


Abstract
In rat hepatic C9 cells, angiotensin II (Ang II)-induced activation of angiotensin type 1 (AT1) receptors (AT1-Rs) stimulates extracellular signal-regulated kinase (ERK) 1/2 phosphorylation via transactivation of the endogenous epidermal growth factor (EGF) receptor (EGF-R) by a protein kinase C (PKC) δ/Src/Pyk2-dependent pathway. This leads to phosphorylation of the EGF-R as well as its subsequent internalization. On the other hand, EGF-induced activation of the EGF-R in C9 cells was found to cause phosphorylation of the AT1-R. This was prevented by selective inhibition of the intrinsic tyrosine kinase activity of the EGF-R by AG1478 [4-(3′-chloroanilino)-6,7-dimethoxy-quinazoline] and was reduced by inhibition of PKC and phosphoinositide 3-kinase. EGF-induced AT1-R phosphorylation was associated with a decrease in membrane-associated AT1-Rs and a reduced inositol phosphate response to Ang II. Agonist activation of endogenous AT1-Rs and EGF-Rs induced the formation of a multireceptor complex containing both the AT1-R and the transactivated EGF-R. The dependence of these responses on caveolin was indicated by the finding that cholesterol depletion of C9 cells abolished Ang II-induced inositol phosphate production, activation of Akt/PKB and ERK1/2, and AT1-R internalization. Confocal microscopy demonstrated that caveolin-1 was endogenously phosphorylated and was distributed on the plasma membrane in patches that undergo redistribution during Ang II stimulation. Agonist-induced phosphorylation and association of caveolin 1 with the AT1-R was observed, consistent with a scaffolding role of caveolin during transactivation of the EGF-R by Ang II. The EGF-induced AT1-R/caveolin association was abolished by AG1478, suggesting that activation of the EGF-R promotes the association of caveolin and the AT1-R.
G protein-coupled receptors (GPCRs) and receptor tyrosine kinases (RTKs) are two major receptor families that transduce extracellular stimuli into intracellular signaling and phenotypic responses, including cell growth and proliferation. The signals generated by GPCRs and RTKs were originally thought to be compartmentalized, with little or no cross-talk between receptors or sharing of their individual signaling pathways. However, GPCR-mediated transactivation of RTKs has proven to be an important factor in the regulation of cellular differentiation, growth and proliferation, in particular via activation of the ERK1/2 members of the mitogen-activated protein kinase family. This process has been demonstrated for GPCRs activated by adrenaline, angiotensin II (Ang II), bombesin, bradykinin, carbachol, endothelin-1, lysophosphatidic acid, and thrombin (Daub et al., 1996; Luttrell et al., 1999; Zwick et al., 1999; Maudsley et al., 2000; Ushio-Fukai et al., 2001; Shah and Catt, 2002). Furthermore, RTKs such as the EGF-R, platelet-derived growth factor receptor (PDGF-R), and insulin receptor have been found to exhibit cross-talk with GPCRs during signal initiation, propagation, and regulation (Medina et al., 2000; Doronin et al., 2002; García-Sáinz et al., 2004).
Ang II is a multifunctional octapeptide hormone that exerts major regulatory actions on the cardiovascular system and stimulates target cells in the heart, blood vessels, kidney, brain, and adrenal cortex via activation of the Gq/11 protein-coupled AT1-R (de Gasparo et al., 2000). In many of these tissues, activation of the AT1-R leads to cell growth and differentiation responses through downstream signaling mechanisms that include phospholipase C, protein kinase C (PKC), the Ras-Raf-MEK-ERK signaling pathway, and signal transducers and activators of transcription (STATs) (Eguchi et al., 1998; Shah et al., 2004). Many of the growth-related effects of Ang II are mediated by the transactivation of RTKs, including EGF-Rs and PDGF-Rs, that serve as scaffolds for several of the signaling molecules involved in Ang II-induced tyrosine kinase signaling pathways (Eguchi et al., 1998; Ushio-Fukai et al., 2001).
Hepatic clone 9 cells (C9 cells), which are derived from the normal rat liver and retain an epithelial phenotype, express endogenous AT1-Rs that are coupled to both Gq and Gi proteins and endogenous RTKs, such as the EGF-R, that activate the ERK kinase cascades (García-Sáinz et al., 1998; García-Caballero et al., 2001; Shah and Catt, 2002). Agonist activation of AT1-Rs in C9 cells induces ERK1/2 activation by a PKCδ-dependent mechanism that is mediated by the Src/Pyk2 complex and matrix metalloproteinase, with the subsequent release of heparin-binding EGF, which acts as an endogenous ligand and activates the EGF-R (Shah et al., 2004). These liver-derived cells provide a useful model in which to study Ang II-induced changes in cell growth and proliferation. Recent reports indicate that cholesterol-rich microdomains, such as caveolae and their structural caveolin proteins, have important roles in regulating transactivation of the EGF-R by Ang II in smooth muscle cells (Ushio-Fukai et al., 2001).
As noted above, several studies have demonstrated that cross-talk between RTKs and GPCRs can also occur in the opposite direction, and in some cases, RTKs exert negative modulation on the activity of GPCRs. Treatment with EGF and bFGF causes down-regulation of the AT1R (Nickenig and Murphy, 1994; Ullian et al., 1997) and consequent reduction of Ang II-induced InsP production (Ullian et al., 2004). In addition, activation of EGF-R, PDGF-R, and insulin receptor signaling initiates intracellular pathways that inhibit the actions of α1b-adrenergic receptors (α1b-ARs) by promoting their phosphorylation and decreasing their coupling to heterotrimeric G proteins (Medina et al., 2000; García-Sáinz et al., 2004). In the present study, both functional and physical interactions between the EGF-R and AT1-R were observed, as well as a requirement for cholesterol-rich microdomains in the spatial and temporal organization of Ang II signaling and regulation in C9 hepatic cells.
Materials and Methods
Materials. Recombinant human EGF was obtained from Invitrogen (Carlsbad, CA). Tissue culture media, fetal bovine serum, and the clone 9 (C9) rat liver epithelial cell line were from American Type Culture Collection (Manassas, VA), Ang II was from Peninsula Laboratories (Belmont, CA), and goat polyclonal anti-EGF-R antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-phospho-p44/42 ERK antibody was from Cell Signaling Technology (Beverly, MA). Mouse monoclonal anti-caveolin and anti-phospho-caveolin (P-Tyr14) antibodies were from BD Transduction Laboratories (Lexington, KY). Rabbit polyclonal anti-phospho-Akt (P-Ser472/473/474) was from BD Bioscience PharMingen (San Diego, CA). Secondary antibodies conjugated to horseradish peroxidase were from Kirkegaard and Perry Laboratories (Gaithersburg, MD). [myo-2-3H]inositol and 125I-EGF were from PerkinElmer Life and Analytical Sciences (Boston, MA).
