


Abstract
Estrogen receptor regulation of nitric oxide production by vascular endothelium may involve rapid, membrane-initiated signaling pathways in addition to classic genomic mechanisms. In this study, we demonstrate using intact cerebral blood vessels that 17β-estradiol rapidly activates endothelial nitric-oxide synthase (eNOS) via a phosphoinositide-3 (PI-3) kinase-dependent pathway. The effect is mediated by estrogen receptors (ERs), consistent with colocalization of ERα and caveolin-1 immunoreactivity at the plasma membrane of endothelial cells lining cerebral arteries. Treatment with 10 nM 17β-estradiol for 30 min increased NO production, as measured by total nitrite assay, in cerebral vessels isolated from ovariectomized rats. This effect was significantly decreased by membrane cholesterol depletion with β-methyl-cyclodextrin, the ER antagonist ICI 182,780 [fulvestrant (Faslodex)], and two inhibitors of PI-3 kinase: wortmannin and LY294002 [2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride]. In parallel with NO production, 17β-estradiol treatment rapidly increased phosphorylation of both eNOS (p-eNOS) and Akt (p-Akt). PI-3 kinase inhibitors also blocked the latter effects; together, these data are consistent with ER activation of the PI-3 kinase-p-Akt-p-eNOS pathway. ERα protein (66 and 50 kDa) coimmunoprecipitated with eNOS as well as with the p85α regulatory subunit of PI-3 kinase, further implicating ERα in kinase activation of eNOS. Little is known regarding the effects of estrogen on cellular kinase pathways in vivo; therefore, we compared cerebral blood vessels isolated from ovariectomized rats that were either untreated or given estrogen replacement for 4 weeks. Long-term estrogen exposure increased levels of cerebrovascular p-Akt and p-eNOS as well as basal NO production. Thus, in addition to the rapid activation of PI-3 kinase, p-Akt, and p-eNOS, estrogen signaling via nontranscriptional, kinase mechanisms has long-term consequences for vascular function.
Up-regulation of endothelial nitric oxide (NO) production plays an important role in the vasoprotective effects of estrogen (Nilsson et al., 2001). The cerebral vasculature is a significant target tissue for this hormone, and in vivo exposure to estrogen increases NO-mediated vasodilation in rodent cerebral arteries (Geary et al., 1998, 2001). This effect can be explained in part by an increase in the expression of endothelial NO synthase (eNOS) in cerebral blood vessels (McNeill et al., 1999, 2002; Stirone et al., 2003a). Estrogen receptor α (ERα) is present in the endothelium of cerebral arteries (Stirone et al., 2003b), and studies with estrogen receptor antagonists (McNeill et al., 2002) and ERα knockout mice (Geary et al., 2001) suggest that estrogen acts on ERα to modulate levels of cerebrovascular eNOS protein. Long-term estrogen treatment also increases the level of eNOS mRNA in cerebral vessels (Stirone et al., 2003a). Together, these effects are consistent with the classic genomic mechanism of estrogen receptor action that involves transcription of target genes in the nucleus.
However, there is a growing consensus that estrogen receptors also mediate nongenomic effects by activating cell signaling pathways outside the nucleus (Haynes et al., 2002; Ho and Liao, 2002). Much of the current knowledge is based on studies using a variety of transfected and tissue-derived cells in culture, including endothelial cells (Haynes et al., 2000, 2002; Chambliss and Shaul, 2002; Ho and Liao, 2002). In particular, estrogen was found to stimulate NO production within minutes of treatment (Haynes et al., 2000; Hisamoto et al., 2001). Because multiple signal transduction pathways converge to regulate eNOS activity by phosphorylation (Dimmeler et al., 1999; Fulton et al., 1999; Gallis et al., 1999; Shaul, 2002), it was proposed that estrogen activates one or more of these kinase cascades. A number of cell culture studies suggest estrogen rapidly increases NO release by stimulating the phosphoinositide-3 (PI-3) kinase-Akt kinase (protein kinase B) pathway (Simoncini et al., 2000, 2003; Hisamoto et al., 2001; Haynes et al., 2002). Activation of Src kinase has been suggested to play a role in mediating the effects of estrogen on this pathway (Haynes et al., 2003). In contrast, in ovine uterine endothelial cells, estrogen rapidly affects NO production without stimulating Akt phosphorylation (Chen et al., 2004). Other studies with cultured endothelial cells demonstrate that estrogen activation of mitogen-activated protein kinase cascades, in particular extracellular signal-regulated protein kinase 1/2, modulates eNOS activity (Chen et al., 1999, 2004). At this point, it is not known whether culture conditions or cell origins, e.g., species and/or vascular bed, contribute to discrepancies in the apparent mechanism of rapid estrogen-stimulated increases in NO production.
It is hypothesized that receptors associated with the plasma membrane are responsible for rapid signaling in response to estrogen (Chambliss et al., 2000; Haynes et al., 2002; Ho and Liao, 2002; Levin, 2002; Li et al., 2003). The exact nature of these receptors is still controversial, but data suggest that the traditional nuclear receptors ERα (Chen et al., 1999; Chambliss et al., 2000) and ERβ (Chambliss et al., 2002; Levin, 2002) or truncated splice variants of ERα (Figtree et al., 2003; Li et al., 2003) may associate with plasma membranes and, in particular, endothelial caveolae (Kim et al., 1999; Chambliss et al., 2000, 2002). 17β-Estradiol has been shown to activate eNOS in caveolae fractions isolated from immortalized ovine pulmonary artery endothelial cells (Chambliss et al., 2000), suggesting that all the necessary signaling components for rapid estrogen action are associated with membrane caveolae.
