


Abstract
Many drugs inhibit the human ether-a-go-go-related gene (HERG) cardiac K+ channel. This leads to action potential prolongation on the cellular level, a prolongation of the QT interval on the electrocardiogram, and sometimes cardiac arrhythmia. To date, no activators of this channel have been reported. Here, we describe the in vitro electrophysiological effects of (3R,4R)-4-[3-(6-methoxyquinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5-trifluoro-phenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid (RPR260243), a novel activator of HERG. Using patch-clamp electrophysiology, we found that RPR260243 dramatically slowed current deactivation when applied to cells stably expressing HERG. The effects of RPR260243 on HERG channel deactivation were temperature- and voltage-dependent and occurred over the concentration range of 1 to 30 μM. RPR260243-modified HERG currents were inhibited by dofetilide (IC50 = 58 nM). RPR260243 had little effect on HERG current amplitude and no significant effects on steady-state activation parameters or on channel inactivation processes. RPR260243 displayed no activator-like effects on other voltage-dependent ion channels, including the closely related erg3 K+ channel. RPR260243 enhanced the delayed rectifier current in guinea pig myocytes but, when administered alone, had little effect on action potential parameters in these cells. However, RPR260243 completely reversed the action potential-prolonging effects of dofetilide in this preparation. Using the Langendorff heart method, we found that 5 μM RPR260243 increased T-wave amplitude, prolonged the PR interval, and shortened the QT interval. We believe RPR260243 represents the first known HERG channel activator and that the drug works primarily by inhibiting channel closure, leading to a persistent HERG channel current upon repolarization. Compounds like RPR260243 will be useful for studying the physiological role of HERG and may one day find use in treating cardiac disease.
Voltage-dependent ion channels control electrical activity in the human heart. The activation of voltage-dependent Na+ and Ca2+ channels leads to excitation and contraction of the myocardium, whereas repolarization is largely controlled by the activity of voltage-dependent K+ channels. Several voltage-dependent K+ channels exist in the human myocardium. The human ether-a-go-go-related gene (HERG) encodes the K+ channel that carries the rapid component of the delayed rectifier current (IKr) in the human heart (Sanguinetti et al., 1995; Trudeau et al., 1995). HERG channel activity contributes to cardiac repolarization and is an important determinant of the QT interval on the electrocardiogram (ECG). Mutations in HERG leading to a reduction in its activity are responsible for the type 2 form of congenital long QT syndrome (Curran et al., 1995) and can lead to ventricular arrhythmia and sudden cardiac death.
It is now well established that many drugs can specifically block the HERG channel, leading to a condition known as acquired long QT syndrome. In this case, drugs, usually as a side effect, inhibit HERG, leading to QT prolongation on the ECG and to the development of the life-threatening ventricular arrhythmia known as torsades de pointes. Indeed, a staggering array of drugs including antihistamines, antibiotics, and anti-psychotics are known to produce acquired long QT syndrome via a specific inhibition of HERG (Pearlstein et al., 2003; Redfern et al., 2003). Despite extensive pharmacological study, no activators of HERG have yet been documented. The present report describes the in vitro electrophysiological effects of the first HERG channel activator, RPR260243.
Materials and Methods
Cell Culture and Isolation. Chinese hamster ovary (CHO) cells (American Type Culture Collection, Manassas, VA) were stably transfected with the cDNA encoding the HERG cardiac K+ channel as described previously (Rampe et al., 1997). Cells were grown in Ham's F-12 media supplemented with 10% fetal bovine serum and 500 μg/ml G418 (Invitrogen, Carlsbad, CA) in an atmosphere of 95% air/5% CO2. KvLQT1 and minK were cloned from human heart and stably expressed in CHO cells as described previously (Kang et al., 2000). The cDNA encoding SCN5A, the human Na+ channel, was stably transfected into human embryonic kidney 293 cells (American Type Culture Collection) as described previously (Kuryshev et al., 2000). The cDNA encoding the erg3 human brain K+ channel was transiently transfected into CHO cells using LipofectAMINE Plus Reagent kit (Invitrogen) as reported previously (Kang et al., 2001).
Single ventricular myocytes were isolated from guinea pigs and were used to record action potentials, IKr, and Ca2+ channel currents. Single ventricular myocytes were isolated from guinea pigs using a method modified from that described by Salata et al. (1995). In brief, male guinea pigs (Hartley) were anesthetized with 5% isoflurane (Baxter Healthcare Corporation, Deerfield, IL) in a mixture of nitrous oxide and oxygen (1:1). Then a thoracotomy was performed, and the heart was removed and immediately transferred to oxygenated (100% O2) cold saline. The heart was perfused retrogradely at 10 ml/min through the aorta with an oxygenated Ca2+-free saline at 37°C in three stages: first with standard Ca2+-free saline for 5 min, then with the same solution containing 280 U/ml type II collagenase (Worthington Biochemicals, Freehold, NJ) plus 0.75 U/ml type XIV protease (Sigma-Aldrich, St. Louis, MO) for 8 min, and finally with saline containing 0.2 mM CaCl2 for an additional 7 min. The left ventricle was cut into small pieces and was gently shaken at room temperature for approximately 5 min to disperse single myocytes. The isolated myocytes were then maintained at 10°C for electrophysiological recording within 4 to 5 h after isolation.