Cell Culture. C9 cells were cultured in Ham's F-12 nutrient mixture, Kaighn's modification, supplemented with 10% fetal bovine serum, 100 μg/ml streptomycin, 100 U/ml penicillin, and 0.25 μg/ml amphotericin B at 37°C under a 95% air/5% CO2 atmosphere. For all studies, C9 cells were used between passages 3 and 12, when there is maximum expression of their endogenous AT1-Rs. Before each experiment, C9 cells were serum-deprived for a period of 24 h.
Western Blotting. C9 cells were grown in six-well plates in Ham's F-12 nutrient mixture, Kaighn's modification, supplemented with 10% fetal bovine serum. At 60 to 70% confluence, cells were serum-starved for 24 h before treatment at 37°C with selected agents. To determine ERK1/2 and Akt phosphorylation in whole-cell lysates, media were removed by aspiration, and the cells were washed once with ice-cold PBS. After lysis in 100 μl of Laemmli sample buffer, proteins were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Phosphorylated ERK1/2 and Akt were detected by specific antibodies as described previously (García-Caballero et al., 2001; Shah and Catt, 2002).
AT1-R and EGF-R Phosphorylation Assay. Confluent C9 cells cultured in 10-cm Petri dishes were serum-deprived for 24 h before metabolic labeling for 4 h at 37°C in phosphate-free Dulbecco's modified Eagle's medium containing 100 μCi/ml [32P]Pi. After three washes in Krebs-Ringer-HEPES buffer, cells were incubated in the same medium for 10 min in a 37°C water bath and stimulated with vehicle, Ang II (100 nM), or EGF (1 ng/ml) for an additional 5 min. This time was selected on the basis of preliminary experiments. When required, specific PKC or phosphatidylinositol 3-kinase (PI3K) inhibitors were added for 30 min at 37°C before agonist treatment. In Ang II-stimulated cells, AT1-R phosphorylation was determined as described previously (García-Caballero et al., 2001). In EGF-R phosphorylation experiments, the plates were washed with ice-cold PBS and drained before scraping the cells into radioimmunoprecipitation assay lysis buffer containing protease and phosphatase inhibitors (García-Caballero et al., 2001), then probe-sonicated for 40 s. After removal of nonsolubilized material by centrifugation, the cell lysate was incubated with 2% (v/v) protein G plus/protein A-agarose for 1 h at 4°C. The precleared supernatant was incubated overnight at 4°C with 5 μg of anti-EGF-R antibody and 2% (v/v) protein G plus/protein A-agarose. The agarose-bound immune complexes were washed with radioimmunoprecipitation assay buffer lacking protease inhibitors; Laemmli sample buffer was then added, and the mixtures were heated at 100°C for 5 min. The denatured complexes were resolved by SDS-PAGE on an 8 to 16% gradient resolving gel. Phosphorylated EGF-Rs were visualized with a Storm 860 PhosphorImager (Amersham Biosciences, Piscataway, NJ), and the images were exported to the graphs. The level of receptor phosphorylation was assessed in the gels with ImageQuant software.
[3H]Inositol Phosphate Measurements. C9 cells cultured in 24-well plates were labeled by overnight incubation in inositol-free Dulbecco's modified Eagle's medium containing 0.1% (w/v) bovine serum albumin, antibiotics, and 20 μCi/ml [2-myo-3H]inositol. After washing and preincubation with 10 mM LiCl for 30 min, 100 nM Ang II was added for a further 30 min. InsP were extracted as described previously (Olivares-Reyes et al., 2001) and applied to AG 1-X8 columns (Bio-Rad Laboratories, Hercules, CA). After washing three times with water and twice with 0.2 M ammonium formate/0.1 M formic acid, the combined [3H]InsP2 + [3H]InsP3 fractions were eluted with 1 M ammonium formate/0.1 M formic acid, and radioactivities were determined by liquid scintillation spectrometry. Desensitization experiments were performed as described previously (Olivares-Reyes et al., 2001). Labeled C9 cells were preincubated in medium alone (time 0) or with 10 ng/ml EGF for the indicated times and washed with PBS before treatment with 150 mM NaCl, and 50 mM glycine, pH 3, for 30 s at 4°C. After washing three times with ice-cold PBS, the cells were incubated for 15 min in medium containing 10 mM LiCl with 100 nM Ang II. [3H]Inositol phosphates were extracted and measured as described above.
AT1-R and EGF-R Internalization Assays. AT1Rs were measured by addition of 125I-Ang II in serum-free Medium 199 containing 1 mg/ml bovine serum albumin to C9 cells in 24-well plates maintained at 37°C for the required times. Incubations were stopped by rapid washing with ice-cold PBS, and acid-released and acid-resistant radioactivities were separated and measured by γ-spectrometry as described previously (Olivares-Reyes et al., 2001). The percentage of internalized ligand at each time point was calculated from the ratio of the acid-resistant specific binding to the total (acid-released + acid-resistant) specific binding. Nonspecific binding was determined in the presence of an excess (10 μM) of unlabeled [Sar1,Ile8]Ang II. The effect of EGF on AT1-R internalization was examined by adding 10 ng/ml EGF to C9 cells for the indicated times at 37°C, followed by radioligand binding assay (5 h at 4°C) to measure AT1 receptors remaining at the cell surface. Internalized receptors were expressed as a percentage of cell surface binding compared with levels in cells not exposed to EGF. Similar experiments were performed to examine the effect of 100 nM Ang II on EGF-R internalization. After stimulation for the indicated times at 37°C, radioligand binding assay was performed with 125I-EGF (5 h at 4°C) to measure EGF receptors remaining at the cell surface.
Immunocytochemistry. C9 cells were plated at low density (100-500 cells) on poly-l-lysine-coated glass coverslips for 24 h, then rinsed and incubated in serum-free medium for 24 h. Cells were then stimulated with 200 nM Ang II for 0, 15, or 60 min. After stimulation, cells were rinsed in warm PBS and fixed in 3.7% formaldehyde for 15 min. Cells were again washed with PBS and permeabilized by immersing the coverslips in 100% methanol for 2 min. C9 cells were then incubated at 37°C in blocking medium (3% bovine serum albumin and 3% normal goat serum) for 60 min, followed by incubation in anti-phospho-caveolin antibody diluted 1:50 or anti-EGF-R antibody diluted 1:200 in blocking medium. After incubation with primary antibody, the cover slips were washed in PBS (three times for 5 min each) and incubated with Texas Red-labeled goat anti-mouse antibody diluted in blocking medium. After the final incubation, cells were gently washed with PBS (three times), mounted in ProLong mounting medium (Molecular Probes, Eugene, OR), and analyzed with a Bio-Rad confocal microscope.
