


Abstract
Inositol-1,4,5-trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) often coexist within the endoplasmic/sarcoplasmic reticulum (ER/SR) membrane and coordinate precise spatial and temporal coding of Ca2+ signals in most animal cells. Xestospongin C (XeC) was shown to selectively block IP3-induced Ca2+ release and IP3R-mediated signaling (Gafni et al., 1997). We have further studied the specificity of xestospongin structures possessing ring hydroxyl (-OH) substituents toward IP3R, RyR, and ER/SR Ca2+-ATPase (SERCA) activities. XeC potently inhibits IP3R, weakly inhibits RyR1, and lacks activity toward SERCA1 and SERCA2. XeD (9-OH XeC), 7-OH-XeA, and araguspongin C isolated from the marine sponge Xestospongia species also inhibit IP3-mediated Ca2+ release and lack activity toward SERCA. However, these hydroxylated derivatives possess a unique activity in that they enhance Ca2+-induced Ca2+ release from SR vesicles by a mechanism involving the sensitization of RyR1 channels within the same concentration range needed to block IP3-induced Ca2+ release. These results show that xestospongins and related structures lack direct SERCA inhibitory activity, as suggested by some previous studies. A new finding is that XeD and related structures possessing a hydroxylated oxaquinolizidine ring are IP3R blockers that also enhance Ca2+-induced Ca2+ release mediated by RyRs. In intact cells, the actions of XeD are blocked by ryanodine pretreatment and do not interfere with thapsigargin-mediated Ca2+ mobilization stemming from SERCA block. Hydroxylated bis-oxaquinolizadine derivatives isolated from Xestospongia species are novel bifunctional reagents that may be useful in ascertaining how IP3Rs and RyRs contribute to cell signaling.
Inositol-1,4,5-trisphosphate (IP3) is a universal secondary messenger. As a product of receptor-activated hydrolysis of phosphatidylinositol 4,5-bisphosphate, IP3 rapidly diffuses within cells activating IP3 receptors (IP3Rs) and release of Ca2+ stored in endoplasmic/sarcoplasmic reticulum (ER/SR). Activation of IP3R changes intracellular Ca2+ concentration, which can lead to the activation of RyRs, magnifying the effect of Ca2+ signaling (Endo et al., 1970; Streb et al., 1983; Berridge, 1993; Bootman and Berridge, 1995; Berridge et al., 2000). To study the Ca2+ signals orchestrated by activation of IP3Rs and RyRs, xestospongin C (XeC) has been widely used as a pharmacological tool to selectively block IP3R (Bootman et al., 2002; Verkhratsky, 2005).
Xestospongins are marine natural products that were first isolated from the Pacific sponge Xestospongia exigua and have been shown to have vasodilatory properties (Nakagawa and Endo, 1984). Gafni and coworkers (1997) discovered that XeC potently blocks IP3-induced Ca2+ release from cerebellar ER membranes without directly competing with IP3 at its binding sites on IP3Rs. Because XeC is membrane-permeable, it has been widely used to explore the role of IP3 signaling in intact cells (Bootman et al., 2002; Verkhratsky, 2005). However, among those studies, three reports have indicated that XeC may also potently block SR/ER Ca2+-ATPase (SERCA pumps) and therefore limit its usefulness (De Smet et al., 1999; Castonguay and Robitaille, 2002; Solovyova et al., 2002). These conclusions were determined from increases in intracellular Ca2+ observed in intact or permeabilized cell preparations subsequent to the addition of XeC, although no direct measure of SERCA activity or SR/ER transport function was made to establish mechanism. Paradoxically, other studies that directly measured SERCA activity in isolated ER/SR membrane fractions have indicated that neither XeC (Haak et al., 2001) nor XeB (Jaimovich et al., 2005) inhibits tharpsigargin (TG)-sensitive Ca2+-ATPase.