The recent data on estrogen membrane receptors and rapid signal transduction are compelling; however, little is known about the presence and significance of these phenomena in intact vascular tissue. In contrast to cells grown in culture, endothelium under normal physiological conditions is relatively quiescent and subjected to a variety of mechanical and chemical stimuli that differ among vascular beds. Thus, the goal of the current study was to determine whether 17β-estradiol activates rapid NO release from native blood vessels in an important vascular target: the cerebral circulation. The presence of membrane receptors and the role of kinase pathways also were addressed using intact vessels. Moreover, because vascular consequences of nongenomic mechanisms have not been explored in vivo, we determined the long-term impact of exposure to estrogen at physiologically relevant levels on Akt and eNOS activation in rat cerebral blood vessels.
Materials and Methods
In Vivo Treatments. All animal procedures were approved by the University of California Irvine Institutional Animal Care and Use Committee. Three groups of three-month-old Fischer-344 female rats (Harlan, Indianapolis, IN) were used: unoperated (intact female; INTF), ovariectomized (OVX), or ovariectomized and treated with 17β-estradiol (OE) as described previously (Geary et al., 1998; Stirone et al., 2003b). Rats were anesthetized with 46 mg/kg ketamine and 4.6 mg/kg xylazine (i.p.) for all surgical procedures. For OE animals, hormone treatment was started at the time of ovariectomy with the use of 17β-estradiol-filled Silastic tubing (1.57-mm inner diameter × 3.18-mm outer diameter; 10 mm in length) implanted subcutaneously (dorsally at the neck). Implants were left in place for 4 weeks; animals were then anesthetized by CO2 and killed by decapitation. We have previously demonstrated using this procedure that serum estrogen levels in OE animals are within the physiological range (Geary et al., 1998; McNeill et al., 2002). Brains were removed, and the blood vessels were isolated. Body weights were 183 ± 1 g for OVX and 164 ± 1 g for OE (P ≤ 0.05). Uterine weights were 34 ± 2 mg for OVX and 125 ± 4 mg for OE (P ≤ 0.05).
Cerebral Vessel Isolation. Blood vessels were isolated from whole brain as described previously (McNeill et al., 1999; Stirone et al., 2003b). In brief, four brains from each animal group were pooled, gently homogenized in phosphate-buffered saline (PBS), and centrifuged at 720g for 5 min at 4°C. The pellet was resuspended in PBS and layered over 16% dextran (molecular mass = 35–45 kDa; Sigma-Aldrich, St. Louis, MO), followed by centrifugation at 4500g for 20 min at 4°C. The blood vessel pellet was resuspended in ice-cold PBS and washed over a 50-μm nylon mesh. This preparation contains both pial and intraparenchymal vessels that, when examined using light microscopy, are a mixture of arteries, arterioles, capillaries, veins, and venules. Vessels were either used immediately for functional experiments or homogenized in ice-cold lysis buffer and stored at -80°C for later use.
Assay for NO Production. Freshly isolated cerebral blood vessels were pre-equilibrated at 37°C in PBS with 95% O2/5% CO2 for 30 min before initiation of the assay. Blood vessels were then incubated for 30 min at 37°C in 100 μl of PBS containing 10 nM 17β-estradiol (encapsulated in 2-hydroxy-propyl-β-cyclodextrin; Sigma-Aldrich) or an equivalent concentration of 2-hydroxy-propyl-β-cyclodextrin alone (vehicle control; Sigma-Aldrich). In some cases, vessels were pretreated with a much higher concentration of β-methyl-cyclodextrin (5 mM; 30 min; Sigma-Aldrich) to deplete membrane cholesterol (Kaiser et al., 2002). In other experiments, the estrogen receptor antagonist ICI 182,780 (10 μM; Tocris Cookson Inc., Ellisville, MO), PI-3 kinase inhibitors wortmannin or LY294002 (10 μM; Calbiochem, San Diego, CA), or the src-kinase inhibitor PP2 (10 μM; Calbiochem) was included during the 30-min pre-equilibration period and maintained during 17β-estradiol treatment. After incubation, samples were centrifuged at 5000g for 1 min to pellet vessels, and the supernatant was removed for nitrite measurement. Total protein content of the pelleted vessels was determined using a bicinchoninic acid protein assay kit (Pierce, Rockford, IL). Before nitrite measurement, supernatants were centrifuged at 12,000g for 2 min to remove any remaining particulate matter. Total nitrite levels were measured using a Nitric Oxide Quantitation Kit (Active Motif Inc., Carlsbad, CA) per the manufacturer's protocol. Total nitrite values in each sample were normalized to protein content, and this ratio was expressed as -fold difference versus vehicle control.
Immunoblot Analysis. Blood vessels were glass-homogenized at 4°C in lysis buffer (50 mM β-glycerophosphate, 100 μM NaVO3, 2 mM MgCl2, 1 mM EGTA, 0.5% Triton X-100, 1 mM dl-dithiothreitol, 20 μM pepstatin, 20 μM leupeptin, 0.1 U/ml aprotinin, and 1 mM phenylmethylsulfonyl fluoride) and incubated on ice for 20 min. Samples were then centrifuged at 4500g for 10 min at 4°C, the supernatant was collected, and protein concentrations were determined. Lysates were used immediately or stored at -80°C.