Coimmunoprecipitation Assay for AT1-R and Associated Proteins. C9 cells grown in 10-cm dishes were serum-starved for 16 h and treated with agonists (100 nM Ang II or 1-10 ng/ml EGF) for the indicated times at 37°C. Cells were washed twice with ice-cold DPBS and lysed in Nonidet P-40 solubilization buffer (25 mM NaCl, 50 mM HEPES, 0.5% Nonidet P-40, 10% glycerol, 2 mM EDTA, pH 8.0, containing protease and phosphatase inhibitors) (García-Caballero et al., 2001). After immunoprecipitation of AT1-Rs as described, the proteins were resolved by SDS-PAGE, Western-blotted, and probed with EGF-R, caveolin, or phospho-caveolin antibodies, followed by a horseradish peroxidase conjugate to identify coimmunoprecipitated proteins. Blots were developed and immunoreactive bands were visualized using enhanced chemiluminescence reagent and autoradiography, and blots were quantified in an imaging densitometer (GS-700; Bio-Rad) using Molecular Analyst software. The procedure for immunoprecipitation of endogenous AT1-Rs in C9 cells was modified to include a cross-linking step for coimmunoprecipitation of AT1 and EGF-Rs using the reversible cross-linker dithiobis(succinimidyl propionate) (DSP).
All data were analyzed and plotted using Prism version 4.00 for Windows (GraphPad Software, San Diego, CA). Statistical analysis between comparable groups used analysis of variance with Bonferroni's analysis and was performed with software included in GraphPad Prism.
Results
Agonist-Induced MAP Kinase Activation and EGF-R Phosphorylation and Regulation. As previously observed, stimulation of C9 cells with 100 nM Ang II led to a rapid and transient increase of ERK1/2 activity (Fig. 1A). Likewise, treatment with 10 ng/ml EGF caused rapid activation of ERK1/2 (Fig. 1B). Treatment with the EGF-R kinase inhibitor AG1478 abolished ERK1/2 activation induced by both Ang II and EGF (Fig. 1C). These data indicate that Ang II-induced ERK1/2 activation in C9 cells depends primarily on transactivation of the EGF-R. Such transactivation of RTKs by GPCRs is mediated by multiple signaling pathways that culminate in RTK phosphorylation, downstream signaling, and receptor endocytosis and regulation (Maudsley et al., 2000; Grewal et al., 2001). Consistent with these results, Ang II stimulated EGF-R phosphorylation in a concentration- and time-dependent manner (Fig. 2A). To determine the functional consequences of Ang II-induced EGF-R phosphorylation, internalization of the EGF-R was analyzed by radioligand binding assay with 125I-EGF. C9 cells were stimulated with 100 nM Ang II for 15 or 60 min at 37°C and then chilled on ice to terminate receptor trafficking. As shown in Fig. 2B, Ang II stimulation caused time-dependent internalization of EGF-R (∼70%) at 15 min that was sustained over the next 60 min. On confocal microscopy, in the absence of Ang II, the EGF-Rs were largely located at the plasma membrane and around the nucleus (Fig. 2C). However, after 15 min of stimulation with Ang II, a significant fraction of the receptors had translocated to intracellular vesicles, consistent with the binding data shown in Fig. 2B. After 60 min, the cytoplasmic staining was reduced, consistent with degradation of the internalized receptors and partial recycling to the region of the cell membrane.
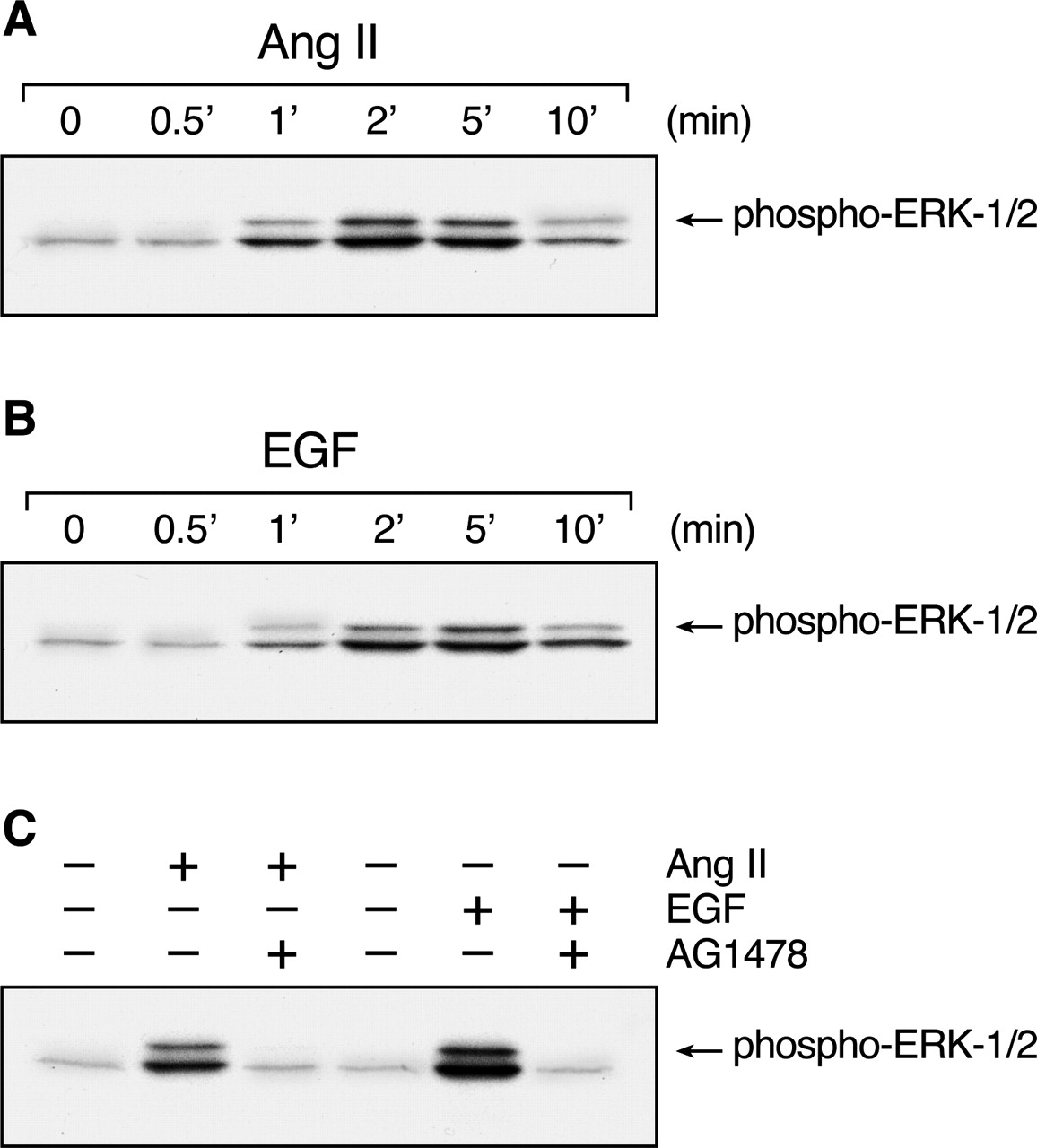
Ang II-induced MAP kinase activation in C9 cells. C9 cells were treated with 100 nM Ang II (A) or 10 ng/ml EGF (B) for the indicated times, washed twice with ice-cold PBS, and collected in Laemmli sample buffer for analysis and immunoblotting with anti-phospho-ERK1/2 antibody. The effects of EGF-R kinase inhibition by 100 nM AG1478 on ERK activation by 100 nM Ang II and 10 ng/ml EGF are shown in C. Cells were treated with AG1478 for 20 min before stimulation. The data shown are representative of two independent experiments.