Patch-Clamp Recording. All ionic currents were recorded using the whole-cell configuration of the patch-clamp technique (Hamill et al., 1981). Electrodes (2–4 MΩ resistance) were made from TW150F glass capillary tubes (WPI, Sarasota, FL). For all K+ channel recordings, electrodes were filled with the following solution: 120 mM potassium aspartate, 20 mM KCl, 4 mM Na2ATP, 5 mM HEPES, and 1 mM MgCl2, pH 7.2, with KOH. The external solution contained 130 mM NaCl, 5 mM KCl, 2.8 mM sodium acetate, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, and 1 mM CaCl2, pH 7.4, with NaOH. The internal and external solutions for Na+ and Ca2+ channel recordings have been described previously (Kang et al., 2004). All ionic currents were recorded using an Axopatch 200B amplifier (Axon Instruments Inc., Union City, CA). Spontaneous rundown of HERG currents averaged approximately 10% over the course of 10-min recording. Currents were analyzed using the pCLAMP suite of software (Axon Instruments). Deactivation was fitted to a biexponential process: y = C + A1 exp(–t/τ1) + A2 exp(–t/τ2), where τ1 and τ2 are the time constants, A1 and A2 are the amplitudes of each component of the exponential, and C is the baseline value. Data are presented as mean ± S.E.M., and statistical comparisons were made using a paired Student's t test. P values of less than 0.05 were considered significant. Time constants for the envelope of tails test were derived from a single exponential fit of the data using Origin software (OriginLab Corp, Northampton, MA).
Action Potential Recording. Myocytes were placed in a temperature-controlled (37 ± 1°C) chamber and perfused with a modified Tyrodes solution containing 132 mM NaCl, 4 mM KCl, 1.2 mM MgCl2, 1.8 mM CaCl2, 10 mM glucose, and 10 mM HEPES, pH 7.4, with NaOH. Action potentials were recorded using a standard glass microelectrode filled with 3 M KCl (resistance, 20–45 MΩ). Action potentials were amplified using an AxoClamp 2B amplifier (Axon Instruments), and data were stored and analyzed using the pCLAMP suite of software (Axon Instruments). Myocytes were allowed to equilibrate at a stimulation rate of 1 Hz for 30 min before action potential traces were recorded. Data are presented as the mean ± S.E.M., and statistical comparisons were made using a paired Student's t test with P values of less than 0.05 considered statistically significant.
Langendorff Heart Preparation. Male guinea pigs weighing 450 to 550 g were used. Under isofluorane anesthesia (4.5%), the hearts were rapidly removed and were set up for perfusion of the coronary artery according to the Langendorff method in Locke's solution. The Locke's solution was saturated with O2 at 32.5°C and had the following composition: 153.6 mM NaCl, 5.6 mM KCl, 2 mM CaCl2, 6 mM NaHCO3, 11.1 mM dextrose, 2 mM sodium pyruvate, and 0.05 mM Na2 EDTA. The hearts were paced at 3 Hz via a pair of bipolar electrodes on the right atrium at a site close to the sinoatrial node using a 1-ms pulse that was 20% above the threshold voltage. ECG waveforms were recorded and analyzed using silver chloride electrodes, as described by Zabel et al. (1995), and an ECG amplifier and recording software (Buxco Electronics, Wilmington, NC). The hearts were set up and allowed to equilibrate for 2 to 3 h before testing. Control ECG tracings were then recorded for 20 min followed by perfusion of the heart with RPR260243 for an additional 20 min. Five ECG waveforms were analyzed for PR interval, QRS duration, QT interval, and T-wave amplitude at 5-min intervals. The data were then averaged and expressed as the mean ± S.E.M., and statistical comparisons were made using a paired Student's t test with P values of less than 0.05 considered statistically significant.
Chemicals. RPR260243 was synthesized at Aventis Pharmaceuticals (Paris, France). All other chemicals were obtained from commercial sources. Drugs were made as stock solutions in dimethyl sulfoxide, which was present in all solutions as a 0.1% final concentration.
Results
During the course of screening compounds for HERG channel-blocking activity, we came across a compound known as RPR260243 that displayed peculiar effects on HERG channel currents. Figure 1 illustrates these effects in CHO cells stably transfected with HERG. Cells were held at –80 mV, and 2-s depolarizing pulses to +20 mV were applied followed by repolarization to –50 mV to produce large, slowly deactivating tail currents characteristic of HERG (Sanguinetti et al., 1995). The pulses were delivered at 30-s intervals. Upon addition of RPR260243 (10 μM), HERG tail currents failed to decay, and a large persistent current developed, making further recordings difficult (Fig. 1A). This response was completely reversible upon washing the cell with drug-free solution for several minutes. Figure 1B illustrates the mechanism underlying these findings. In these experiments, cells were held at –80 mV and depolarized to +20 mV for 2 s. The cells were then returned to –80 mV, and the tail currents were observed for approximately 40 s. Once control currents were recorded, 10 μM RPR260243 was added to the cells for 3 min without pulsing. We observed no increase in the holding current during this time. After the drug had washed in, we delivered one more depolarizing step. We found that RPR260243 had no significant effect on the initial current amplitude recorded at +20 mV. Currents measured at the end of the +20-mV depolarizing pulses were increased by an average of 15 ± 9% after exposure to 10 μM RPR260243 (P > 0.05, paired t test; n = 5). However, a consistent and dramatic slowing of current deactivation at –80 mV was observed. Under control conditions, tail current decay was fit to a biexponential function. Fast and slow time constants (τF and τS) measured 129 ± 15 and 534 ± 60 ms, respectively (n = 5; r2 = 0.99). After the addition of 10 μM RPR260243, tail current decay was slowed, with τF and τS measuring 637 ± 122 and 12,706 ± 1513 ms, respectively (n = 5; r2 = 0.96). Indeed, it took approximately 2 min for the currents to return to the prepulse holding potential. Examples of the fits to the tail currents are illustrated in Fig. 1C. The effect of RPR260243 on HERG channel deactivation was voltage-dependent. The slow time constants observed in the tail currents after exposure to 10 μM RPR260243 measured 5638 ± 909 and 2402 ± 298 ms when cells were returned to –100 and –120 mV, respectively (n = 4). The slow time constants in the presence of drug are plotted as a function of voltage in Fig. 1C (inset). Because increasing temperature greatly speeds both activation and deactivation of HERG, we repeated the experiment described for Fig. 1B at 37°C (Fig. 1D). As was the case for the room-temperature recordings, current amplitude measured at the end of the +20 mV depolarizing pulse was not significantly altered in the presence of the drug and measured 11 ± 9% higher than the predrug value (P > 0.05, paired t test). In the absence of drug, HERG tails currents measured at –80 mV were fit to a biexponential function. τF and τS measured 28 ± 2 and 107 ± 4 ms, respectively (n = 6; r2 = 0.98). After the addition of 10 μM RPR260243, tail current decay was again dramatically slowed, although not to the extent observed at room temperature. Tail currents in the presence of RPR260243 decayed with a fast time constant measuring 207 ± 58 ms and a slow time constant measuring 2176 ± 169 ms (n = 6; r2 = 0.95). Tail currents took approximately 5 s to fully decay.

