 64Copper Accumulation. Copper uptake measurements were made using cells grown in medium lacking geneticin to 80% confluence in the 35-mm wells of a six-well plate. After the addition of prewarmed media containing 2 μM 64CuSO4, the plates were incubated at 37°C on 5% CO2 for 0 to 24 h. At the end of the incubation period, the plates were placed on ice and rinsed three times with 6 ml of ice-cold phosphate-buffered saline. Cell lysis buffer (0.1% Triton X-100 and 1% SDS in phosphate-buffered saline) in a volume of 500 μl was added to the wells, and the lysate was harvested by scraping the dish twice before it was transferred to tubes for γ counting on a Gamma 5500B counter (Beckman Coulter, Inc., Fullerton, CA). Six cultures were harvested for each time point. Lysates from a set of identical cultures not exposed to 64CuSO4 were used to measure protein concentration.
Drug-Sensitivity Assay. Six-well plates were seeded with 3000 cells per well, and 24 h later the medium was replaced with medium containing various concentrations of either cisplatin or copper. After 7 days of continuous exposure, cultures were trypsinized and stained with trypan blue. The live cell number per well was determined by counting cells that excluded trypan blue with a hemocytometer. Each drug concentration had six samples per cell line, and the experiment was repeated three times.
Real-Time PCR. Real-time PCR was performed on both the hCTR1 transfect and empty vector cells to verify the overexpression of hCTR1 in the transfected cells. The real-time PCR was performed as detailed previously (Katano et al., 2002). Briefly, 10 μg of total RNA, extracted with TRIzol reagent (Invitrogen), was treated with DNase and converted to cDNA using random hexamer primers with the SuperScript First-Strand Synthesis System (Invitrogen). An ABI Prism 7700 and Sequence Detection System software was used for real-time PCR and primer design, respectively (PerkinElmer Life and Analytical Sciences, Boston, MA). Triplicate PCR amplifications of 10 ng of the cDNA were performed using the Taqman Master Mix provided by PerkinElmer. Fold change in RNA abundance was calculated using the standard curve method for quantification (ABI Prism 7700 SDS User Bulletin 2 P/N 4303859 Revision A). The GenBank sequence number (U83460) was used for the hCTR1 primer design. The primers are as follows: forward, 5′-AGGACTCAAGATAGCCCGAGAGA-3′; reverse, 5′-CCTGGGACAGGCATGGAA-3′; and probe, 5′-CTGCGTAAGTCACAAGTCAGCATTCGCTACA-3′.
Platinum Accumulation Assays. Cells were plated in 145-cm2 dishes in media without any drugs. Once plates were 80% confluent, 2 μM cisplatin was added, and plates were incubated at 37°C in 5% CO2 for various periods of time. The plates were then placed on ice and rinsed three times with 3 ml of ice-cold phosphate-buffered saline. Cells were harvested by scraping, followed by centrifugation for 10 min at 3000 rpm and 4°C. The resulting pellet was dissolved in 70% nitric acid at 65°C for at least 2 h. Samples were diluted to 5% nitric acid with water containing 1 ppb indium and 0.1% Triton X-100. Platinum content was determined using inductively coupled plasmon mass spectroscopy (Element2; PerkinElmer) on an instrument available through the Analytical Facility at the Scripps Institute of Oceanography (University of California at San Diego, San Deigo, CA). Lysates from a set of identical cultures suspended in 0.1% Triton X-100 and 1% SDS in phosphate-buffered saline were used to measure protein concentration.
Measurement of Basal Copper Levels. Cells were grown in 145-cm2 dishes until 80% confluence, after which they were washed three times with ice-cold phosphate-buffered saline, harvested by scraping with a rubber policeman, and pelleted at 3000 rpm for 10 min at 4°C. The pellet was resuspended in 70% nitric acid at 65°C for a minimum of 2 h. After dilution of the nitric acid to 5%, copper content was assayed using an inductively coupled plasma optical emission spectroscopy apparatus (model 3000DV; PerkinElmer) available at the Analytical Facility at the Scripps Institute of Oceanography.