Effects of EGF on Phosphorylation and Regulation of AT1Rs. Many studies on GPCR/RTK cross-talk have addressed the mechanism and role of transactivation of RTKs in the mediation and regulation of promitogenic pathways stimulated by GPCRs (Luttrell et al., 1999; Maudsley et al., 2000; Shah and Catt, 2002). However, recent evidence has indicated that RTKs can also act as regulators of GPCR signaling (Medina et al., 2000; Casas-González et al., 2003; García-Sáinz et al., 2004). In C9 cells, EGF induced phosphorylation of the AT1-R in a dose-dependent manner by 70 to 80% over basal (Fig. 3A), reaching a maximum at 5 to 15 min and decreasing over the next 45 min (Fig. 3B). Furthermore, inhibition of EGF-R kinase by treatment of C9 cells with AG1478 reduced EGF-induced AT1-R phosphorylation by ∼70% (Fig. 3C).
PI3K is an important regulator of the cellular processes activated by EGF and insulin. In Rat-1 cells, EGF causes phosphorylation of the α1b-AR through a PI3K- and PKC-dependent pathway (Medina et al., 2000). Moreover, previous studies have demonstrated PI3K-dependent activation of PKC (Wymann and Pirola, 1998). Wortmannin (WT) and LY294002 (LY) are two selective PI3K inhibitors with different mechanisms of action. WT is a potent noncompetitive inhibitor of PI3K, whereas LY competes at the kinase binding site for ATP. Pretreatment of C9 cells with these agents reduced the EGF-induced AT1-R phosphorylation by ∼50% (Fig. 3C).
EGF has been shown to stimulate the phosphoinositide/Ca2+ signaling pathway through activation of phospholipase C-γ (Todderud et al., 1990), which can lead to activation of PKC. To evaluate the ability of EGF to stimulate InsP production, [2-myo-3H]inositol-labeled C9 cells were treated with 10 ng/ml EGF for the indicated times, and inositol phosphates were extracted and measured. This revealed that EGF caused a time-dependent increase in InsP production that reached a peak at 15 to 30 min (40% increase) and began to decline at 45 min (Fig. 4A). To evaluate the role of PKC in EGF-induced AT1-R phosphorylation, we examined the effect of the specific PKC inhibitor bisindolylmaleimide I (BIM). As shown in Fig. 3C, treatment of C9 cells with BIM reduced the effect of EGF on AT1-R phosphorylation by 50%.
The functional consequences of EGF-induced AT1-R phosphorylation were determined by analyzing AT1-R internalization and Ang II-induced InsP signaling responses. In C9 cells stimulated with EGF (10 ng/ml) for up to 45 min at 37°C, EGF stimulation caused modest internalization of AT1-R that reached a maximum of 30% at 15 min and was sustained over the next 45 min (Fig. 4B). Evaluation of the effect of EGF treatment on AT1-R signaling revealed a comparable reduction in the maximal Ang II-stimulated InsP production (Fig. 4C).

Ang II-induced transactivation of the EGF-R in C9 cells. A, cells labeled with [32P]Pi for 4 h were exposed to the indicated concentrations of Ang II for 5 min. After solubilization, EGF-Rs were immunoprecipitated and resolved by SDS-PAGE (right). The time dependence of Ang II-stimulated EGF-R phosphorylation is shown on the left. Results are expressed as the percentage of EGF-R basal phosphorylation. Mean values are plotted, and vertical lines represent the S.E.M. for three independent experiments, of which representative autoradiographs are shown. B and C, cellular distribution of EGF-R after pre-exposure to Ang II. B, internalization of EGF-Rs in response to 100 nM Ang II for 15 or 60 min at 37°C was determined by measuring cell surface receptor density (125I-EGF binding, 5 h at 4°C). The means are plotted, and vertical lines represent the S.E.M. of three to four determinations using different cell preparations. *, P < 0.01 versus control. C, C9 cells were plated at low density (100-500 cells) on poly-l-lysine-coated glass coverslips for 24 h, then rinsed and incubated in serum-free medium for 12 to 24 h. The cells were then incubated without or with 200 nM Ang II for the indicated times. EGF-R distribution was determined by immunocytochemistry as described under Materials and Methods. Representative examples of three independent experiments are shown.
Coimmunoprecipitation of AT1 and EGF receptors. To determine whether the reciprocal AT1-R/EGF-R cross-talk observed in agonist-stimulated C9 cells reflects the formation of a protein complex involving the AT1-R and the EGF-R, we analyzed AT1-R immunoprecipitates for the presence of coprecipitated EGF-Rs. As shown in Fig. 5, EGF-Rs were present in AT1-R immunoprecipitates prepared from C9 cells. Furthermore, stimulation by both Ang II and EGF increased the amount of EGF-R in AT1-R immunoprecipitates in a time-dependent manner (Fig. 5, A and B). Under our experimental conditions, there was rapid association between the AT1 and EGF-Rs during agonist stimulation. Stabilization of the AT1-R-EGF-R complex before immunoprecipitation by covalent, thiol-sensitive cross-linking with DSP considerably increased the efficacy of this procedure (Fig. 5C).