Effects of RPR260243 on HERG channel currents. A, HERG currents were elicited at 30-s intervals at room temperature according to the pulse protocol shown. Addition of 10 μM RPR260243 produced slowly deactivating currents and the appearance of a persistent current component. These effects were reversible upon washout of the drug. The chemical structure of RPR260243 is shown in the inset. B, control currents were recorded every 30 s at room temperature according to the pulse protocol shown. After this time, RPR260243 was added to the cell for 3 min without pulsing. The first pulse after this incubation period is shown. Note the extremely slow tail current decay. The effects of RPR260243 were reversible upon washout. C, biexponential fits of the tail currents shown in B. The time constants and correlation coefficients (r2) are indicated. The slow time constants observed in the presence of 10 μM RPR260243 are plotted as a function of the return potential in the inset. D, effects of RPR260243 on HERG tail currents recorded at 37°C. Currents were recorded as described for B.
Figure 2 shows the concentration-response relationship for RPR260243 on HERG. Cells were held at –80 mV, and 2-s depolarizations to +20 mV were applied every 30 s followed by repolarization of the cells to –50 mV. Currents were recorded at room temperature. Immediately after the –50 mV step, the cells were hyperpolarized to –120 mV for 10 s. Figure 2A illustrates the effects of RPR260243 on these HERG currents. The most obvious effect of the drug was to produce a slowing of current deactivation. This effect is illustrated by plotting the slow time constants of deactivation measured at –120 mV as a function of drug concentration (Fig. 2B). Drug effects were observed over the concentration range of 1 to 30 μM.

Dose-response relationship for RPR260243 on HERG. A, HERG currents were elicited by 2-s depolarizing pulses to +20 mV followed by repolarization to –50 mV. This was followed by 10-s hyperpolarizing pulses to –120 mV. The concentrations of RPR260243 (in micromoles) are indicated. RPR260243 produced a dose-dependent slowing in tail current decay that was obvious at concentrations of 1 μM and higher. B, the slow time constants derived from biexponential fits of the tail currents at –120 mV are plotted as a function of drug concentration. Error bars denote S.E.M. (n = 5).
To further ensure that the currents evoked by RPR260243 were carried through HERG, we explored the interaction of the potent and selective HERG channel antagonist dofetilide with RPR260243. Using the pulse protocol used in Fig. 2, we stimulated the HERG channel with 10 μM RPR260243 (Fig. 3A). As expected, this treatment resulted in the removal of current deactivation at –50 mV along with a slight increase in tail current amplitude. These RPR260243-stimulated tail currents were inhibited by dofetilide in a dose-dependent manner (Fig. 3B). The IC50 values for dofetilide inhibition of RPR260243-modified currents measured 58 nM (34–98 nM, 95% confidence limits). This value was somewhat higher than the 11 nM IC50 value that we have reported previously for dofetilide inhibition of HERG (Kang et al., 2004). Although dofetilide abolished current through these channels, it did not reverse the effects of RPR260243 on tail current decay.

Effects of dofetilide on RPR260243-altered HERG currents. HERG currents were elicited as described in Fig. 2, and the tail currents obtained at –50 mV are shown. Currents were evoked at 30-s intervals. The effects of RPR260243 on HERG currents and their subsequent inhibition by dofetilide are shown in A. B, summary of the current measured at the end of the –50-mV pulse under various treatment conditions. Error bars indicate S.E.M. (n = 5).
From the data in Fig. 1D, it seemed that RPR260243 slowed the time to peak current upon depolarization from –80 to +20 mV. To examine this further, we conducted an envelope of tails test (Trudeau et al., 1995). In this experiment, tail currents were generated at –80 mV after the application of voltage steps of increasing duration to +20 mV at a temperature of 37°C. Currents in the absence and presence of 10 μM RPR260243 are shown in Fig. 4, A and B, respectively. Peak tail currents were normalized to the value obtained for the last pulse under control conditions. These normalized values were then fitted with a single exponential and are plotted in Fig. 4C. The time constant measured in the absence of drug measured 60 ms (49–70 ms, 95% CL). This value was increased to 91 ms (75–107 ms, 95% CL) in the presence of 10 μM RPR260243.