In the present study, we examined the activity of XeC and several structurally related natural products isolated from Xestospongia species for their activity toward IP3R, SERCA, and RyR. We found that neither XeC nor several hydroxylated derivatives (≤50 μM) inhibited TG-sensitive Ca2+-ATPase rates nor interfered with TG's ability to inhibit SERCA activity. A new finding was that hydroxylated structures including XeD, ArC, and 7-OH-XeA, although potent inhibitors of IP3-induced Ca2+ release, also sensitized RyR1-mediated Ca2+-induced Ca2+ release (CICR). Hydroxylated xestospongins, therefore, provide cell-permeable bifunctional pharmacological tools to effectively block IP3R and amplify CICR. These findings may also explain the Ca2+-mobilizing activity of xestospongin preparations reported in a few studies (De Smet et al., 1999; Castonguay and Robitaille, 2002; Solovyova et al., 2002).
Materials and Methods
Materials. [3H]Ryanodine was purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Xestospongins were isolated and purified from Xestospongia species as described previously (Gafni et al., 1997; Moon et al., 2002).
Membrane Preparations. Microsomes enriched in IP3R were isolated from the cerebellum of male Sprague-Dawley rats as described previously (Gafni et al., 1997). In brief, dissected cerebella were homogenized with ice-cold buffer consisting of 5 mM HEPES, 320 mM sucrose, 100 μM phenylmethylsulfonyl fluoride, and 10 μg/ml leupeptin at pH 7.4. The microsomal fraction was isolated by differential centrifugation, resuspended in a buffer containing 5 mM HEPES and 320 mM sucrose at pH 7.4, portioned in 100-μl aliquots, rapidly frozen in liquid nitrogen, and stored at –80°C until thawed for experimentation. Junctional SR membrane vesicles enriched in the skeletal isoform of ryanodine receptor, RyR1, were prepared from fast-twitch skeletal muscle obtained from 3- to 4-kg male New Zealand White rabbits according to a method described previously (Saito et al., 1984; Pessah et al., 1986). Porcine cardiac SR membrane vesicles were enriched in the type 2 isoform of ryanodine receptor, RyR2, as described previously for rat preparations (Pessah et al., 1990; Cherednichenko et al., 2004). The Lowry method was used to determine the protein concentrations with a bovine serum albumin standard curve (Lowry et al., 1951).
IP3-Triggered Ca2+ Release. The rate of calcium release from the intravesicular of rat cerebellar microsomal membrane was measured as a function of changes in absorbance of the metallochromic dye Antipyrylazo III (APIII) over time using the diode arrays spectrophotometer. Cerebellar microsomes were incubated with Ca2+ for 20 h at 4°C in transport buffer as described previously (Gafni et al., 1997). Calcium-loaded microsomes were then diluted 1:3 in Ca2+ transport buffer to a final volume of 1.2 ml/assay/cuvette. To this solution, aliquots of XeC, XeD, 7-OH-XeA, or ArC were added to the cuvettes from 100-fold concentrated stocks dissolved in methanol. Assays were performed at 37°C with constant stirring. IP3-induced Ca2+ release was initiated by the addition of 5 μM d-IP3, and the initial rates of release were compared with methanol solvent control.
Calcium-Induced Calcium Release. The rate of Ca2+-induced calcium release (CICR) from junctional SR membrane vesicles enriched in the skeletal isoform of ryanodine receptor, RyR1, was measured in transport buffer containing the Ca2+-sensitive dye APIII. Skeletal SR vesicles (60 μg/ml) were preincubated in XeD (0–15 μM) for 2 min in 1.4 ml of ATP-regenerating buffer consisting of 92.5 mM KCl, 18.5 mM potassium MOPS, 7.5 mM sodium pyrophosphate, 0.250 mM APIII, 20 μg/ml creatine phosphokinase, 0.005 mM phosphocreatine, and 1 mM MgATP, pH 7.0 (final volume, 1.5 ml). Measurements were performed at 37°C with constant stirring. SR vesicles were loaded with four sequential additions of 45 nmol CaCl2 that constituted approximately 80% of their total (100%) loading capacity (2.605 ± 0.19 mmol Ca2+/mg protein). The rate of Ca2+ flux across SR vesicles upon bolus addition of 100 nmol (67 μM) of CaCl2 to induce Ca2+ release was measured by obtaining the liner slope of extravesicular changes in free Ca2+. At the end of each experiment, the total intravesicular Ca2+ was determined by the addition of 3 μM concentration of the Ca2+ ionophore A23187, and the absorbance signals were calibrated by the addition of 15 nmol CaCl2 from a National Bureau of Standard stock solution. The dose-response relationship between rates of CICR and XeD were fitted with a sigmoidal equation using Origin software (OriginLab Corp, Northampton, MA).