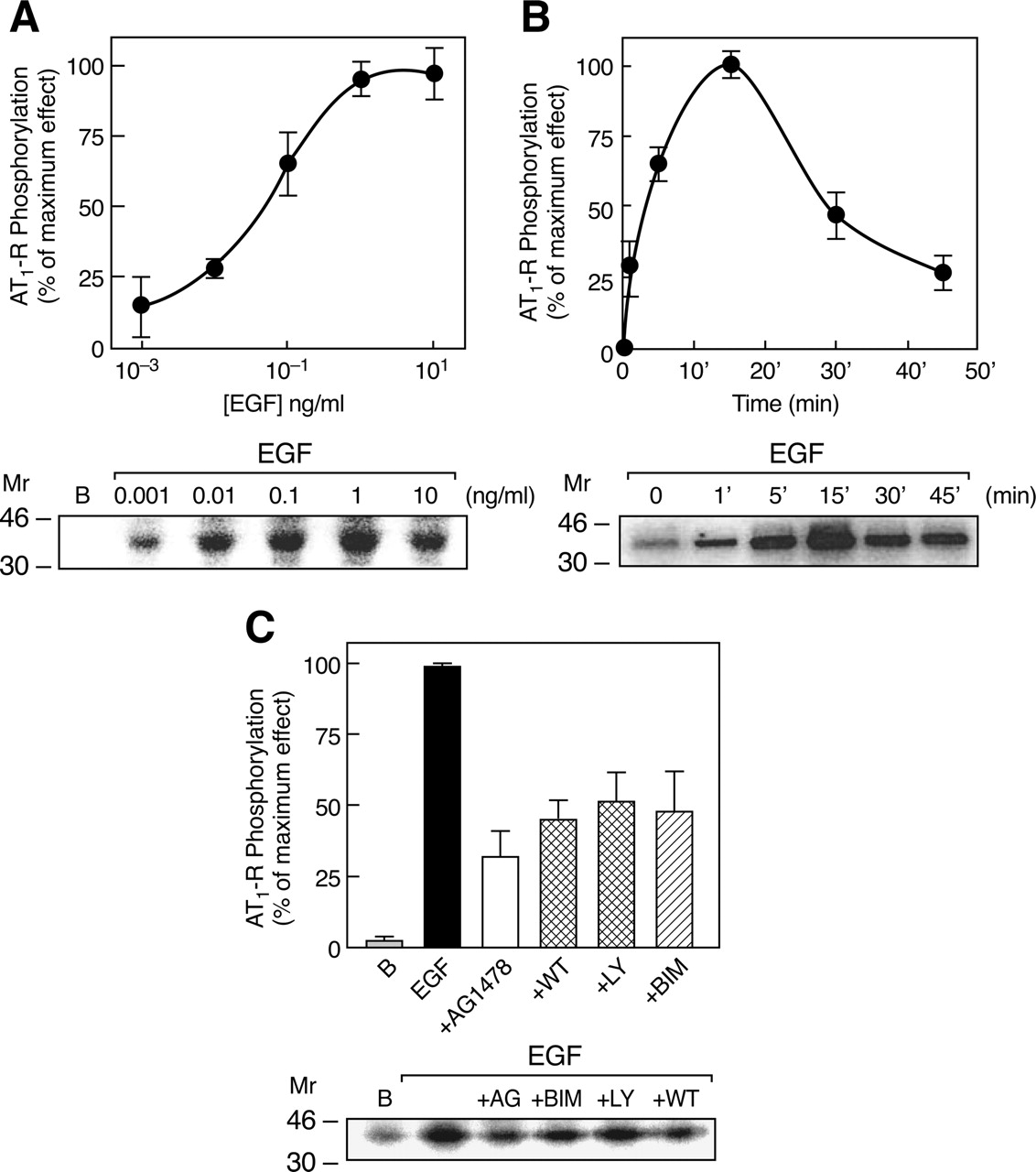
EGF-induced phosphorylation of the AT1-R. A, C9 cells were labeled with [32P]Pi for 4 h before addition of the indicated concentrations of EGF for 5 min. AT1-Rs were immunoprecipitated from the solubilized membranes by a specific anti-AT-R antibody and resolved by SDS-PAGE. B, time course of EGF-stimulated AT1-R phosphorylation. C, C9 cells were preincubated for 30 min without or with 5 μM AG1478, 100 nM WT, 1 μM LY, or 1 μM BIM and then stimulated with 1 ng/ml EGF. Data are expressed as percentage of maximum AT1-R phosphorylation during EGF stimulation. Representative autoradiographs are shown on the right. The means are plotted, and vertical lines represent the S.E.M. of three to four determinations using different cell preparations.

Effects of EGF treatment on AT1-R internalization and Ang II-induced [3H]InsP production. A, effect of EGF on InsP production. C9 cells were incubated for the time indicated with 10 ng/ml EFG. [3H]InsP-labeled C9 cells were preincubated with 10 mM LiCl before the addition of 10 ng/ml EGF for the indicated times. [3H]Inositol phosphates were extracted and measured as described under Materials and Methods. The means and vertical lines represent the S.E.M. of three independent experiments performed with different cell preparations. B, internalization of AT1-Rs in response to 10 ng/ml EGF for the indicated time periods at 37°C was determined by measuring cell-surface receptor density (125I-Ang II binding, 5 h at 4°C). The means are plotted, and vertical lines represent the S.E.M. of three determinations using different cell preparations. *, P < 0.001 versus control (time 0). C, C9 cells were preincubated in medium alone (time 0) or with 10 ng/ml EGF for the indicated times and treated with 150 mM NaCl and 50 mM glycine, pH 3, for 30 s, and washed three times with PBS. The cells were then incubated for 15 min in medium containing 10 mM LiCl with 100 nM Ang II. [3H]InsP was extracted and measured. The data represent mean values (± S.E.) from four independent experiments. *, P < 0.05 versus control (time 0).
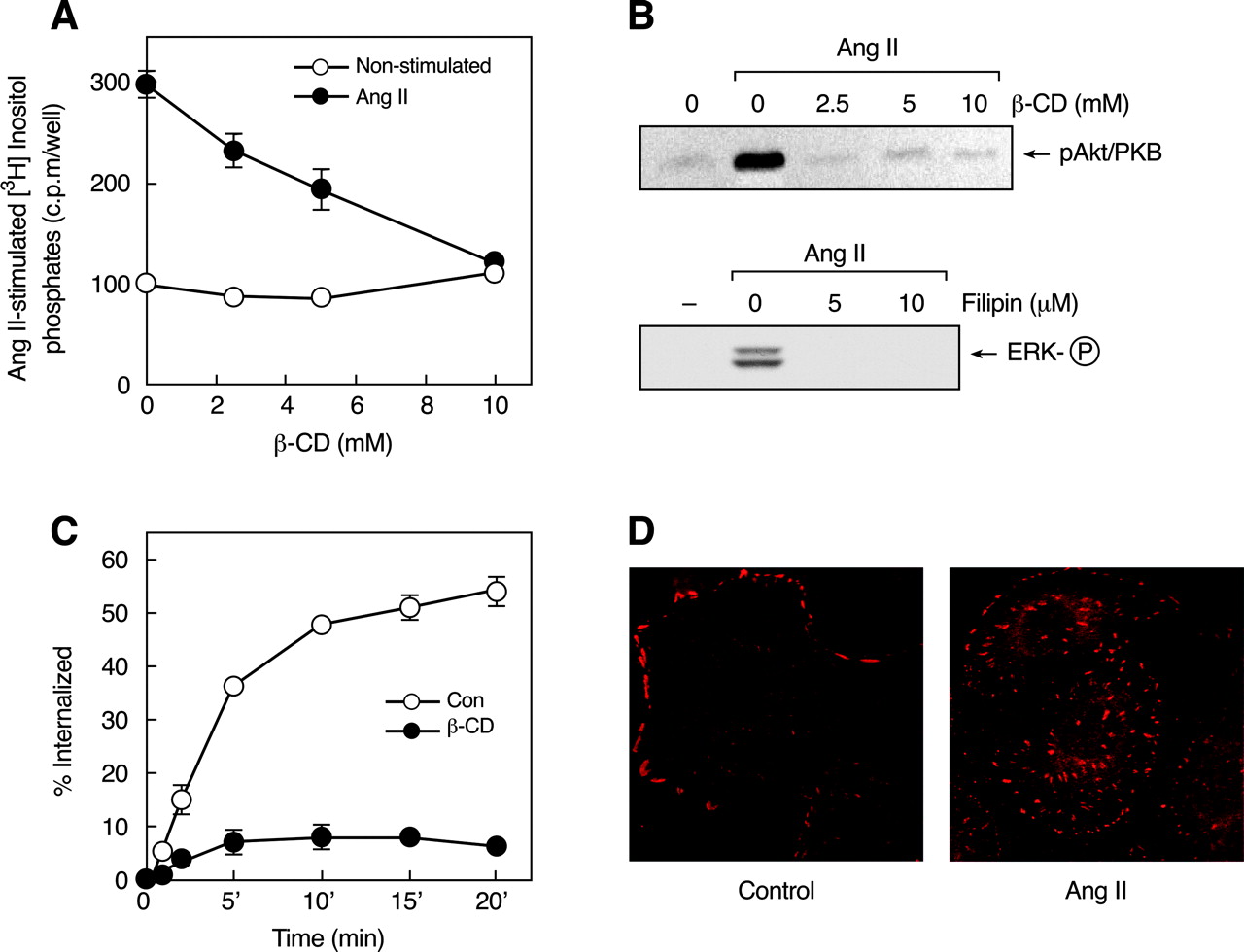
Role of Caveolin in Ang II-Induced Signaling. In rat hepatocytes, caveolae and caveolin have been shown to serve as functional microdomains for the internalization of cholera toxin B and retinal-binding protein (Calvo et al., 2001). Moreover, Ang II stimulates the movement of AT1-Rs into a caveolin-enriched membrane fraction and induces their association with caveolin in vascular smooth muscle cells (Ishizaka et al., 1998). Lipid rafts and caveolae contain high concentrations of sphingolipids and cholesterol, which contributes to the structural integrity of caveolae and their functional importance in signaling. Caveolae are disrupted by treatment with cholesterol binding agents such as β-cyclodextrin (β-CD), filipin, or nystatin, which extract cholesterol from the plasma membrane of intact cells (Yancey et al., 1996; Ushio-Fukai et al., 2001). Analysis of the role of caveolae in AT1-R signaling by treatment of C9 cells with increasing concentrations of β-CD before stimulation with Ang II (Fig. 6, A and B, top) revealed that coupling to inositol phosphate signaling and Akt/PKB activation was abolished. In addition, inhibitory effects on Ang II-induced ERK1/2 activation were observed after pretreatment of the cells with another cholesterol binding agent, filipin (Fig. 6B, bottom). The ability of these maneuvers to inhibit Akt/PKB and ERK1/2 activation indicates that Ang II-induced EGF-R transactivation in C9 cells requires intact cholesterol-rich microdomains.