Statistics. Tests of significance used a two-sided paired Student's t test with the assumption of unequal variance; p values of <0.05 were considered significant. All p values are for the comparison of the values for the A2780/EV versus the A2780/hCTR1 cells.
Results
Characterization of Ovarian Carcinoma Cells Engineered to Overexpress hCTR1. The ovarian carcinoma cell line A2780 was transfected with either an empty vector (A2780/EV cells) or a vector containing the hCTR1 cDNA under the control of a CMV promoter (A2780/hCTR1 cells). The hCTR1 mRNA level was 50-fold higher in the A2780/hCTR1 than in the A2780/EV, cells as determined by RT-PCR (data not shown). Figure 1 shows that the level of the 28-kDa form of the hCTR1 protein was 20-fold higher in the A2780/hCTR1 cells, as determined by Western blot analysis on denaturing gels. To document that the exogenously expressed hCTR1 was correctly localized to membrane structures in the cell, the distribution of hCTR1 in the A2780/hCTR1 cells was compared with that of endogenous hCTR1 in the parental A2780 cells using immunoflourescent staining and deconvoluted confocal digital microscopy. As shown in Fig. 2, although the A2780/hCTR1 cells contained much more total hCTR1, the distribution of hCTR1 in the transfected cells mimicked the localization and distribution observed for endogenous hCTR1 in the parental cells, indicating that the exogenously expressed hCTR1 was correctly localized to both the plasma membrane and intracellular membranous structures. As also shown in Fig. 2, exposure of the A2870/hCTR1 cells to 100 μM copper for 5 min caused extensive redistribution of hCTR1, as has been reported previously (Petris et al., 2003; Guo et al., 2004).

hCTR1 expression in A2780/EV and A2780/hCTR1 cells. Western blot analysis of hCTR1 expression. hCTR1 is detected at 28 kDa, whereas tubulin is detected at 55 kDa.

Subcellular localization of hCTR1 in A2780/EV and A2780/hCTR1 cells. A, A2780 cells. B, A2780/hCTR1 cells. C, A2870/hCTR1 cells exposed to 100 μM copper for 5 min. hCTR1 was visualized using a rabbit anti-hCTR1 antibody and a goat anti-rabbit fluorescein isothiocyanate-conjugated secondary. Hoescht 33342 dye was used to label the nucleus. Each image is representative of three images taken from each of three independent experiments. Images are normalized to the autofluorescence of unstained A2780/EV cells and cells stained with only the secondary antibody.
Effect of hCTR1 Expression on the Cellular Pharmacokinetics of Copper. The amount of copper present in cells when they are grown in medium containing no added copper is a sensitive measure of the extent to which copper homeostasis has been perturbed. Figure 3A shows that the steady-state level of copper was 6.5-fold higher in the A2780/hCTR1 than in the A2780/EV cells (23.9 ± 6.9 versus 3.7 ± 1.18 ng copper/mg protein). This result suggests that the exogenous hCTR1 expressed from the transfected vector is functional and that it augments the effect of endogenous hCTR1 on the influx of copper. To document this effect further, the A2870/EV and A2780/hCTR1 cells were exposed to 2 μM 64CuSO4 for time periods varying from 0 to 24 h, washed thoroughly, and the cell-associated 64copper was quantified by γ counting. After a 5-min exposure to 64Cu, the copper accumulation in the A2780/hCTR1 cell line was 13.7-fold higher than in the A2870/EV cells. As shown in Fig. 3B, after 5 min, the A2780/hCTR1 cells had accumulated 7.4 ± 0.71 (S.E.M.) pmol Cu/mg protein, whereas the empty vector-transfected A2780/EV cells had accumulated only 0.54 ± 0.56 (S.E.M.) pmol Cu/mg protein. At all time point up to 24 h, the uptake in the A2780/hCTR1 cells exceeded that in the A2780/EV cells. By 24 h, at which time accumulation of 64Cu had reached steady state in the A2780/EV cells, the A2780/hCTR1 cells contained 17-fold more copper [40.8 ± 3.2 versus 2.37 ± 0.07 (S.E.M.) pmol Cu/mg protein). Thus, the exogenously expressed hCTR1 not only localized correctly to membraneous structures in the cell, it also was functional in modulating the cellular pharmacology of copper.