Envelope of tail current test. Depolarizing pulses to +20 mV of increasing duration (30–300 ms) were applied every 10 s at 37°C. Current traces in the absence and presence of 10 μM RPR260243 are shown in A and B, respectively. The peak tail currents upon return to –80 mV were normalized to the last pulse in the absence of drug and are plotted in Fig. 4C. Single exponential fits of the data are shown. Time constants measured 60 and 91 ms in the absence and presence of drug, respectively. Error bars indicate S.E.M. (n = 5).
We next examined the effects of RPR260243 over a wide range of test potentials. In these experiments, cells were held at –80 mV, and currents were elicited by 2-s depolarizing pulses to potentials ranging from –50 to +30 mV in 10-mV increments. The membrane potential was then returned to –80 mV, and the peak tail currents were measured and used to construct steady-state activation curves. These experiments were carried out at 37°C. Current traces in the absence and presence of 10 μM RPR260243 are shown in Fig. 5, A and B, respectively. RPR260243 produced long-lasting tail currents upon repolarization of the cells to –80 mV. Otherwise, the drug produced only minor effects on the current waveforms, including a small slowing of activation, especially at more depolarized potentials, and an approximately 10 to 20% increase in current amplitude when measured at the end of the depolarizing step (Fig. 5C). The drug did not significantly alter the steady-state activation curve generated from the peak tail currents. Under control conditions, the midpoint (V0.5) and the slope (k) of the curve measured –28 mV (–30 to –27 mV, 95% CL) and 8.0 (6.8–9.2, 95% CL), respectively. After the addition of 10 μM RPR260243, V0.5 and k measured –27 mV (–29 to –24 mV, 95% CL) and 9.3 (7.1–11.5, 95% CL), respectively.
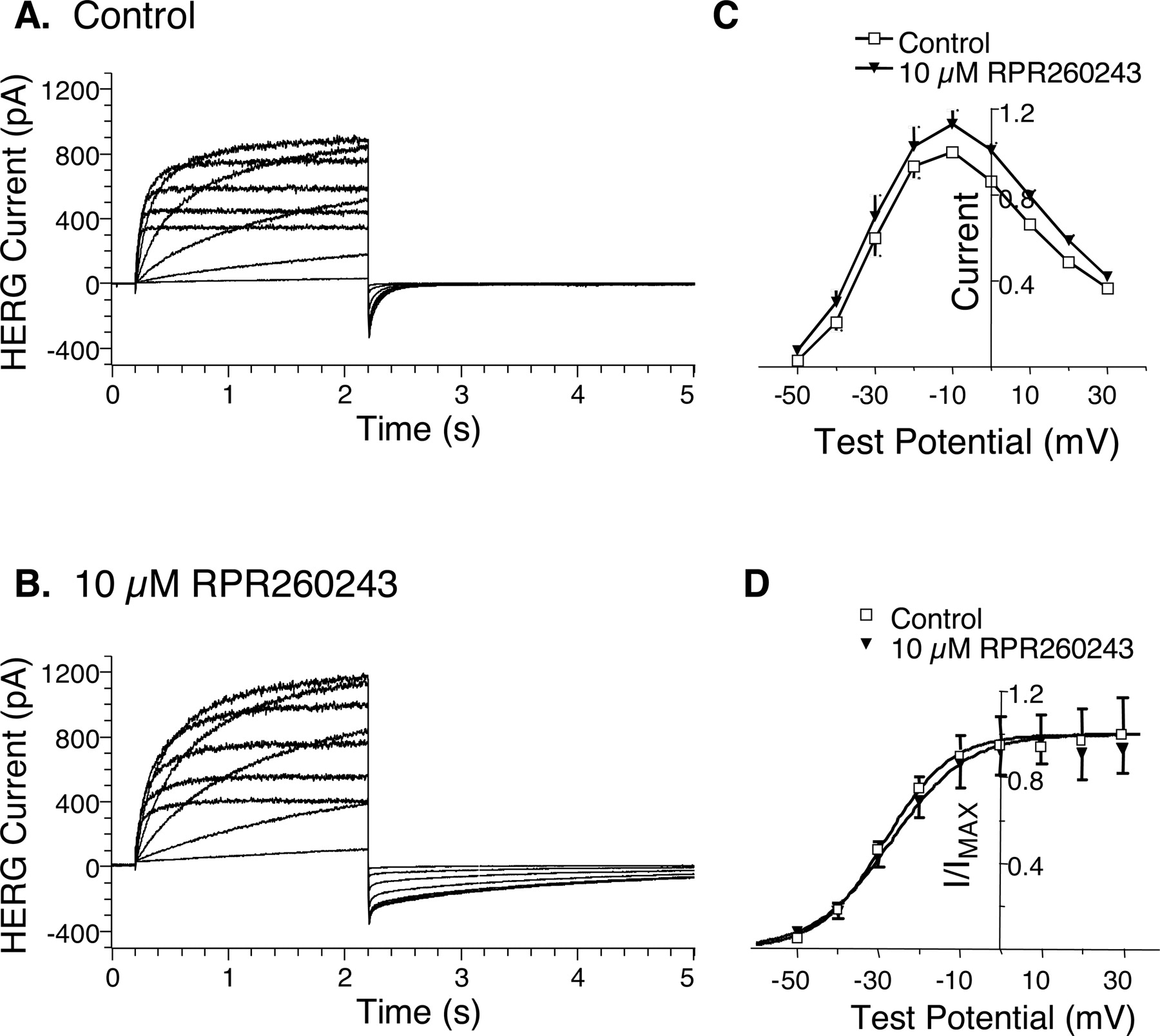
Effects of RPR260243 on HERG currents at various membrane potentials. Cells were held at –80 mV and depolarized for 2 s to potentials ranging from –50 to +30 mV in 10-mV increments. The cells were then returned to –80 mV to produce inward tail currents. Currents were recorded at 37°C. Current traces in the absence and presence of 10 μM RPR260243 are illustrated in A and B, respectively. C, current-voltage relationships in the absence and presence of 10 μM RPR260243 are indicated. Current was sampled at the end of the 2-s depolarizing pulse and was normalized (current amplitude = 1.0) to the value obtained at –10 mV in the absence of drug. Error bars indicate S.E.M. (n = 5). D, steady-state activation curves derived from the peak inward tail currents at –80 mV are shown. Current was normalized to the value obtained after the +30-mV pulse in the absence of drug and fit according to the equation Y = 1/[1 + exp{–(V – V0.5)/k}], where V0.5 is the midpotential of the curve and k is the slope. Error bars denote S.E.M. (n = 5).