In all immunoblot experiments, equal amounts of protein [50 μgin 1× SDS sample buffer (Invitrogen, Carlsbad, CA), boiled for 4 min] were loaded in each lane of an 8% Tris-glycine gel (Novex, San Diego, CA) and separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were then transferred to nitrocellulose membranes (Amersham Biosciences Inc., Piscataway, NJ), incubated in blocking buffer (0.01 M PBS, 0.1% Tween 20, and 6.5% nonfat dry milk), and treated with primary antibodies [p-Ser-473-Akt, HC-20 (ERα), p-Ser-1177-eNOS, p85α, and cav-1; Santa Cruz Biotechnology, Inc., Santa Cruz, CA] at dilutions from 1:500 to 1:200. Blots were then incubated in the appropriate secondary antibody (goat anti-rabbit IgG-HRP, donkey anti-goat IgG-HRP, or goat anti-mouse IgG-HRP; Santa Cruz Biotechnology, Inc.) at a 1:10,000 dilution, and the bands were visualized using an enhanced chemiluminescence reagent (ECL; Amersham Biosciences Inc.) and Hyperfilm (Amersham Biosciences Inc.). UN-SCAN-IT software (Silk Scientific Inc., Orem, UT) was used for densitometric analysis of immunoreactive bands. In all applicable Western blot experiments, α-actin protein levels were determined to verify equal protein loading of the gel.
Immunoprecipitation. Cerebral vessel lysates from intact or ovariectomized females with and without 30 min estrogen treatment were centrifuged at 12,000g for 10 min to reduce particulate matter. Lysate equivalent to 100 μg of total protein and 5 μg of antibody were added to a total volume of 500 μl of lysis buffer and mixed end-over-end for 1 h at 4°C. Fifty microliters of Protein G (50% slurry) was added to the sample and then mixed end-over-end for 1 h at 4°C. Samples were centrifuged at 12,000g for 30 s, and the pellet was washed four times with 1 ml of lysis buffer, with a 30-s 12,000g centrifugation between each wash. The final pellet was suspended in 30 μlof2× SDS sample buffer, boiled for 3 min, and then centrifuged at 12,000g for 30 s to remove the supernatant. The entire 30-μl supernatant was loaded onto an 8% Tris-glycine polyacrylamide gel and analyzed by SDS-PAGE and immunoblotting as described above. As a negative control, samples of each lysate also were incubated with normal IgG antibodies, followed by the immunoprecipitation protocol and immunoblotting.
Confocal Microscopy. Cerebral blood vessels were dissected from the surface of the brain, cut into small segments, fixed in 3% formaldehyde for 30 min, and permeabilized using 0.1% Triton X-100 for 5 min. Vessels were then incubated for 30 min in 1% bovine serum albumin/PBS, followed by overnight incubation at 4°C and a 1:50 dilution in primary antibodies: rabbit anti-ERα H-184 (Santa Cruz Biotechnology, Inc.) and mouse anti-caveolin-1 (Zymed Laboratories, South San Francisco, CA) or mouse-anti eNOS (Transduction Laboratories, Lexington, KY) and rabbit-anti caveolin-1 (Santa Cruz Biotechnology, Inc.). Vessels were then washed for 30 min in PBS and incubated overnight at 4°C with the secondary antibodies at 10 μg/ml (goat anti-rabbit Oregon Green 488 and goat anti-mouse Texas Red, Molecular Probes), followed by a final wash for 30 min in PBS. Vessels were laid on slides and covered with mounting medium containing DAPI (VECTASHIELD + DAPI; Vector Laboratories, Burlingame, CA), and coverslips were applied. Images were obtained using a Bio-Rad model 1024 laser scanning confocal microscope equipped with standard and UV lasers (Bio-Rad, Hercules, CA).
Statistics. All data values are given as mean ± S.E.M. Statistical differences in immunoblot band densities and total nitrite assays were determined by one-way analysis of variance with repeated measures followed by Dunnett's multiple comparison test (GraphPad Prism 2.0 software; GraphPad Software Inc., San Diego, CA) and displayed as -fold difference relative to OVX or vehicle control. Where appropriate, statistical differences were determined by Student's t test. In all cases, statistical significance was set at P ≤ 0.05.