We and others have shown previously that AT1-R internalization is a dynamin- and β-arrestin-dependent process and occurs primarily via clathrin-coated vesicles (Anborgh et al., 2000; Gaborik et al., 2001; Qian et al., 2001; Shah et al., 2002). However, recent reports have suggested an important role of noncoated pits, such as caveolae, in AT1-R internalization (Ishizaka et al., 1998; Ushio-Fukai et al., 2001). An examination of the effect of caveolar disruption on Ang II-induced AT1-R endocytosis in C9 cells (Fig. 6C) showed that receptor internalization was completely inhibited by β-CD treatment, consistent with the proposed role of caveolae in AT1-R internalization. Such inhibition of AT1-R internalization by β-CD may be attributable to the ability of cholesterol depletion to inhibit clathrin-mediated endocytosis through a physical restraint on membrane curvature (Subtil et al., 1999), an action that probably affects endocytosis of the AT1-R by both clathrin-coated vesicles and caveolae. Immunocytochemical/confocal microscopy studies (Fig. 6D) demonstrated that caveolin-1 is endogenously phosphorylated and is localized as clusters on the plasma membrane of C9 cells. During Ang II stimulation, phospho-caveolin is redistributed and seems to form aggregates within the cells, suggesting cointernalization of caveolae and AT1-Rs.
Agonist-Induced Phosphorylation and Association of Caveolin-1 with the AT1-R. Treatment of C9 cells with 100 nM Ang II (Fig. 7A) and 10 ng/ml EGF (Fig. 7B) induced phosphorylation of caveolin in a time-dependent manner, and stimulation with either agent caused significant association between the AT1-R and caveolin (Fig. 7C). These results also showed that the AT1-R is associated with caveolin after EGF stimulation, indicating its possible role in the regulation of receptor function and cellular localization. It is possible that ligand-dependent phosphorylation of the AT1-R may also modulate the activities of downstream signaling molecules, as well internalization of the receptor, but this has not been clearly demonstrated. However, we have recently observed that AT1-R phosphorylation by G protein-coupled receptor kinase may not be essential for Ang II-induced receptor desensitization and endocytosis (Olivares-Reyes et al., 2001). An evaluation of the role of EGF-induced phosphorylation of the AT1-R in its association with caveolin, by treatment with increasing concentrations of AG1478, revealed progressive inhibition of EGF-induced association of the AT1-R with caveolin (Fig. 7D).

Coimmunoprecipitation of AT1 and EGF receptors. C9 cells were incubated with 100 nM Ang II (A) or 10 ng/ml EGF (B) for the indicated times, and cell lysates were immunoprecipitated (IP) with anti-AT1-R antibody. After SDS-PAGE, polyvinylidene difluoride membranes were immunoblotted (IB) with anti-EGF-R antibody. Mean values are plotted, and vertical lines represent the S.E.M. for three independent experiments. Representative blots are presented. A, *, P < 0.01; **, P < 0.001 versus control (time 0). B, *, P < 0.05 versus control (time 0). C, cells treated without (-) or with (+) 100 nM Ang II for 5 min were incubated with 2.5 mM DSP cross-linker and solubilized. Cell lysates were immunoprecipitated with anti-AT1-R antibody before SDS-PAGE analysis and immunoblotted with anti-EGF-R antibody. Data are representative of three similar experiments.
Discussion
Hepatic C9 cells express endogenous AT1-Rs and EGF-Rs and provide a useful model for studies on the individual and interacting signaling mechanisms of GPCRs and RTKs (García-Sáinz et al., 1998; García-Caballero et al., 2001; Shah and Catt, 2002; Shah et al., 2002, 2004). Our investigation of the reciprocal cross-interaction between the endogenous AT1-Rs and EGF-Rs expressed in C9 cells revealed that whereas Ang II-induced ERK1/2 phosphorylation occurs via transactivation of the EGF-R, EGF stimulates phosphorylation of AT1-Rs through activation of PI3K and PKC. Moreover, immunoprecipitation studies revealed a physical association and functional interaction between these receptors, as well as with caveolin.
The PI3K family of enzymes has important roles in signaling and regulation of cellular activity (Wymann and Pirola, 1998) and has been implicated in GPCR-mediated ERK1/2 activation (Hawes et al., 1996; Smith et al., 1999) and other downstream signaling pathways from the GPCR-transactivated EGF-R. However, the role of PI3K in EGF-induced AT1-R phosphorylation has not been investigated. Although Ang II-induced transactivation of the EGF-R in C9 cells causes recruitment of several adaptor molecules, including Shc, Grb, and Sos, and PI3K/Akt, PI3K/Akt has no role in ERK1/2 activation by Ang II and EGF in these cells (Shah and Catt, 2002; Shah et al., 2004). In contrast, our observation that treatment with LY294002 and wortmannin impairs EGF-induced AT1-R phosphorylation indicates a role of PI3K in this process (Fig. 3). In C9 cells, Ang II causes EGF-R transactivation through PKC-dependent induction of metalloproteinase activity (Shah et al., 2004). However, EGF-induced downstream signaling, such as ERK1/2 phosphorylation, is PKC-independent (Shah and Catt, 2002). In contrast, the present study shows that EGF-induced AT1-R phosphorylation is significantly reduced by PKC inhibition (Fig. 3C). EGF is known to cause activation of PKC through PLCγ (Toker and Newton, 2000) and is also a potent activator of PI3K/Akt in C9 cells (Shah and Catt, 2002). Moreover, PI3K causes activation of several PKC isoforms (δ, ϵ, η, ζ, and λ) (Wymann and Pirola, 1998). Therefore, it is likely that EGF-induced AT1-R phosphorylation occurs through activation of the PI3K/PKC cascade in C9 cells. The mechanism of EGF action in C9 cells resembles that of lysophosphatidic acid, which induces phosphorylation of the α1B-adrenergic receptor through sequential activation of PI3K and PKC (Casas-González et al., 2003).