Figure 6 shows the effects of RPR260243 on HERG channel inactivation parameters. These studies were carried out in a manner similar to that described previously for HERG (Smith et al., 1996; Spector et al., 1996; Walker et al., 1999). To assess the effects of RPR260243 on recovery from inactivation, cells were depolarized to +30 mV for 500 ms at room temperature. Cells were then returned to –100, –90, or –80 mV for 100 ms to generate inward tail currents. Recovery from inactivation was determined by fitting the initial rising phase of these tail currents, before deactivation occurred, to a single exponential function. Figure 6A shows the tail currents obtained in the presence and absence of 10 μM RPR260243. Figure 6B plots the time constants measured in these experiments in the presence and absence of drug. RPR260243 produced no significant effect on the rate of recovery from inactivation at these potentials (p > 0.05, paired t test). To examine the voltage dependence of inactivation, cells were depolarized for 1 s to +30 mV. This was followed by 20-ms pulses to potentials between –110 and +30 mV followed by a second 200-ms step to +30 mV. The amplitudes of the peak currents elicited by the second step to +30 mV, in the absence and presence of drug, were then determined. The currents were normalized to the first pulse obtained in the absence of drug (i.e., after the –110-mV step), and the V0.5 and k of the curves were calculated. In the absence of drug, V0.5 and k measured –70 mV (–79 to –62 mV, 95% CL) and –25 (–29 to –21, 95% CL), respectively. After the addition of 10 μM RPR260243, V0.5 and k measured –69 mV (–75 to –62 mV, 95% CL) and –25 (–28 to –21, 95% CL), respectively.
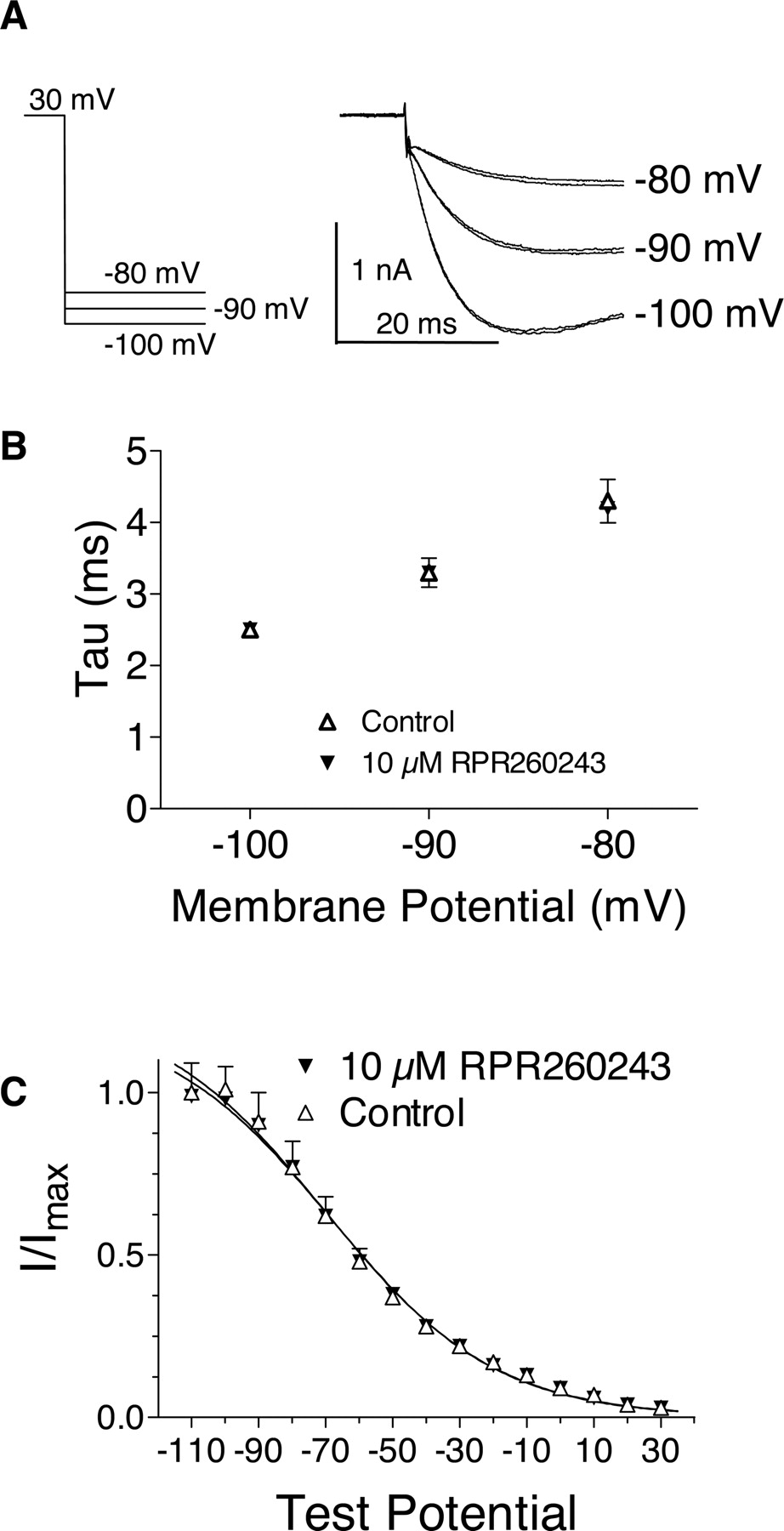
Effects of RPR260243 on recovery from inactivation and the voltage-dependence of inactivation. A, inward HERG tail currents were recorded at –100, –90, and –80 mV after a 500-ms pulse to +30 mV. The initial portions of the tail currents at these potentials are shown in the presence and absence of 10 μM RPR260243. B, the initial rising phase preceding slower deactivation of the tail currents were fit to a single exponential and plotted as a function of voltage. Error bars denote S.E.M. (n = 5). C, voltage dependence of inactivation. After a 1-s pulse to +30 mV, 20-ms pulses were applied to potentials between –110 and +30 mV followed by a second 200-ms depolarizing pulse to +30 mV. Peak current amplitudes obtained during the second pulse to +30 mV were recorded. These currents were normalized to the amplitude obtained after the –110 mV step in the absence of drug. Curves were fit according to the equation Y = 1/[1 + exp{(V – V0.5)/k}], where V0.5 is the midpotential of the curve and k is the slope. Error bars indicate S.E.M. (n = 4). All recordings were carried out at room temperature.