[3H]Ryanodine Binding Analysis. Equilibrium measurements of specific high-affinity [3H]ryanodine ([3H]Ry) binding were determined according to the method of Pessah et al. (1987). SR vesicles enriched in RyR1 were incubated in the absence and presence of test compound (100 nM to 50 μM) in binding assay buffer containing 250 mM KCl, 20 mM HEPES, pH 7.4, 0.001 mM CaCl2, and 1 nM [3H]Ry for 3 h at 37°C. The stock buffer contained 20 μM CaCl2, to which 27.4 μM EGTA was added to obtain the final free Ca2+ of 1 μM, calculated using the “Bound and Determined” software program. Nonspecific binding was determined by incubating SR vesicles with 1000-fold excess unlabeled ryanodine. The presence of xestospongin that gave maximum binding did not alter nonspecific binding. Binding reactions were quenched by rapid filtration through Whatman GF/B glass fiber filters using a cell harvester (Brandel, Gaithersburg, MD). The filters were then washed twice with 3 ml of ice-cold harvest buffer consisting of 20 mM Tris, 250 mM KCl and 15 mM NaCl, and 0.05 mM CaCl2 at pH 7.1 and placed in 5 ml of scintillation cocktail (ScintiVerse; Fisher Scientific Co., Pittsburgh, PA) overnight. The radioactivity on each filter disk was measured by scintillation counting (model LS 6000IC; Beckman Coulter, Inc., Fullerton, CA). Specific [3H]Ry binding was determined by subtracting the nonspecific binding from the total binding. The IC50 and EC50 values and their respective S.D. of each test compound were analyzed by sigmoidal curve-fitting. Logit-log analysis was used to calculate the logit slope for inhibition.
Measurement of SERCA Activity. Activity of SERCA from skeletal (type 1 isoform) or cardiac (type 2 isoform) SR was measured using a coupled enzyme assay that monitors the rate of oxidation of NADH at 340 nm as described previously (Haak et al., 2001). In brief, assay buffer consisted of 7 mM HEPES, pH 7.0, 143 mM KCl, 7 mM MgCl2, 0.085 mM EGTA, 0.43 mM sucrose, 0.57 mM NADH, 0.0028 mM phosphoenolpyruvate, 24 μl of coupling enzyme mixture (700 units of pyruvate kinase II and 1000 units of lactate dehydrogenase), and 20 μg of SR protein. To measure SERCA type 2 isoform from cardiac SR preparations, rotenone (10 nM) was included in the assay medium to block the NADH oxidase activity associated with RyR2 (Cherednichenko et al., 2004). Once the baseline was established, 1 mM Na2ATP was added to initiate the reaction. Tharpsigargin (TG, 200 nM) was added to the negative control to measure the Ca2+-independent (non-SERCA) component of ATPase activity. Each experimental condition was repeated three to six times, and data represent the mean ± S.D.
Recordings of Single-Channel Current. Bilayers were composed of phosphatidylethanolamine/phosphatidylserine/phosphatidylcholine (5:3:2 w/w/w) dissolved in decane at a final concentration of 50 mg/ml across a 200-μm aperture on a polysulfone cup (Warner Instruments, Hamden, CT). Bilayer partitioned two chambers (cis and trans) with symmetric buffer solution of 500 mM CsCl, defined Ca2+, and 20 mM HEPES-Tris, pH 7.4. The cis chamber was where protein was added and was connected to the head stage input of an amplifier (Bilayer Clamp BC 525C; Warner Instruments). The trans chamber was virtually grounded. The purified RyR1 preparations were preincubated with FKBP12 at 3.4/4 μM before being reconstituted into bilayer lipid membrane (BLM). For incorporation of RyR1 channel into BLM by inducing SR vesicle fusion, 10:1 CsCl (cis/trans) gradient was used. Upon fusion of SR vesicle with bilayer, cis chamber was immediately perfused to halt further fusion. Single-channel gating was monitored and recorded at a holding potential of –40 mV (applied to the cis side). The amplified current signals, filtered at 1 kHz (Low-Pass Bessel Filter 8 Pole; Warner Instruments) were digitized and acquired at a sampling rate of 10 kHz (Digidata 1320A; Molecular Devices, Sunnyvale, CA). The channel Po open was calculated using pClamp software 9.0 (Molecular Devices) (Buck et al., 1992).