Results
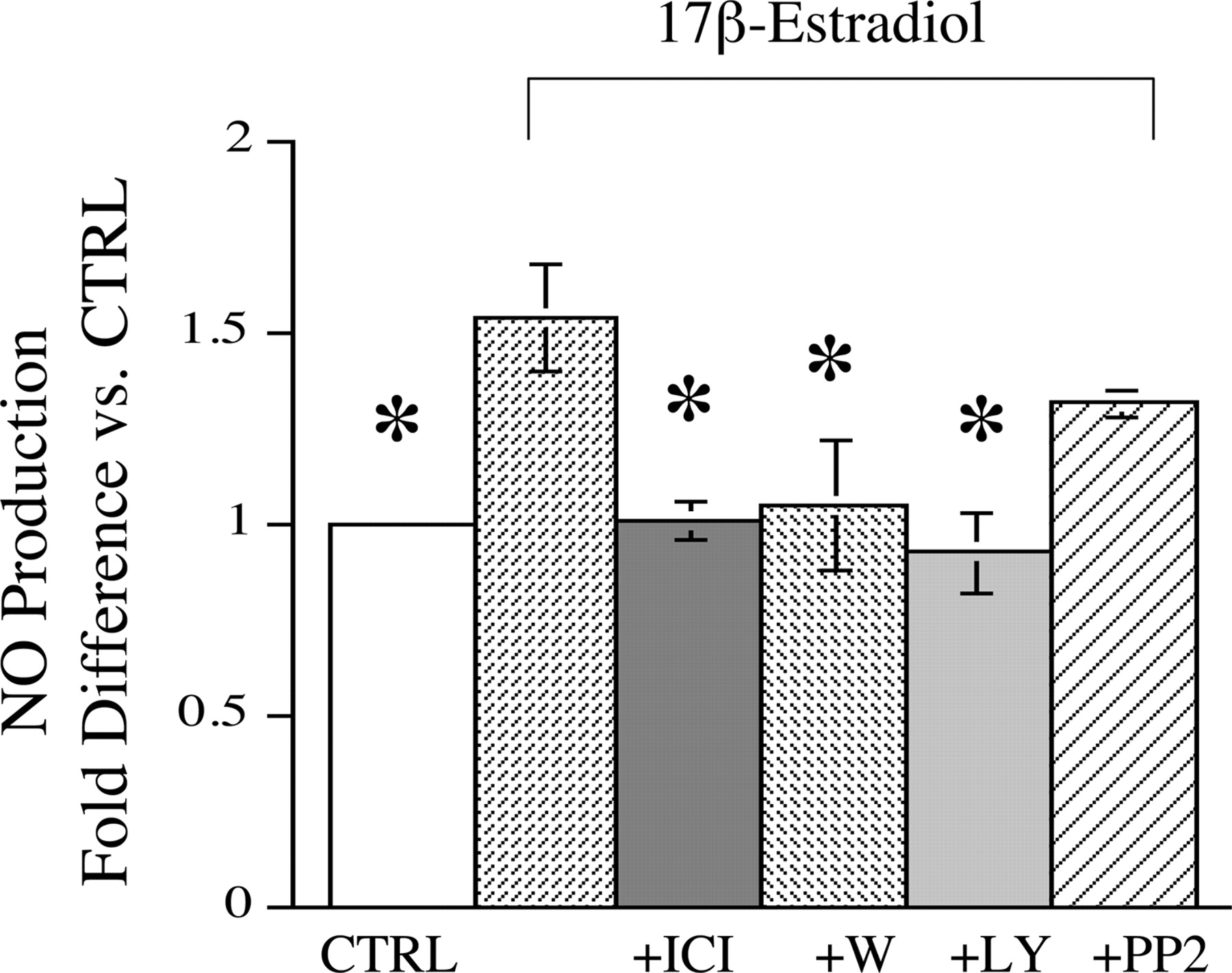
Estrogen Mediates Rapid Increases in NO Production. Cerebral blood vessels isolated from OVX animals were incubated with 10 nM 17β-estradiol for 30 min at 37°C. NO levels in the media were measured by a total nitrite assay. Estrogen caused a significant increase in cerebrovascular NO production versus vehicle-treated control (Fig. 1). Preliminary experiments indicated that estrogen increased cerebrovascular NO production within 15 min of exposure; however, because of limitations of the sensitivity of the nitrite assay, the earliest time point at which data could be consistently reproduced was 30 min. For this reason, we used the 30-min time point for further studies. To test whether the estrogen-mediated increase in NO production was mediated by estrogen receptors, the estrogen receptor inhibitor ICI 182,780 was used. ICI 182,780 (10 μM) fully inhibited estrogen-mediated increases in NO production (Fig. 1). To test whether the effect of estrogen on NO production involved the PI-3 kinase pathway, two inhibitors of PI-3 kinase, wortmannin and LY294002 (10 μM), were used. As shown in Fig. 1, both inhibitors blocked the ability of estrogen to increase NO production.

Effect of short-term estrogen treatment on NO production. Isolated cerebral blood vessels from OVX females were incubated in PBS at 37°C for 30 min in the presence of 10 nM 17β-estradiol or vehicle (CTRL). As indicated, antagonists for estrogen receptors (ICI 182,780), PI-3 kinase (wortmannin and LY294002), or src kinase (PP2) were added (each at a concentration of 10 μM) in the presence of 17β-estradiol. Nitric oxide production was determined by total nitrite assay and expressed as a ratio of total nitrite levels to vessel protein concentration. Values are normalized to vehicle control [n = 9 for estradiol (E2); n = 4 for E2 + ICI; n = 4 for E2 + W; n = 4 for E2 + LY; n = 5 for E2 + PP2]; *P ≤ 0.05 versus E2 alone.
A recent study suggested that src kinase may play an intermediary role between estrogen receptor activation and PI-3 kinase activation (Haynes et al., 2003). Therefore, we tested this hypothesis with the src kinase inhibitor PP2; however, PP2 failed to significantly inhibit estrogen-mediated increases in NO (Fig. 1). To test whether inducible NO synthase (iNOS) was contributing to NO production, aminoguanidine, an iNOS inhibitor, was added in the presence of estrogen. Aminoguanidine (10 μM) failed to significantly inhibit estrogen-mediated increases in NO production (aminoguanidine + estrogen treatment resulted in a 49 ± 5% increase in NO production versus control, compared with a 54 ± 7% increase for 30 min of estrogen treatment alone, n = 3). In addition, no iNOS was detected by immunoblot analysis in cerebral vessels before or after incubation.
Short-Term Estrogen Exposure Increases Ser-473-Phosphorylated Akt. To determine whether estrogen activates the PI-3 kinase/Akt pathway, Western blots of lysates from OVX cerebral vessels exposed to 30 min of estrogen treatment (10 nM) were probed for p-ser-473-Akt. Immunoblot analysis demonstrated that 30 min of estrogen treatment led to significant increases in p-Akt with no change in total Akt levels (Fig. 2). Furthermore, to demonstrate that the increase in phosphorylation was mediated by estrogen receptors acting through the PI-3 kinase pathway, an estrogen receptor blocker, ICI 182,780, and a PI-3 kinase inhibitor, LY294002, were used. Each inhibitor prevented the effect of estrogen on p-Akt levels (Fig. 2, A and B). Another PI-3 kinase inhibitor, wortmannin, also blocked the estrogen-induced increase in p-Akt levels (91 ± 3% decrease versus estrogen treatment alone, n = 4). Although a physiologically relevant concentration of estrogen was used throughout this study (10 nM), we also verified that concentrations of estrogen from 0.1 nM to 100 μM caused a concentration-dependent increase in p-Akt levels at the 30-min time point (n = 3, data not shown).