Figure 7 examines the effects of RPR260243 recorded at different pulse frequencies. In these experiments cells, were depolarized for 300 ms to +20 mV from a holding potential of –80 mV at 37°C. Thirty such pulses were delivered at 1 Hz in the absence of drug (Fig. 7A, inset). After this, RPR260243 was added to the cells for approximately 3 min without pulsing. After this equilibration period, cell were again stimulated for 30 s at a rate of 1 Hz. We found that the effects of the drug were immediately apparent upon the first pulse of this train. Figure 7A compares the first trace obtained with this pulse train in the absence and presence of RPR260243. Both the rising time of the current and current deactivation were slowed in the presence of the drug compared with control. All 30 traces obtained at a 1-Hz frequency in the presence of drug are shown in Fig. 7B. In addition to this pulse train, we also examined the effects of the drug at stimulation frequencies of 0.5 and 0.1 Hz. Currents obtained at these frequencies are shown for comparison in Fig. 7, C and D. We observed no differences in the effects of the drug at these various stimulation rates, with the exception of the fact that the 0.1-Hz rate was sufficiently slow to allow for channel closure between pulses.

Effects of RPR260243 at various stimulation frequencies. HERG channel currents were elicited by 300-ms depolarizing pulses to +20 mV from a holding potential of –80 mV at 37°C. Cells were then returned to –80 mV. Thirty control currents were first recorded at a 1-Hz stimulation frequency (A, inset). Then RPR260243 (10 μM) was equilibrated with the cell without pulsing. After this time, 30 additional pulses were applied at a rate of 1 Hz. A, the first pulse of these trains in the absence and presence of drug is shown. All 30 pulses of this train in the presence of RPR260243 are shown in B. Stimulation rates of 0.5 and 0.1 Hz (30-s duration) in the presence of drug are also shown in C and D, respectively.
We also studied the effects RPR260243 on other voltage-dependent ion channels. Figure 8 shows the effects of RPR260243 on the human cardiac Na+ channel, the Kv-LQT1/mink cardiac K+ channel, the L-type Ca2+ channel recorded from guinea pig myocytes, and the human brain K+ channel erg3. RPR260243 had no significant effects on the human cardiac Na+ channel or on KvLQT1/minK at concentrations up to 30 μM (Fig. 8, A and B). RPR260243 was a weak inhibitor of the L-type Ca2+ channel, producing 10 ± 3 and 30 ± 6% inhibition at 10 and 30 μM, respectively (Fig. 8C; n = 5–6). Unlike its effects on HERG, RPR260243 failed to demonstrate any agonist-like effects on the erg3 K+ channel, despite the fact that erg3 is in the same family as HERG (Warmke and Ganetzky, 1994). In fact, RPR260243 was an antagonist of erg3, inhibiting peak tail currents measured at –100 mV by 42 ± 8 and 68 ± 5% at concentrations 10 and 30 μM, respectively (Fig. 8D; n = 6).

Effects of RPR260243 on other voltage-dependent ion channels. A, effects of RPR260243 on the human cardiac Na+ channel. Na+ currents were elicited at a frequency of 1 Hz according to the pulse protocol indicated. Currents in the absence and presence of 10 and 30 μM RPR260243 are shown. B, effects of 10 and 30 μM RPR260243 on KvLQT1/minK currents are shown. Currents were recorded every 10 s according to the pulse protocol shown. C, effects of 10 and 30 μM RPR260243 on Ca2+ currents recorded at 10-s intervals from guinea pig myocytes are shown. D, Effects of 10 and 30 μM RPR260243 on erg3 currents recorded at 10-s intervals are shown.