Effect of increased hCTR1 expression on steady-state copper content and 64Cu accumulation. A, picomoles of copper present in A2780/hCTR1 and A2780/EV cell lines under basal conditions. ▪, A2780/hCTR1 cells; □, A2780/EV cells. Each bar represents the mean of three independent experiments, each performed with sextuplet cultures. ⋆⋆⋆, p < 0.0003 for comparison of accumulation in A2780/EV versus A2780/hCTR1 cells. Vertical bars, S.E.M. B, accumulation after 1, 5, 60, and 1440 min of exposure to 64CuSO4; ▪, A2780/hCTR1 cells; □, A2780/EV cells. Each bar represents the mean of three independent experiments each performed with sextuplet cultures. ⋆⋆, p < 0.005; ⋆⋆⋆, p < 0.0005 for comparison of accumulation in A2780/EV versus A2780/hCTR1 cells. Vertical bars, S.E.M.
Effect of hCTR1 Expression on the Cellular Pharmacokinetics of Cisplatin. The effect of increased expression of hCTR1 on the uptake of cisplatin was determined by exposing the A2780/EV and A2780/hCTR1 cells to 2 μM cisplatin, a concentration attained in the plasma of patients receiving standard doses of the drug, and quantifying the total cellular platinum at various time points by inductively coupled plasmon mass spectroscopy. As shown in Fig. 4, after a 1-h exposure to 2 μM cisplatin, the A2780/hCTR1 cells had accumulated 165 ± 3 (S.E.M.) pg Pt/mg protein, whereas the A2780/EV cells had accumulated 113 ± 26 (S.E.M.) pg Pt/mg protein. Thus, at this time point, the hCTR1-overexpressing cells contained 46% more platinum. Figure 4 shows that after a 24-h exposure, the A2780/hCTR1 cells had accumulated 55% more platinum than had the A2780/EV cells. Even initial uptake was increased in the hCTR1-transfected cells. After a 5-min exposure to 2 μM cisplatin, the A2780/hCTR1 transfected line had accumulated 81% more platinum than the empty vector-transfected line (134 ± 8 versus 74 pg Pt/mg protein, respectively) (data not shown). Thus, the increased expression of hCTR1 in the A2780/hCTR1 cells resulted in increased accumulation of both copper and cisplatin; however, the magnitude of the effect was much greater for copper than for cisplatin. This suggests that cisplatin is not as good a substrate for hCTR1 as copper and that there are substantial differences in the ability of this transporter to mediate the influx of these two compounds.

Effect of increased hCTR1 expression on cisplatin accumulation. Accumulation was measured after 1 and 24 h of exposure to 2 μM cisplatin. ▪, A2780/hCTR1 cells; □, A2780/EV cells. Each bar represents the mean of three independent experiments each performed with triplicate cultures. ⋆, p < 0.03 for comparison of accumulation in A2780/EV versus A2780/hCTR1 cells. Vertical bars, S.E.M.