Effect of estrogen treatment on levels of ser-473-phosphorylated Akt. Representative Western blots for p-Akt and Akt (A) or p-Akt and α -actin (B) are shown for vessels from OVX females incubated for 30 min with vehicle (CTRL), estrogen (E2, 10 nM), or estrogen plus the antagonists (A) ICI 182,780 (10 μM) or (B) LY294002 (10 μM). Mean density values are presented as -fold difference compared with control (n = 4). *P ≤ 0.05 versus CTRL.
Short-Term Estrogen Exposure Increases Ser-1177-Phosphorylated eNOS. Western blot analysis was used to determine whether estrogen also increased phosphorylation of eNOS at serine-1177. Short-term estrogen exposure in vitro (30 min) led to significant increases in p-eNOS levels in isolated cerebral blood vessels with no change in total eNOS protein (Fig. 3). To determine whether the increase in p-eNOS required PI-3 kinase activation, vessels were pretreated with LY294002 before estrogen exposure. As demonstrated in Fig. 3, LY294002 treatment inhibited the estrogen-mediated increase in p-eNOS levels. Because p-eNOS was difficult to detect by Western blotting of cerebral vessel lysates, a second approach was also used. Total eNOS was first immunoprecipitated from vessels with and without 30 min of estrogen exposure. The samples were then analyzed by SDS-PAGE and probed for phosphorylated eNOS. Immunoprecipitation of eNOS from vessels treated with estrogen also demonstrated significant increases in the phosphorylated form of eNOS (47 ± 4% increase versus OVX controls, n = 4).

Effect of short-term estrogen treatment on phosphorylated eNOS. Cerebral blood vessels from OVX females were treated for 30 min with vehicle (CTRL), estrogen, or estrogen plus LY294002 and then probed by Western blot analysis for ser-1177-phosphorylated eNOS and total eNOS. Mean density values are presented as -fold difference versus control (n = 6). *P ≤ 0.05 versus CTRL.
ERα Coimmunoprecipitates with p85α and eNOS. To further explore the mechanism by which estrogen rapidly mediates increases in NO production, the interaction of ERα with proteins involved in eNOS activation was explored through immunoprecipitation experiments. As shown in Fig. 4A, immunoprecipitation of ERα reveals an association with the p85α regulatory subunit of PI-3 kinase as well as eNOS in vessel lysates from intact female rats. Immunoprecipitation of eNOS confirms the association of this protein with at least two forms of ERα, one at 66 and the other at 50 kDa (Stirone et al., 2003b). Likewise, immunoprecipitation of ERα from vessels of OVX females also indicated an association of the receptor with eNOS and p85α (data not shown). Estrogen treatment in vitro (10 nM, 30 min) significantly increased the amount of eNOS protein associated with ERα (Fig. 4B), but under these conditions we did not detect any change in the levels of ERα associated with p85α (data not shown).

ERα coimmunoprecipitates with the p85α subunit of PI-3 kinase and with eNOS. A, lysate of intact female cerebral blood vessels used. From the top, immunoprecipitation of ERα reveals coimmunoprecipitation with both p85α and eNOS as determined by Western blot. Immunoprecipitation of eNOS shows coimmunoprecipitation of ERα. B, vessel lysates from OVX females with and without 30 min of estrogen exposure compared. Mean data are shown for immunoprecipitation of ERα, followed by Western blot for eNOS (n = 5).
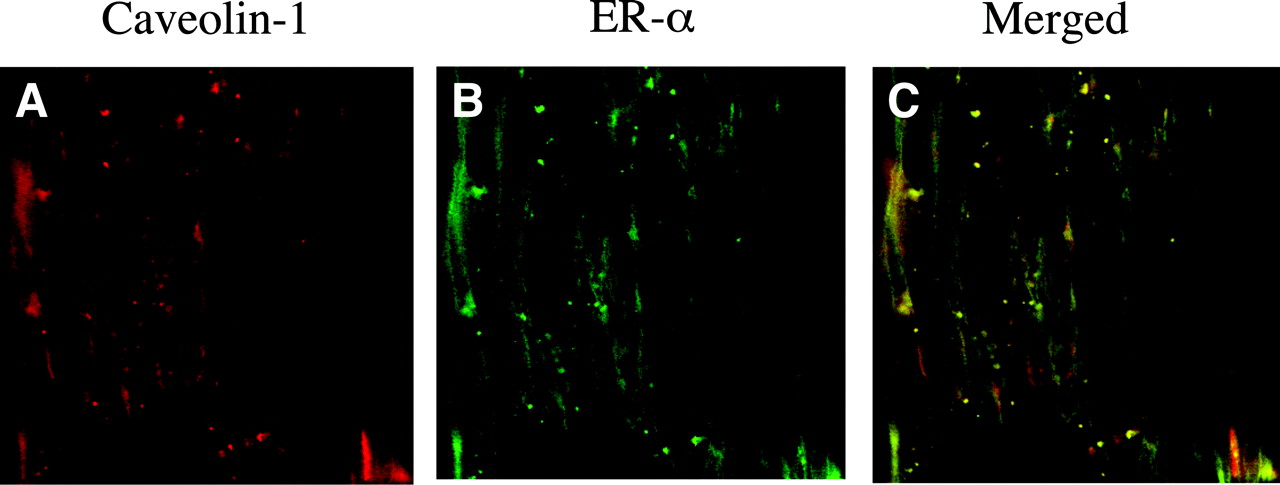
ERα Is Colocalized with Caveolin-1 in Cerebrovascular Endothelium. We previously visualized ERα immunoreactivity in the endothelium of cerebral blood vessels (Stirone et al., 2003b). In the current study, we addressed the possibility that ERα may be localized at the endothelial cell membrane by using immunohistochemistry and confocal microscopy at a higher magnification (100×). Antibodies against ERα and a marker for caveolae, caveolin-1, were used to stain rat cerebral arteries dissected off the surface of the brain. Figure 5 shows that both ERα (Fig. 5B) and caveolin-1 (Fig. 5A) are present in endothelial cells. The merged image reveals colocalization of these two proteins at the membrane, which can be seen outlining the individual endothelial cells, oriented in the direction of blood flow (Fig. 5C).