Calcium Imaging. Mouse primary myotubes were cultured and differentiated as described previously (Cherednichenko et al., 2004). Cells were loaded with 5 μM Fluo-4AM (Invitrogen, Carlsbad, CA) at 37°C with 5% CO2 for 20 min in loading buffer containing 0.5% bovine serum albumin. The cells were then washed three times with imaging buffer containing 125 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 25 mM HEPES, 6 mM dextrose, and 2 mM CaCl2 at pH 7.4. Myotubes were then transferred to a Nikon Diaphot microscope (Nikon, Tokyo, Japan), and Fluo-4 was excited at 494 nm using a random access monochrometer (Photon Technology International, Lawrenceville, NJ). Fluorescence emission was measured at 525 nm using a 40× quartz objective. Data were collected with an IC300 intensified charge-coupled device camera from four to eight individual cells and analyzed using ImageMaster software (Photon Technology International). XeD (10 μM) was prepared in imaging buffer 1 h before the experiment. Calcium mobilization in response to 2.5 mM caffeine stimulation was measured in the presence and absence of XeD.
Results
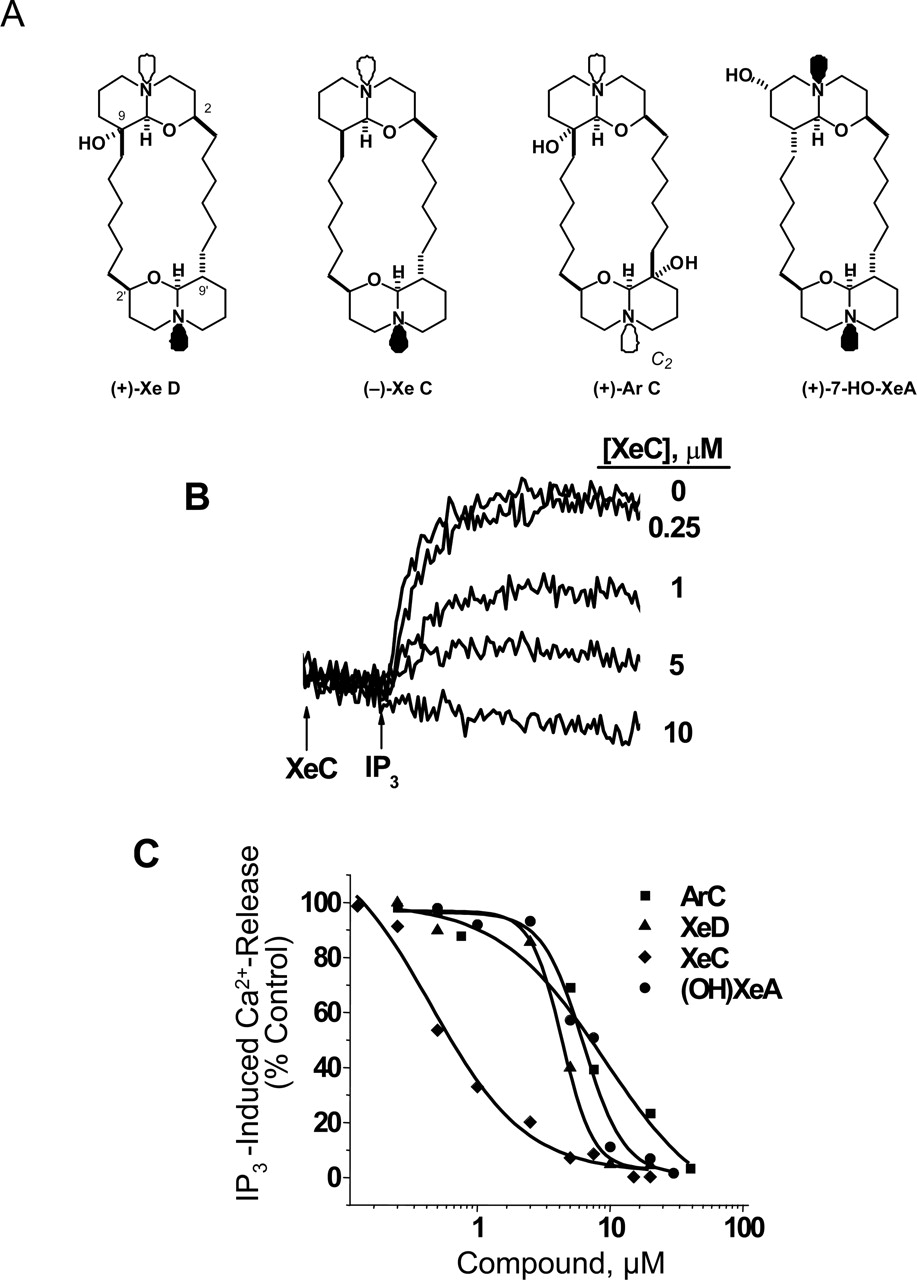
Isolation and Characterization of Xestospongins. Compounds were purified according to literature procedure, briefly summarized here. Xestospongia species (93-09-147) was collected at Benett's Shoal (Exmouth Gulf, Western Australia) at a depth of –10 m (January 1993) and kept frozen until required. The specimen was identified by Mary Kay Harper (University of Utah, Salt Lake City, UT). A preserved type sample is archived at the University of California, Davis (Davis, CA). The lyophilized sponge (107.3 g) was exhaustively extracted with MeOH (×3), and the combined MeOH extracts were concentrated. The water content (% v/v) of the MeOH extract was adjusted before sequentially partitioning against n-hexane (10% H2O), CHCl3 (40%), and n-BuOH (100%). The n-BuOH and aqueous MeOH extracts were combined and concentrated to give a reddish-brown oil, which was concentrated onto silica gel before loading onto the top of a silica column. Elution of the column [10% MeOH (saturated with NH3)/CHCl3] gave eight fractions. Fractions 1 and 2 were combined (929 mg) and triturated with EtOAc to produce a white solid, which was further purified by preparative HPLC (condition A, see below) to give (–)-XeC (32 mg) and (+)-XeD (38 mg) after crystallization of each from acetone. The filtrate was further separated by flash chromatography (Clark Still et al., 1978) under similar conditions to give nine fractions (f1–9). Preparative purification of compounds was carried out using normal-phase (silica) HPLC chromatography (5-μm particle, 25 × 300 mm), using two different conditions and differential refractive index detection. Condition A: 3:7 isopropanol/hexane/0.5% triethylamine, 12 ml/min; condition B: 1:9 isopropanol/hexane/0.5% triethylamine, 12 ml/min. Preparative HPLC (condition B) of combined f5 and f6 afforded (+)-XeA (31.3 mg) and additional (–)-XeC (40.3 mg). Additional quantities of (–)-XeC were obtained from f7 (34 mg) and f8 (26 mg) by HPLC. HPLC purification of f8 (condition A) also yielded pure (>99% purity) (+)-DMXeB (10 mg), (+)-ArC (12 mg), and (+)-7-hydroxyxestospongin A (7-OH-XeA, 12 mg) after crystallization from acetone (Moon et al., 2002). Low-resolution mass measurements were made on an LC Deca ion trap (Finnigan-Thermoquest, San Jose, CA). To further verify the purity of compounds identified, 1H NMR and 13C NMR were measured at 300 or 400 MHz and 75 and 100 MHz, respectively, on Mercury or Inova instruments (Varian, Inc., Palo Alto, CA). The compounds were identified by comparison with literature spectroscopic data (Nakagawa and Endo, 1984; Kobayashi et al., 1989) (Fig. 1A).