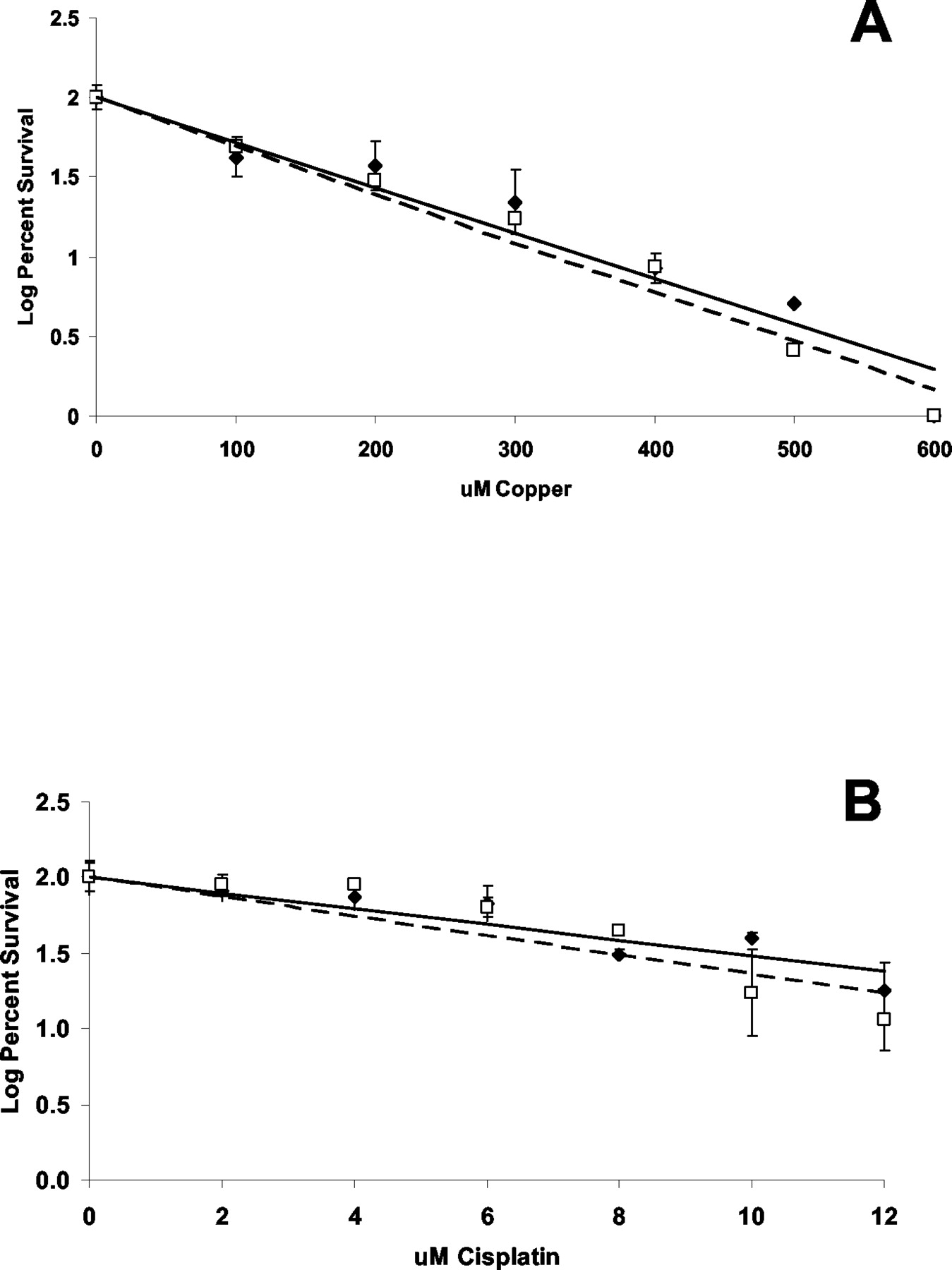
Effect of hCTR1 Expression on Sensitivity to the Cytotoxic Effects of Copper and Cisplatin. The cytotoxicity of both copper and cisplatin is a function of how much of the compound enters the cell. However, cisplatin toxicity is additionally proportional to the amount of drug entering the cell that actually forms adducts in DNA. From the observation that expression of exogenous hCTR1 increased the whole-cell uptake of copper and cisplatin, one might anticipate enhanced sensitivity to the cytotoxic effect of both agents, and because the magnitude of the increase in copper was substantially greater than cisplatin, one might expect a greater effect on sensitivity to copper than to cisplatin. Growth rate as a function of copper or cisplatin concentration was determined for each of the cell lines under conditions of continuous drug exposure. As shown in Fig. 5A, the IC50 for copper was only 6% lower in the A2780/hCTR1 cells (160.7 ± 5.7 μM) that for the A2780/EV cells (171.2 ± 12.0 μM) (S.E.M.). Likewise, as shown in Fig. 5B, the IC50 for cisplatin was only 1.8% lower for the A2870/hCTR1 cells (7.17 ± 0.23 μM) than for the A2780/EV cells (7.30 ± 0.053 μM) (S.E.M.). Thus, a large change in the level of expression of hCTR1, although it was associated with a clear increase in copper and cisplatin uptake, produced only a very small change in sensitivity to either compound. This suggests that much of the copper and cisplatin entering the A2780/hCTR1 cells is not reaching key targets in the cell. To examine this question further, the extent of cisplatin adduct formation in DNA was assessed by measuring the platinum content of DNA extracted from A2780/EV and A2780/hCTR1 cells after a 1-h exposure to 2 μM cisplatin. As shown in Fig. 6, despite the fact that forced expression of hCTR1 in the A2780/hCTR1 cells increased whole-cell cisplatin accumulation, there was no difference in the extent of DNA adduct formation. Thus, the hCTR1-mediated enhanced cisplatin uptake delivers this compound to one or more compartments in the cell from which it does not have ready access to its major cytotoxic target, and the same thing seems to be true for copper.