Colocalization of caveolin-1 and ERα in the endothelium of a cerebral artery. Laser scanning confocal microscopy was used to image a small segment of a pial artery isolated from intact female rat brain. The artery was dual-stained with an N-terminal ERα antibody (H-184, green) and anti-caveolin-1 antibody (red). The merged image shows areas of colocalization (orange). Images represent a single focal plane through the endothelial cell layer as identified by orientation of cell nuclei stained with DAPI.
Caveolar Disruption by β-Methyl-cyclodextrin Inhibits Short-Term Estrogen-Mediated NO Production but Not Akt Phosphorylation. To test the functional significance of ERα localized to membrane caveolae of cerebrovascular endothelial cells, isolated cerebral arteries were pretreated for 30 min with 5 mM β-methyl-cyclodextrin (β-mCD), a concentration and form of cyclodextrin previously shown to significantly deplete cholesterol and disrupt the structure of caveolae from aortic rings within 30 min (Kaiser et al., 2002). As shown in Fig. 6A, 30 min of β-mCD pretreatment significantly inhibited the effect of estrogen to rapidly increase vessel NO production. However, this regimen of β-mCD pretreatment failed to inhibit the effect of estrogen on cerebrovascular Akt phosphorylation (Fig. 6B).

Effect of disruption of caveolae on estrogen-stimulated NO production and Akt phosphorylation. Cerebral vessels from OVX females were pretreated with or without 5 mM β-mCD to remove membrane cholesterol and disrupt caveolae. A, NO production as measured by total nitrite assay from vessels incubated in vehicle (CTRL) or 10 nM estrogen (E2) for 30 min (n = 4). B, Western blot for p-Akt and α-actin (loading control) from vessels exposed to 10 nM estrogen or vehicle for 30 min. Protein levels are expressed as -fold difference from vehicle control (n = 4).
In Vivo Estrogen Treatment Increases NO Production. To determine whether long-term treatment with estrogen to maintain physiologically relevant hormone levels also increases NO production in the cerebral circulation, vessels were isolated from OVX and OE animals and assayed for total nitrite levels after 30 min of incubation in PBS at 37°C. As shown in Fig. 7, basal NO production was significantly higher in vessels from OE animals compared with OVX controls. However, in contrast to what was found in OVX vessels, the addition of estrogen (10 nM) during the 30-min incubation period did not further increase NO production in vessels from females treated with estrogen (data not shown).

Effect of long-term in vivo estrogen treatment on cerebrovascular NO production. Basal NO production was measured by total nitrite assay in vessels isolated from OVX and OE females. Values were calculated as the ratio of total nitrite production to vessel total protein content and expressed as -fold difference versus the OVX control (n = 4).
Long-Term in Vivo Estrogen Treatment Increases p-Akt and p-eNOS Levels. To determine whether estrogen-mediated phosphorylation of Akt and eNOS occurs under conditions of prolonged exposure to estrogen in vivo, immunoblot analysis of cerebral vessels from OVX animals exposed to estrogen for one month (OE) were compared with vessels from OVX females and intact females. As shown in Fig. 8A, cerebrovascular p-Akt levels were significantly elevated in cerebral vessels from animals undergoing long-term exposure to estrogen as well as from intact females compared with the OVX group. Total Akt protein levels did not change with long-term estrogen treatment.

Effect of long-term in vivo estrogen exposure on levels of p-Akt, Akt, eNOS, and p-eNOS. Cerebral blood vessels from OE, OVX, and INTF rats were probed for p-Akt and Akt (A) and p-eNOS and eNOS (B) immunoreactivity by Western blot analysis. A representative blot is shown for each protein or phosphoprotein. The optical density for each OE and INTF band is expressed as -fold difference relative to the OVX band on the same blot, and the mean values ± S.E.M. are plotted (n = 4). *, P ≤ 0.05 compared with the OVX value for the relevant protein or phosphoprotein.
As we have previously demonstrated (McNeill et al., 1999), total eNOS protein was increased after long-term estrogen exposure. Levels of p-eNOS were also significantly greater in vessels from intact females and OVX animals undergoing long-term exposure to estrogen compared with OVX controls (Fig. 8B).
Discussion
The major finding of this study is that estrogen receptors activate a PI-3 kinase cascade in rat cerebral blood vessels that leads to both rapid and long-term increases in levels of p-Akt and p-eNOS as well as increases in NO production. This study is an important verification of rapid, nongenomic signaling by estrogen in intact vasculature. Furthermore, it demonstrates that long-term exposure to physiologically relevant hormone levels in vivo also stimulates kinase activation of cerebrovascular Akt and eNOS. Confocal imaging of cerebral arteries reveals a significant pool of ERα colocalized with caveolin-1 at the plasma membrane of endothelial cells in situ. Furthermore, cerebrovascular ERα coimmunoprecipitates with the p85α regulatory subunit of PI-3 kinase and eNOS. Both the full-length ERα (66 kDa) and a truncated ERα (about 50 kDa) are found in the immunoprecipitates, suggesting that multiple receptor types might mediate nongenomic signaling by estrogen in native tissue. Disruption of vascular caveolae inhibits estrogen-stimulated NO production, but, interestingly, this perturbation did not prevent estrogen-mediated phosphorylation of Akt. Together, these data suggest that nongenomic actions of estrogen, mediated by caveolae-associated ERα, contribute to cerebrovascular protective effects of estrogen.