Figure 9 shows the effects of RPR260243 on IKr and action potentials recorded from guinea pig myocytes. To record IKr, myocytes were held at –50 mV and depolarized every 5 s for 300 ms to +10 mV. The cells were then returned to –50 mV, and outward tail currents were recorded. The experiments were conducted at 37°C and used the same external and internal solutions used for the cloned HERG channel studies. As was the case with the cloned HERG channel, IKr deactivation was slowed by the addition of 10 and 30 μM RPR260243, resulting in a nondeactivating current under these conditions (Fig. 9A). This effect was completely abolished when 1 μM dofetilide was further added to the cells (Fig. 9A). Peak dofetilide-sensitive tail currents measured 46 ± 6 pA under control conditions and 78 ± 9 and 95 ± 11 pA after the addition of 10 and 30 μM RPR260243, respectively. To record action potentials, myocytes were stimulated at a rate of 1 Hz, and the resting membrane potential (RMP), action potential amplitude (APA), and the action potential duration at 50% and 90% repolarization (APD50 and APD90, respectively) were recorded. Typical action potential recordings in the absence and presence of RPR260243 are shown in Fig. 9B. RPR260243 (1–30 μM) had no effect on RMP or APA. RPR260243 at concentrations up to 10 μM also had no significant effect on APD50 or ADP90. Only at 30 μM did RPR260243 change action potential duration, shortening both APD50 and APD90 by 12 ± 3% (P < 0.05, paired t test; n = 6). However, the effects of RPR260243 on action potential duration were more pronounced in the presence of dofetilide. APD90 was increased by 23 ± 5% (n = 6) by the addition of 10 nM dofetilide. This prolongation in APD90 was reversed in a dose-dependent manner with the addition of RPR260243 (3–30 μM; Fig. 9, C and D). Similar results were obtained for APD50 (data not shown).

Effects of RPR260243 on IKr and action potentials recorded from guinea pig myocytes. A, effects of RPR260243 on IKr are shown. Myocytes were held at –50 mV and depolarized to +10 mV every 5 s. Cells were then returned to –50 mV to evoke outward tail currents characteristic of IKr. Trace 1 indicates control current tracing, whereas traces 2 and 3 represent the addition of 10 and 30 μM RPR260243, respectively. Trace 4 indicates the further addition of 1 μM dofetilide in the presence of 30 μM RPR260243. B, the dose-dependent effects of RPR260243 on the action potential waveform are shown. No significant effects were observed on APA or RMP over the concentration range of 1 to 30 μM. APD50 and APD90 were reduced by 12% after the addition of 30 μM. C, effects of RPR260243 on action potentials prolonged by dofetilide. Trace 1 indicates the control tracing, whereas trace 2 shows the effects of 10 nM of dofetilide. Traces 3, 4, and 5 indicate the subsequent addition of 3, 10, and 30 μM RPR260243 to the cell. D, summary of the APD90 values under various treatment conditions. Error bars indicate S.E.M. (n = 6).
We also examined the effects of RPR260243 on the ECG waveform measured in guinea pig hearts in vitro using the Langendorff method. The hearts were allowed to stabilize for 2 to 3 h after which time control ECG traces were recorded for 20 min. RPR260243 (5 μM) was then added to the perfusate, and ECGs were recorded for an additional 20 min. RPR260243 had multiple effects on the ECG waveform. Most prominent was a large increase in T-wave amplitude that began immediately after drug exposure and continued throughout the recording time. Compared with control tracings, T-wave amplitude increased by a maximum of 82 + 24% (n = 6) after the addition of RPR260243 (Fig. 10A). QRS duration was unaffected by the drug (Fig. 10B). Other changes in the ECG waveform included an 11 ± 3% prolongation in the PR interval (Fig. 10C) and a 6 ± 1% reduction in the QT interval (Fig. 10D). Because some of these effects are similar to those produced by elevated extracellular K+ levels, we examined the effects of increased levels of extracellular K+ on the ECG recordings. When extracellular K+ concentration was increased from 5.4 to 10 mM, T-wave amplitude was increased by a maximum of 52 ± 20% (n = 6). In addition, QRS and PR intervals were prolonged by 31 ± 5 and 32 ± 5%, respectively (Fig. 10, B and C), whereas QT interval was reduced by 3 ± 1% (Fig. 10D).

Effects of RPR260243 on ECG waveforms recorded from guinea pig hearts. A, ECG tracings under control conditions and 20 min after the addition of 5 μM RPR260243 are shown. Note the increase in the T-wave amplitude. B, C, and D, effects of 5 μM RPR260243 or 10 mM extracellular K+ on the QRS, PR, and QT intervals, respectively. Arrows indicate the addition of RPR260243 or K+. Error bars indicate S.E.M. and asterisks denote statistical significance compare with control values (P < 0.05, paired t test; n = 6).
Discussion
As a rule, small-molecule activators of voltage-dependent ion channels are rare. For example, despite decades of intensive pharmacological study of cardiac Na+ and Ca2+ channels, only a handful of activators of these proteins are known to exist. Drugs that activate voltage-dependent ion channels can work through many different mechanisms. For example, by promoting long-lasting channel openings, certain dihydropyridines and benzoylpyrroles enhance L-type Ca2+ channel currents and prolong tail current decay (Lacerda and Brown, 1989; Rampe et al., 1993). Piperazine derivatives typified by DPI 201–106 effectively enhance Na+ currents by removing channel inactivation (Nilius et al., 1987). All of these compounds display marked effects on cardiac function and have been valuable tools for exploring the contribution of these channels to cardiac physiology. Like cardiac Na+ and Ca2+ channels, the pharmacology of the HERG cardiac K+ channel is the subject of intense study. Inhibition of the HERG channel leads to prolongation of the action potential duration, QT interval prolongation, and the development of torsades de pointes ventricular arrhythmia. It is now clear that a wide variety of drugs from numerous structural and pharmacological classes act as antagonists of the channel (Pearlstein et al., 2003; Redfern et al., 2003). Although many antagonists of HERG are known, to date, no activators of this channel have been reported. The present study describes some of the in vitro electrophysiological effects of what we believe to be the first known activator of HERG, RPR260243.