XeC and Its Derivatives Selectively Inhibit IP3R. It has been reported by Gafni et al. (1997) that XeC potently blocked IP3-induced Ca2+ release from cerebellar ER/SR membranes without directly competing with IP3 at its binding sites on IP3R. In the present study, we confirmed that new preparations of XeC isolated from Xestospongia species potently inhibited IP3-induced Ca2+ release with an IC50 of 458 ± 62 nM (Fig. 1, B and C). In the present study, we extended the structure-activity determination to three congeners possessing hydroxyl substituents. XeD, ArC, and 7-OH-XeA were all less potent inhibitors of IP3-induced Ca2+ release than XeC (IC50 values of 4.4 ± 0.31, 6.6 ± 0.45, and 6.4 ± 0.88 μM, respectively; Fig. 1C). The logit-log slopes for inhibition of IP3-induced Ca2+ release by XeC, XeD, 7-OH-XeA, and ArC were –0.89 ± 0.36, –0.77 ± 0.24, –1.13 ± 0.3, and –0.54 ± 0.07, respectively.

Inhibition of IP3R by XeC and hydroxylated derivatives. A, naturally occurring structures isolated from Xestospongin species and analyzed in the present study. B, original recordings show the inhibitory effect of XeC on IP3-induced calcium release from vesicles preincubated in a various XeD concentrations. C, structure-activity relationship for inhibition of IP3-induced Ca2+ release for four Xestospongin congeners. The amount of calcium released initiated by an addition of 5 μM IP3 to the cuvette in the presence or absence of the test compound was measured as a function of changes in APIII absorbance. XeC and its hydroxylated derivatives inhibit IP3-induced calcium release in a dose-dependent manner, and their IC50 values are reported in the text.
XeC and Hydroxylated Analogs Are Not Inhibitors of SR/ER Ca2+-ATPase. A few studies with intact or permeabilized cells have reported that XeC releases Ca2+ from stores in a manner similar to that elicited by TG and have concluded that XeC lacks selectivity by blocking both IP3R and SERCA (De Smet et al., 1999; Castonguay and Robitaille, 2002; Solovyova et al., 2002). We therefore tested whether XeC and hydroxylated congeners directly alter SERCA activity. Data summarized in Fig. 2A show that none of the congeners tested inhibited TG-sensitive SERCA1 activity at concentrations up to 50 μM. Moreover, Fig. 2B showed that the compounds tested did not interfere with the ability of TG to inhibit SERCA1 activity. The study was extended to SERCA2 isolated from cardiac muscle. Neither XeC nor the hydroxylated congeners were found capable of inhibiting SERCA2 activity at concentrations as high as 50 μM (Fig. 2D). Collectively, these data indicate that xestospongins lack direct inhibitory activity toward SERCA pumps, and they do not interfere with the TG binding site as suggested previously (Castonguay and Robitaille, 2002) at concentrations ranging from 10- to 100-fold of their IC50 value for block of IP3-induced Ca2+ release.

Xestospongins do not inhibit SERCA activity. Activity of SERCA from skeletal (type 1 isoform) or cardiac (type 2 isoform) SR was measured using a coupled enzyme assay (pyruvate kinase II; lactate dehydrogenase) that records the rate of oxidation of NADH at 340 nm in the absence and presence of 50 μM test compound as described under Materials and Methods. A, activity of SERCA1 was not affected by XeC or its derivatives. Addition of 200 nM TG fully inhibited ATPase activity. B, TG maintained its ability to block SERCA1 activity in the presence of xestospongins. C, summary data for SERCA1 rates (n = 6). D, summary data for SERCA2 rates (n = 3).
XeC Inhibits whereas Hydroxylated Derivatives Enhance [3H]Ry Receptor Binding. The interaction of XeC with RyR1 activity was examined in more detail by measuring its influence on the binding of [3H]Ry to skeletal muscle SR. Because [3H]Ry binds with high affinity to an open state of the RyR channel, occupancy of [3H]Ry-binding sites provides a convenient biochemical indicator of modulation of the open probability of the channel by exogenously added ligands (Pessah et al., 1986, 1987, 1990).