Caveolin-1, the major coat protein of caveolae, interacts directly with, and regulates the activity of, several caveolae-associated signaling proteins (Williams and Lisanti, 2004). In smooth muscle cells, cholesterol-rich microdomains, such as caveolae, and their structural protein, caveolin-1, have been implicated in transactivation of the EGF-R by Ang II (Ushio-Fukai et al., 2001). The AT1-R sequence contains a caveolin-scaffolding domain in its carboxyl-terminal tail region (amino acids 302-312) that could be required for its interaction with caveolin. We observed that Ang II and EGF caused rapid phosphorylation of caveolin at Tyr14 and that both agonists enhanced the interaction of AT1-R with caveolin. This receptor-caveolin interaction was attenuated by inhibition of EGF-R kinase activity (Fig. 7), suggesting that phosphotyrosine-dependent interactions might recruit proteins to these domains after GPCR and RTK stimulation. Caveolins are known to interact with a variety of downstream signaling molecules, including the EGF-R, Src, ERK1/2, and endothelial nitric-oxide synthase, and to maintain these signal transducers in an inactive conformation until their activation by appropriate stimuli. However, caveolins have also been proposed to facilitate Ang II-stimulated EGF-R signaling (Ushio-Fukai et al., 2001; Ringerike et al., 2002; Williams and Lisanti, 2004). In C9 cells, cholesterol depletion and caveolin disruption by β-CD and filipin abolished Ang II effects on ERK1/2 and Akt activation (Fig. 6B), indicating that caveolin could have a positive regulatory role in these cells.
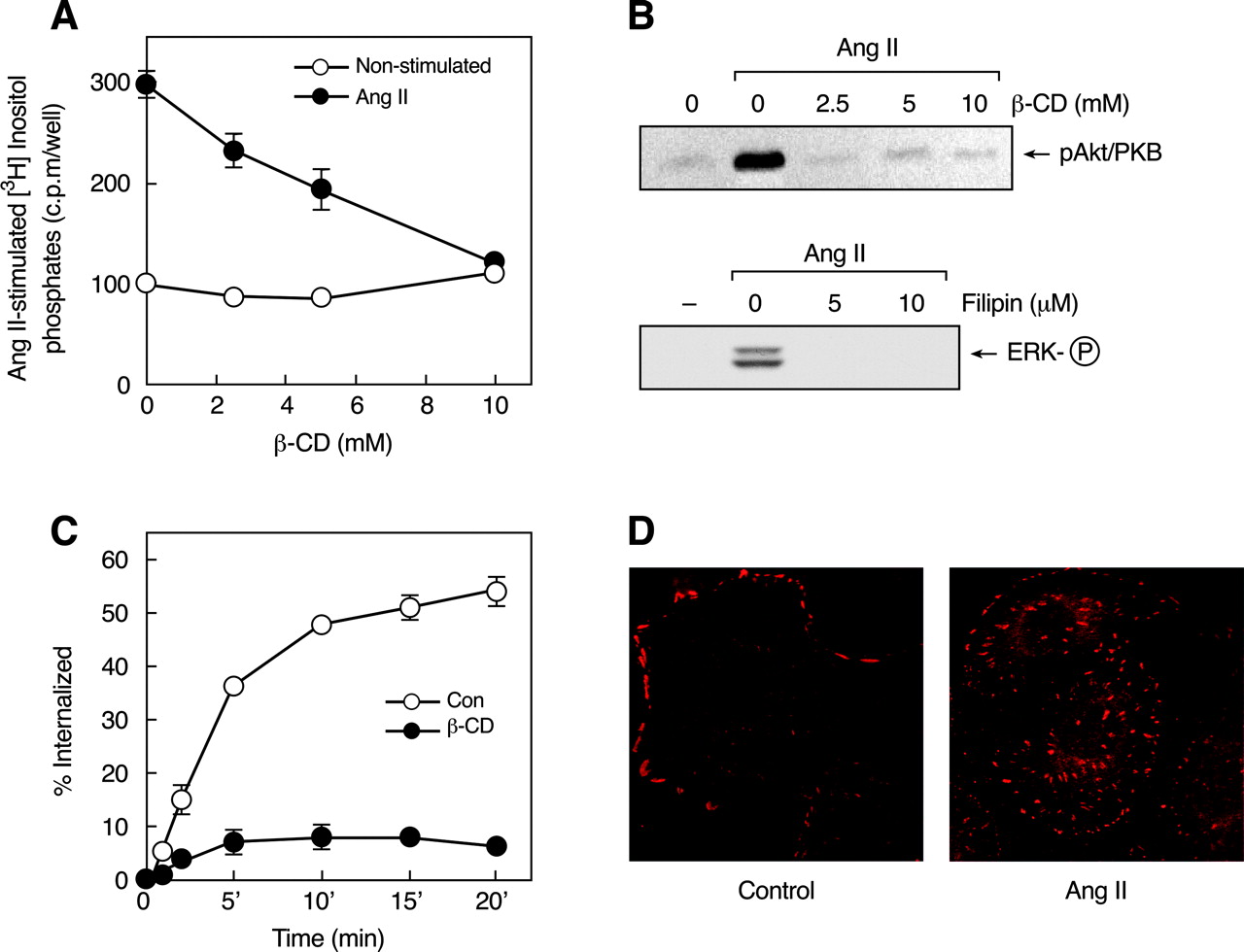
Role of caveolin in Ang II-induced signaling. A, effects of β-CD on Ang II-stimulated inositol phosphate production. [3H]Inositol-labeled C9 cells were preincubated with 10 mM LiCl and increasing concentrations of β-CD for 30 min before the addition of vehicle (○) or 100 nM Ang II (•) for an additional 30 min. [3H]Inositol phosphates were extracted and measured as described under Materials and Methods. Mean values are plotted, and vertical lines represent the S.E.M. for three independent experiments. B, activation of Akt and ERK1/2 in serum-deprived C9 cells incubated with β-CD and filipin, respectively, for 30 min and stimulated with 100 nM Ang II for 5 min. C, internalization of endogenous AT1-Rs. C9 cells preincubated without were with (○) or (•) 10 mM β-CD for 30 min, then incubated with 125I-Ang II at 37°C for the indicated times. Acid-resistant and acid-sensitive binding (counts per minute) were determined as described under Materials and Methods, and the specific internalized (acid-resistant) binding was expressed as a percentage of the total binding at each time point. Mean values are plotted, and vertical lines represent the S.E.M. for three independent experiments. D, cellular distribution of phospho-caveolin (Cav-PY). C9 cells were plated at low density (100-500 cells) on poly-l-lysine-coated glass coverslips for 24 h, then rinsed and incubated in serum-free medium for 12 to 24 h. The cells were then incubated without or with 200 nM Ang II for 15 min. Cav-PY distribution was determined by immunocytochemistry as described under Materials and Methods. The data in A and C represent mean ±S.E.M. from three similar experiments.