Using intact cerebral vessels ex vivo, we demonstrate that physiological concentrations of 17β-estradiol rapidly increase NO production. A similar effect of estrogen is observed using endothelial cells in culture (Chen et al., 1999; Haynes et al., 2000; Chambliss et al., 2002). Estrogen seems to act by triggering eNOS phosphorylation because cerebrovascular levels of p-eNOS, but not total eNOS protein, increase in parallel with NO production. Phosphorylation of eNOS at serine-1117 is known to increase enzyme activity by 10- to 20-fold (Gallis et al., 1999). Mutation of eNOS serine-1177 to aspartate, which mimics the negative charge afforded by phosphorylation, leads to constitutively active eNOS even at low levels of Ca2+ (Dimmeler et al., 1999). This implies that activation of p-eNOS is less calcium-dependent than the unmodified form of eNOS (Dimmeler et al., 1999). Some cell culture studies, in fact, have shown that estrogen elevates NO production even in the absence of a Ca2+ stimulus (Caulin-Glaser et al., 1997; Haynes et al., 2000), which is consistent with an effect of estrogen on eNOS phosphorylation.
We also found that basal levels of p-eNOS are higher in cerebral blood vessels taken from animals exposed to estrogen. Thus, estrogen alters the sensitivity of eNOS in vivo such that the enzyme is more easily activated by a variety of chemical and mechanical stimuli (Shaul, 2002). In addition, there is more basal NO production in cerebral vessels taken from intact females and estrogen-treated animals compared with the ovariectomized group. This correlates with our previous finding that in vivo estrogen treatment decreases cerebral artery tone via endothelial, NO synthase-dependent mechanisms (Geary et al., 1998, 2001; Ospina et al., 2004). It is interesting that ex vivo stimulation of NO release by 17β-estradiol could only be detected in vessels from ovariectomized animals, perhaps because p-eNOS is already elevated in tissue exposed to estrogen in vivo. The in vivo data underscore the physiological relevance of estrogen-induced eNOS phosphorylation. Moreover, this signaling mechanism complements genomic actions of estrogen on vascular eNOS. We previously found that estrogen increases eNOS mRNA (Stirone et al., 2003a) and protein (McNeill et al., 1999; Stirone et al., 2003a) in cerebral vessels. Thus, the higher level of basal NO in vessels from estrogen-exposed animals probably reflects both genomic and nongenomic actions of the hormone.
Several signal transduction mechanisms have been implicated in rapid effects of estrogen, including activation of the PI-3 kinase/Akt kinase cascade (Haynes et al., 2000, 2002; Ho and Liao, 2002; Simoncini et al., 2003). PI-3 kinase phosphorylates Akt at serine-473, and then p-Akt directly phosphorylates eNOS at serine-1177 (Dimmeler et al., 1999; Fulton et al., 1999; Gallis et al., 1999; Shiojima and Walsh, 2002). As illustrated by a study where constitutively active Akt was transfected into rat femoral arteries (Luo et al., 2000), Akt regulates production of NO and affects blood vessel diameter. In isolated cerebral vessels, 17β-estradiol rapidly increases levels of p-Akt but not total Akt protein; these increases in p-Akt, p-eNOS, and NO production are all blocked by two inhibitors of PI-3 kinase, wortmannin and LY294002. These results are consistent with estrogen signaling via the PI-3 kinase/Akt/eNOS pathway, in agreement with some (Haynes et al., 2000; Simoncini et al., 2000), but not all (Chen et al., 1999, 2004; Haynes et al., 2003), studies in cultured endothelium. In particular, we found no evidence in cerebral vessels that estrogen-mediated activation of eNOS involves src kinase (Haynes et al., 2003), and our data contrast with findings from uterine artery endothelial cells that show no effect of estrogen on Akt (Chen et al., 2004). Together, these studies underscore the potential for endothelial differences caused by cell environment and/or tissue origin.
Rapid effects of 17β-estradiol on cerebral blood vessels seem to be mediated by estrogen receptors, in particular ERα and/or a splice variant of ERα. Stimulation of NO production and Akt phosphorylation in intact vessels is blocked by the ER antagonist ICI 182,780, in agreement with findings from cultured endothelial cells (Chen et al., 1999; Kim et al., 1999; Haynes et al., 2000; Stefano et al., 2000). ICI 182,780, however, does not distinguish receptor subtypes; therefore, rapid signaling by estrogen could be mediated by the well defined nuclear receptors ERα and ERβ (Chambliss et al., 2000, 2002; Nilsson et al., 2001; Ho and Liao, 2002), their splice variants (Pendaries et al., 2002; Figtree et al., 2003; Li et al., 2003), or other putative membrane receptors (Russell et al., 2000). Involvement of ERα is indicated by coimmunoprecipitation of this receptor subtype with the p85α regulatory subunit of PI-3 kinase. We have previously shown that ERα is present in both the intimal and medial layers of cerebral vessels (Stirone et al., 2003b); thus, using vessel lysates, we cannot definitively conclude that the ERα-p85α interaction we have identified occurs in the endothelium. However, our data agree with recent studies of cultured endothelial cells that show that ERα, but not ERβ, associates with the p85α subunit of PI-3 kinase (Simoncini et al., 2000, 2003).
We also demonstrate that cerebrovascular ERα coimmunoprecipitates with eNOS, and there is a significant increase in the association of eNOS and ERα after 30 min of estrogen treatment. At this point, we cannot distinguish between a direct protein-protein interaction and coexistence of the two proteins within a tight complex, e.g., caveolae. We previously reported that cerebral vessels express the full-length ERα (66 kDa) as well as a truncated ERα that has a molecular mass of about 50 kDa and lacks the N-terminal region (Stirone et al., 2003b). We detected both the 50- and 66-kDa bands in immunoprecipitates with eNOS. In recent cell transfection studies, a splice variant of human ERα (46 kDa), in which the N-terminal is deleted, was found to be more effective than the full-length ERα (66 kDa) at rapidly stimulating NO release from cultured cells (Figtree et al., 2003; Li et al., 2003). The present study suggests that a truncated form of ERα could play a role in eNOS activation in intact vascular tissue. The full-length ERα, although less effective (Li et al., 2003), may contribute as well.