Effect of copper and cisplatin on cell growth. A, log of percent surviving cells after 7 days of continuous exposure to copper. B, percentage of survival after 7 days of continuous exposure to cisplatin. Solid line, A2780/EV (♦) cells; broken line, A2780/hCTR1 (□) cells. Each point represents the mean of three independent experiments each performed with six cultures. Vertical bars, S.E.M.

Accumulation of platinum in DNA after1 h of exposure to 2, 5, 10, and 50 μM cisplatin. ▪, A2780/hCTR1; □, A2780/EV. Each bar represents the mean of three independent experiments each performed with triplicate cultures. Vertical bars, S.E.M.
Discussion
The results of these studies demonstrate that the level of expression of the copper influx transporter hCTR1 influences the cellular accumulation of cisplatin as well as that of copper in human ovarian carcinoma cells but that the enhanced cellular uptake of these compounds is not accompanied by a significant change in sensitivity to their cytotoxic effect or the delivery of cisplatin to DNA. These observations introduce the novel concept that, although hCTR1 enhances cisplatin uptake into the cell, it delivers it to cellular compartments from which it does not have ready access to key cytotoxic targets. This idea is supported by previous studies showing that exogenously expressed hCTR1 relocalizes from the plasma membrane to vesicles upon copper exposure (Guo et al., 2004), and that exposure to cisplatin produces a similar effect on both endogenous and exogenously expressed hCTR1 (Holzer et al., 2004).
The A2780/hCTR1 cells expressed a 20-fold higher level of hCTR1, as detected by Western blot analysis, and the exogenously expressed hCTR1 correctly localized to membrane structures, as demonstrated by high-resolution immunofluorescent analysis of its subcellular distribution in the A2780/EV and A2780/hCTR1 cells. The functionality of the exogenous hCTR1 was demonstrated by the observation that it relocalized in response to copper exposure and that its expression in the A2780/hCTR1 cells was accompanied by substantial changes in the cellular pharmacology of copper. The basal steady-state level of copper in the A2780/hCTR1 cells was increased by 6.5-fold, and these cells exhibited a marked increase in the uptake rate for 64Cu. Thus, exogenous hCTR1 was functional in A2780 cells and augmented their ability to internalize the small amounts of copper found in standard tissue culture medium and the rate of copper accumulation.