Our data show that the AT1-Rs and the EGF-Rs transiently interact with each other in a ligand-dependent manner and that the timing of their interaction coincides with that of EGF-induced AT1-R phosphorylation. The formation of such an agonist-induced multireceptor complex has also been observed in studies on β2-AR-mediated transactivation of the EGF-R in COS-7 cells (Maudsley et al., 2000). The immunoprecipitation data clearly demonstrate that AT1-Rs and EGF-Rs also colocalize with caveolin and that agonist stimulation further enhances this interaction (Figs. 5 and 7). It seems that ligand-induced movement of receptors into lipid rafts promotes their association with G proteins and effector enzymes, which are enriched in this compartment, and determine their cellular redistribution and internalization (Alderton et al., 2001; Chini and Parenti, 2004). Recent evidence suggests that many signaling molecules and GPCRs, including the α2-AR, AT1-R, sphingosine 1-phosphate EDG-1, m2 muscarinic, and bradykinin receptors, are localized in cholesterol-rich microdomains of the plasma membrane, particularly in lipid rafts and caveolae (Chini and Parenti, 2004). Such compartmentalization could provide the molecular proximity necessary for the rapid, efficient, and specific activation of downstream signaling events.
In general, agonist-induced phosphorylation of GPCRs leads to their internalization and subsequent desensitization of their signaling activity (Gaborik and Hunyady, 2004). Like several other GPCR agonists in receptor-transfected cells (Kim et al., 2003), Ang II stimulated phosphorylation and endocytosis of the endogenous EGF-R in C9 cells (Fig. 2). Endocytosis of the EGF-R is a complex process that involves both caveolae and clathrin-coated pits (Couet et al., 1997; Mineo et al., 1999; Shah et al., 2002), and the internalized receptors are either degraded by lysosomes or recycled to the plasma membrane (Gaborik and Hunyady, 2004). Studies in rat mesangial cells showed that 5-HT stimulation causes profound desensitization of EGF-R by a process associated with the loss of cell-surface EGF-Rs through clathrin-mediated endocytosis (Grewal et al., 2001). In C9 cells, EGF-R internalization likewise occurs through clathrin-mediated pathways (Shah et al., 2002) and EGF-R activation is also associated with caveolin phosphorylation (Fig. 7). It is of interest that EGF-induced endocytosis of the EGF-R is associated with much more rapid receptor degradation than that observed after Ang II stimulation in C9 cells (Shah et al., 2002).

Phosphorylation and association of caveolin-1 with the AT1-R. C9 cells were stimulated for the indicated times in the presence of 100 nM Ang II (A) or 10 ng/ml EGF (B). After washing twice with ice-cold PBS, cells were collected in Laemmli sample buffer, analyzed, and immunoblotted for Cav-PY. Cells were incubated without (C) or with increasing concentrations of AG1478 for 30 min (D) and then stimulated with 100 nM Ang II (C) or 10 ng/ml EGF for 5 min (C and D). Cell lysates were immunoprecipitated (IP) with anti-AT1-R antibody, resolved by SDS-PAGE, and immunoblotted (IB) for Cav-PY (C) or caveolin-1 (D). Data are representative of three similar experiments.
Although the mechanisms involved in agonist-induced GPCR phosphorylation and internalization have been extensively investigated (Gaborik and Hunyady, 2004), relatively little is known about EGF-stimulated GPCR phosphorylation and internalization. Our analysis of the effects of EGF-R activation on AT1-R function showed that EGF causes marked phosphorylation of the AT1-R and that approximately 30% of the AT1-Rs were internalized after EGF treatment (Fig. 4). In contrast, Ang II stimulation caused endocytosis of approximately 55% of the cell-surface AT1-Rs (Fig. 6). To determine the extent to which the signaling events associated with AT1-R phosphorylation are affected by EGF treatment, we measured InsP production in C9 cells, in which Ang II stimulates InsP production through Gq/PLCβ 1 (García-Sáinz et al., 1998). This showed that Ang II stimulation increased InsP production by up to 300%, compared with a 50% increase by EGF. There was a reduction of almost 15% in InsP production after treatment of cells with EGF for 30 to 45 min, followed by stimulation with Ang II for 15 min (Fig. 4). As might be expected, the degree of activation and phosphorylation/internalization of the AT1-R after direct binding of Ang II to its receptors substantially exceeds that mediated by intracellular signaling molecules (PI3K and PKC) after EGF stimulation.
These findings have demonstrated the existence of agonist-dependent physical interactions and reciprocal cross-talk between endogenous AT1-Rs and EGF-Rs in hepatic C9 cells. Whereas PI3K and PKC do not contribute to EGF-induced MAPK activation, these signaling molecules have significant roles in mediating AT1-R phosphorylation. Thus, EGF-R transactivation can contribute to Ang II-induced AT1-R phosphorylation in certain cell types. This process may be a significant determinant of the cellular responses to Ang II and EGF in other endogenous systems.
Acknowledgments
We thank Hiram Rangel-Sánchez and M. Antonio Morquecho-León for technical assistance.
Footnotes
-
This work was supported in part by grants from Consejo Nacional de Ciencia y Tecnología (CONACyT) (36230-N), Programa de Apoyo a Proyecto de Investigación e Inovación Tecnológica (IX247004 and N206302) (to J.A.G.-S.). J.A.O.-R. was supported in part by a grant from CONACyT (39485-Q).
-
ABBREVIATIONS: GPCR, G protein-coupled receptor; RTK, receptor tyrosine kinase; ERK, extracellular signal-regulated kinase; MAP, mitogen-activated protein; Ang II, angiotensin II; EGF, epidermal growth factor; EGF-R, epidermal growth factor receptor; PDGF, platelet derived growth factor; PDGF-R, platelet-derived growth factor receptor; AT1-R, type 1 angiotensin receptor; PKC, protein kinase C; AR, adrenergic receptor; PBS, phosphate-buffered saline; PAGE, polyacrylamide gel electrophoresis; PI3K, phosphoinositide 3-kinase; DSP, dithiobis(succinimidyl propionate); AG 1478, 4-(3′-chloroanilino)-6,7-dimethoxy-quinazoline; WT, wortmannin; LY, LY294002 (2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one); BIM, bisindolylmaleimide I; β-CD, β-cyclodextrin; InsP, inositol phosphate.
- Received December 21, 2004.
- Accepted May 17, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}