Because eNOS and signaling molecules such as PI-3 kinase are localized together within caveolar regions of the plasma membrane, it has been hypothesized that estrogen receptors act at the membrane to mediate rapid hormone signaling (Chambliss and Shaul, 2002; Levin, 2002). Using confocal microscopy, we demonstrate that ERα immunoreactivity colocalizes with the key caveolae scaffolding protein, caveolin-1, at the plasma membrane of endothelial cells lining intact cerebral arteries. Further studies are required to establish the forms of ERα present at the membrane and the mechanisms underlying this localization. Hydrophobicity profiling of native ERα indicates that this receptor does not contain a domain sufficiently hydrophobic to exist as an integral membrane protein, nor does it contain a consensus glycosylation or acylation site. However, ERα has been shown to bind directly to caveolin-1 (Schlegel et al., 2001). In addition, palmitoylation may be involved in translocating the 46-kDa variant of ERα to the caveolar membrane (Li et al., 2003). To date, the evidence for membrane estrogen receptors in endothelial cells has come primarily from cultured cells or immortalized cell lines (Chambliss and Shaul, 2002; Deecher et al., 2003; Figtree et al., 2003; Li et al., 2003; Simoncini et al., 2003; Chen et al., 2004). However, resting cells in culture tend to exhibit primarily intracellular estrogen receptors (Figtree et al., 2003; Li et al., 2003; Simoncini et al., 2003), and transfection techniques are commonly employed to study membrane receptors (Levin, 2002; Li et al., 2003). The present study is one of the first to establish the presence of ERα at the endothelial membrane in native blood vessels, and, in contrast to cultured cells, there seems to be a significant pool of native estrogen receptors localized in or near the plasma membrane.
Using β-methyl-cyclodextrin treatment to disrupt caveolae by sequestration of cholesterol (Parpal et al., 2001; Kaiser et al., 2002), we found that 17β-estradiol activation of NO production in cerebral vessels was prevented. However, short-term 17β-estradiol treatment still resulted in robust phosphorylation of Akt at Ser-473. This was unexpected based on reports that activation of Akt, for example by insulin in adipocytes (Parpal et al., 2001), is inhibited by β-cyclodextrin disruption of caveolae under similar conditions; however, there are some differences between insulin and estrogen activation of Akt (Sasaki et al., 2003). It is also possible that the loss of cholesterol can activate, rather than inhibit, caveolar signaling molecules (Fielding and Fielding, 2003). On the other hand, estrogen stimulates Akt phosphorylation at intracellular sites (Sasaki et al., 2003) that may or may not require caveolar receptors; our pAkt measurements no doubt reflect multiple pools within the blood vessel.
In experimental models, estrogen has a clear protective effect against stroke (Hurn and Brass, 2003). Activation of p-eNOS via PI-3 kinase/Akt also seems to increase cerebral blood flow and decrease cerebral infarct size (Limbourg et al., 2002); thus, this mechanism probably contributes to the effect of estrogen. By increasing cerebrovascular p-eNOS and NO, estrogen can increase vasodilation and inhibit vascular inflammation, leukocyte adhesion, smooth muscle cell proliferation, platelet aggregation, and free radical activity (Geary et al., 1998; Shaul, 2002; Ospina et al., 2004).
Implications for estrogen regulation of the vascular PI-3-kinaseAkt cascade, however, go beyond stimulation of NO production. It was shown recently that stimulation of PI-3 kinase by estrogen up-regulates gene expression in cultured umbilical vein endothelial cells (Pedram et al., 2002). Akt also regulates endothelial cell survival and angiogenesis (Shiojima and Walsh, 2002), a mechanism that may underlie 17β-estradiol inhibition of endothelial apoptosis (Alvarez et al., 1997). Thus, activation by estrogen of the PI-3 kinase/Akt pathway can have far-reaching effects on vascular function.
Much discussion has focused on rapid effects of estrogen, but, in fact, hormone levels do not fluctuate over a period of minutes in the physiological setting. Thus, the significance of these mechanisms may lie not in the speed of estrogen signaling but in the consequences of activating specific signal transduction mechanisms. One of the most interesting findings of the present study is that long-term in vivo estrogen treatment increases basal levels of p-Akt and p-eNOS. This suggests that nongenomic effects are maintained under prolonged exposure to physiological levels of estrogen.
Acknowledgments
The skillful technical assistance of Jonnie Stevens is gratefully acknowledged.
Footnotes
-
This study was supported by National Institutes of Health grant R01-HL50775 and a grant-in-aid from the American Heart Association, National Center (Dallas, TX).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi: 10.1124/mol.104.004465.
-
ABBREVIATIONS: eNOS, endothelial nitric-oxide synthase; ER, estrogen receptor; PI, phosphoinositide; INTF, intact female; OVX, ovariectomized; OE, estrogen-treated ovariectomized; PBS, phosphate-buffered saline; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4,d]pyrimidine; PAGE, polyacrylamide gel electrophoresis; HRP, horseradish peroxidase; DAPI, 4′,6-diamidino-2-phenylindole; iNOS, inducible nitric-oxide synthase; β-mCD, β-methyl-cyclodextrin; ICI 182,780, fulvestrant; LY294002, 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride.
- Received July 1, 2004.
- Accepted October 19, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}