Our prior studies of haploid Saccharomyces cerevisiae demonstrated that the CTR1-mediated uptake of copper was much more rapid than that of cisplatin, suggesting that copper is a better substrate for this import pathway than cisplatin (Lin et al., 2002). Given the high selectivity of hCTR1, as exemplified by its ability to distinguish even between copper(I) and copper(II), this is not surprising. However, disruption of the yCTR1 gene produced marked reductions in the uptake of both compounds, suggesting that although cisplatin may not be a very good substrate, in yeast this transporter accounts for the majority of cisplatin influx. The results of the current study suggest that copper is also a better substrate for hCTR1 than copper in human cells, because the effect of increasing hCTR1 expression was substantially greater for copper than for cisplatin. Nevertheless, hCTR1 clearly increased the cellular uptake of cisplatin as well as copper, and the magnitude of this effect is well within the range in which, if the intracellular drug had ready access to DNA, it would be expected to produce increased cell kill (Katano et al., 2002).
One explanation for the larger relative effect of increased hCTR1 expression on copper than cisplatin may lie in their relative affinity for the metal binding regions of the protein located in the extracellular domain of the protein. hCTR1 is believed to bind two molecules of copper at a time (Zhou and Gitschier, 1997) via MXXXM motifs in the N-terminal domain (Eisses and Kaplan, 2002). Assuming that cisplatin binds to these same motifs, it is likely that the configuration of its binding is different from that of copper. It is also possible that cisplatin and copper are binding at different regions within the N terminal domain of hCTR1, because the beginning of the N terminus is very rich in histidine and methionine. Another plausible explanation for the larger relative effect of increased hCTR1 expression on copper than cisplatin may be that in human cells, other influx transporters account for a larger fraction of total cisplatin influx than in yeast.
Increased hCTR1-mediated copper and cisplatin uptake was not accompanied by increased cytotoxicity, and in the case of cisplatin, it was not accompanied by increased DNA adduct formation. This discrepancy suggests that much of the copper and cisplatin conducted into the cell must be sequestered away from the target through which it triggers cytotoxicity. Petris et al. (2002) reported that hCTR1 is endocytosed when extracellular copper levels are increased, raising the possibility that some or all of the copper and cisplatin brought into the cell via hCTR1 occurs through extracellular binding followed by endocytosis and delivery to a lysosomal or other vesicular compartments. A corollary to this hypothesis is that there must be mechanisms that facilitate the release of copper and cisplatin from the vesicular compartment, and that these, rather than the hCTR1-mediated part of the import pathway, may be rate-limiting. Under this circumstance, the increased expression of the hCTR1 may result in substantial enhanced total cellular uptake but little additional delivery of copper or cisplatin to the nucleus or other key targets. The increase capacity of only one step of the influx mechanism of the copper transport pathway may merely create a backup that is not alleviated unless other downstream steps in the influx pathway are comparably increased. It may be possible to explore this issue further using down-regulation rather than up-regulation of hCTR1 expression.
Now that hCTR1 has been identified as mediating the influx of cisplatin, a central question is whether the reduced uptake of cisplatin observed in most cell lines selected for resistance to this drug is caused by reduced expression or impaired function of hCTR1, as well as whether variations in the expression of hCTR1 in human tumors accounts for the heterogeneity of response to cisplatin-containing chemotherapy regimens. Such studies will require specific and robust reagents with which to quantify hCTR1 expression and access to tumor samples from patients whose clinical response to cisplatin-based therapy is known or can be determined.
Acknowledgments
We thank Dr. Lisbeth Birk Moller for providing the hCTR1 vector and Dr. Kevin Walda and the Analytical Facility at Scripps Institute for Oceanography for technical and theoretical assistance in the cisplatin uptake studies. We also thank C. Zacharia for project management assistance.
Footnotes
-
Supported in part by Grant CA95298 from the National Institutes of Health and grant DAMD17-03-1-0158 from the Department of Defense. This work was conducted in part by the Clayton Foundation for Research–California Division. R. Safaei and S.B.H. are Clayton Foundation investigators.
-
This study was presented in part at the 2003 meeting of the American Association of Cancer Research.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.001198.
-
ABBREVIATIONS: hCTR1, human copper transport protein 1; EV, empty vector; PCR, polymerase chain reaction; prefixes: h, human; y, yeast.
- Received April 6, 2004.
- Accepted June 30, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}