The most obvious effect of RPR260243 that we observed on HERG channel currents was a dramatic slowing of deactivation. This effect was voltage- and temperature-dependent, being less pronounced at more hyperpolarized potentials or at elevated temperature. Even so, tail currents required many seconds to completely decay even at –120 mV (room temperature) or at –80 mV at physiological temperature. The drug also produced a slowly rising current component upon depolarization that was evident by the change in the slope in the envelope of tails test. This test and the “first pulse” effect observed in Fig. 7 suggest that the drug accesses the HERG channel quickly upon depolarization. Some increase in HERG current amplitude could be seen in the presence of drug, but this effect was small (10–20% increase) and not consistently observed. Once HERG currents were modified by the addition of RPR260243, they could be blocked by nanomolar concentrations of dofetilide. RPR260243 had no significant effect on steady-state activation or on the recovery from inactivation or the voltage-dependence of inactivation. In terms of specificity, RPR260243 displayed no activator-like properties on a variety of other voltage-dependent ion channels, including the closely related erg3 K+ channel. Taken together, these results suggest that RPR260243 is an activator of the HERG cardiac K+ channel and that the drug seems to work mainly by slowing closure of the HERG channel once it has opened. However, we do not exclude additional mechanisms of action and believe that more studies, including single-channel analyses, are necessary to fully explore all the molecular properties of this new and unique class of drug.
In addition to its effects on the cloned HERG channel, we also examined the actions of RPR260243 on native IKr and on action potentials, both recorded from guinea pig myocytes. Like its effects on the cloned HERG channel, RPR260243 slowed deactivation of IKr, producing a persistent outward current component. RPR260243 alone had only modest effects on action potential parameters. The only significant effect we observed was a small shortening of action potential duration at the highest concentration tested (30 μM). Because multiple ionic currents contribute to the action potential in these cells, it is possible that the particular actions of RPR260243 on IKr are, by themselves, insufficient to dramatically alter the action potential waveform. However, the effects of RPR260243 were more pronounced when it was administered in addition to the IKr-blocker dofetilide. In this case, the 23% prolongation observed with dofetilide treatment alone was completely reversed, in a dose-dependent manner, by further additions of RPR260243 (3–30 μM). These results suggest that pharmacological activation of HERG/IKr may be a useful mechanism for reversing the acquired long QT syndrome that is a side effect of certain medications.
We used the Langendorff heart method to examine the effects of RPR260243 on the ECG waveform in the intact heart. The most pronounced effect of the drug was to enhance the amplitude of the T wave. This was accompanied by some shortening of the QT interval and a small prolongation in PR interval. The changes observed in the T-wave amplitude and the QT interval suggest that the drug can modify repolarization in the intact heart. Because some of these ECG findings are similar to those observed under conditions of high extracellular K+, we compared the effects of RPR260243 to those produced by 10 mM extracellular K+ in this preparation. Elevated K+ is known to enhance the activity of HERG by reducing channel inactivation and/or relieving channel block by extracellular Na+ (Wang et al., 1996; Mullins et al., 2002). Administration of 10 mM K+ to the hearts also produced an increase in T-wave amplitude but to a lesser extent than that seen with RPR260243. Elevated K+ shortened the QT interval to a lesser extent and prolonged the PR interval to a greater extent than did the administration of RPR260243. Unlike RPR260243, extracellular K+ also significantly prolonged the QRS duration. We believe that RPR260243 administration more specifically highlights the effects of IKr activation on the ECG, whereas the effects of elevated extracellular K+ represent not only an enhancement of IKr, but also multiple other effects including membrane depolarization. It is interesting that increasing serum K+ levels has been shown to improve repolarization parameters in patients who suffer from congenital long QT syndrome caused by HERG mutations (Compton et al., 1996; Etheridge et al., 2003). Provided that at least some functional HERG channels are present, it is possible that HERG agonists could one day be used to treat congenital long QT syndrome via a specific modulation of HERG. Drugs such as RPR260243 may be especially useful in those patients in whom specific HERG mutations result in accelerated channel deactivation (Chen et al., 1999).
In summary, RPR260243 is a prototype for a new class of compounds, HERG channel agonists. We believe that RPR260243 will be a valuable new tool for studying the structural, biophysical, and physiological properties of the HERG cardiac K+ channel. The drug seems to slow the open-to-close transition of the channel, resulting in dramatically prolonged tail currents during repolarization. In cardiac myocytes, RPR260243 can reverse the effects of dofetilide on the action potential. In the intact heart, the drug shortens the QT interval and enhances T-wave amplitude on the ECG. It is therefore possible that HERG channel activators could also someday find use in the treatment of repolarization disorders in the heart, including acquired long QT syndrome, congenital long QT syndrome, and heart failure.
Footnotes
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.104.006577.
-
ABBREVIATIONS: HERG, human ether-a-go-go-related gene; ECG, electrocardiogram; RPR260243, (3R,4R)-4-[3-(6-methoxy-quinolin-4-yl)-3-oxo-propyl]-1-[3-(2,3,5-trifluoro-phenyl)-prop-2-ynyl]-piperidine-3-carboxylic acid; DPI 201–106, (±)-4-[3-(4-diphenylmethyl-1-piperazinyl)-2-hydroxy propoxy]-1H-indole-2-carbonitrile; CL, confidence limit; CHO, Chinese hamster ovary; RMP, resting membrane potential; APA, action potential amplitude; APD, action potential duration; IKr, delayed rectifier current.
- Received August 24, 2004.
- Accepted November 17, 2004.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}