XeC was originally reported to inhibit RyR1 with an IC50 value ∼30-fold larger than the IC50 value for block of IP3-induced Ca2+ release (Gafni et al., 1997). The relative potency of XeC isolated from an independent source toward RyR1 inhibition was corroborated in the present study (XeC IC50 = 22.6 ± 2.2 μM, n = 8) (Fig. 3A). A new and somewhat unexpected finding was that unlike XeC, its hydroxylated derivative XeD (9-OH XeC; Fig. 3B), and related structures ArC and 7-OH-XeA (data not shown) enhanced [3H]Ry binding to RyR1 with an EC50 value of 10.6 ± 1.1 μM (n = 6) for XeD, 7.5 ± 0.9 μM for ArC (n = 4), and 11.4 ± 0.8 μM for 7-OH-XeA (n = 4). Because the amount of specific binding of [3H]Ry to high-affinity sites on RyR1 was shown to directly correlate with an enhanced open probability of the channel, these results suggested that one or more OH substitutions on the oxyquinolizidine ring(s) can promote the open conformation state of RyR1. This hypothesis was explored further using measurements of RyR1 single-channel function reconstituted in BLM.
XeD Enhances CICR and Channel Open Probability. How XeD influences RyR1 and SR Ca2+ transport was further studied by measuring its influence of CICR from actively loaded skeletal SR vesicles, and the result is shown in Fig. 4. SR vesicles were actively loaded with CaCl2 to approximately 80% of their total (100%) loading capacity (2.605 ± 0.19 mmol Ca2+/mg). Approximately 47.1% of the loaded amount is released by 100 nmol CaCl2 bolus addition. The rate of Ca2+ flux across SR vesicles (nmol Ca2+/min/mg protein) was measured by obtaining the liner slope of extravesicular changes in free Ca2+. The results showed that XeD increased the initial rate of CICR in a dose-dependent manner with an EC50 value of 7.4 ± 0.5 μM (n = 4; Fig. 4), consistent with a sensitizing action on CICR.
To confirm a direct interaction of XeD and RyR1, single channels were incorporated into planar lipid bilayers and tested for their response to XeD applied on the cytoplasmic side (cis). Figure 5 showed that XeD directly interacted with RyR1 in a dose-dependent manner to enhance channel Po. In the presence of 10 μM cytosolic Ca2+, the RyR1 channel exhibited moderately low activity, with a Po value of 0.03 (Fig. 5, panel 1). Upon introducing XeD (10 μM) into the cis chamber (cytoplasmic side), the channel Po rapidly increased 6-fold to 0.18 (panel 2). The channel-activating actions of XeD was primarily a result of prolonging open dwell time and shortening closed dwell times. Analysis of channel-gating kinetics revealed that the mean open dwell time doubled, whereas mean closed dwell time decreased approximately 3-fold (panel 6, summary table). The channel remained active with a similar Po for several minutes even after nominally increasing the cis Ca2+ concentration by the addition of 5 μM CaCl2 (Po = 0.16; panel 3). However, upon increasing the cis XeD to 25 μM, the gating activity of the channel was further enhanced 1.6-fold (Po increased to 0.28; panel 4), approximately a 9-fold increase compared with that in the absence of XeD. This XeD-induced channel activation was reflected as a 2-fold increase of open dwell time and a 5-fold decrease of closed dwell time compared with the initial control activity (panel 6, summary table). XeD-activated RyR1 channels remained responsive to ryanodine (100 μM), which locked the channel into a characteristic subconductance state (panel 5).

XeC inhibits whereas XeD enhances the [3H]Ry binding. Equilibrium measurements of specific high-affinity [3H]Ry binding to SR vesicles (50 μg/ml assay) enriched in RyR1 were carried out at 37°C for 3 h. The reaction mixture was incubated in the absence and presence of test compound (100 nM to 50 μM) in binding assay buffer containing 250 mM KCl, 20 mM HEPES, pH 7.4, 0.001 mM CaCl2, and 1 nM [3H]Ry. Non-specific binding was determined by incubating SR vesicles with 1000-fold excess of unlabeled ryanodine. XeC inhibits [3H]Ry binding to RyR1 with an IC50 value of 22.6 ± 2.2 μM (n = 8). In contrast, XeD enhances [3H]Ry binding to RyR1 with an EC50 value of 10.6 ± 1.1 μM (n = 6). Likewise, EC50 values for ArC and 7-OH-XeA are 7.5 ± 0.9 μM (n = 4) and 11.4 ± 0.8 μM(n = 4), respectively. Dose-response curves for ArC and 7-OH-XeA are not shown.
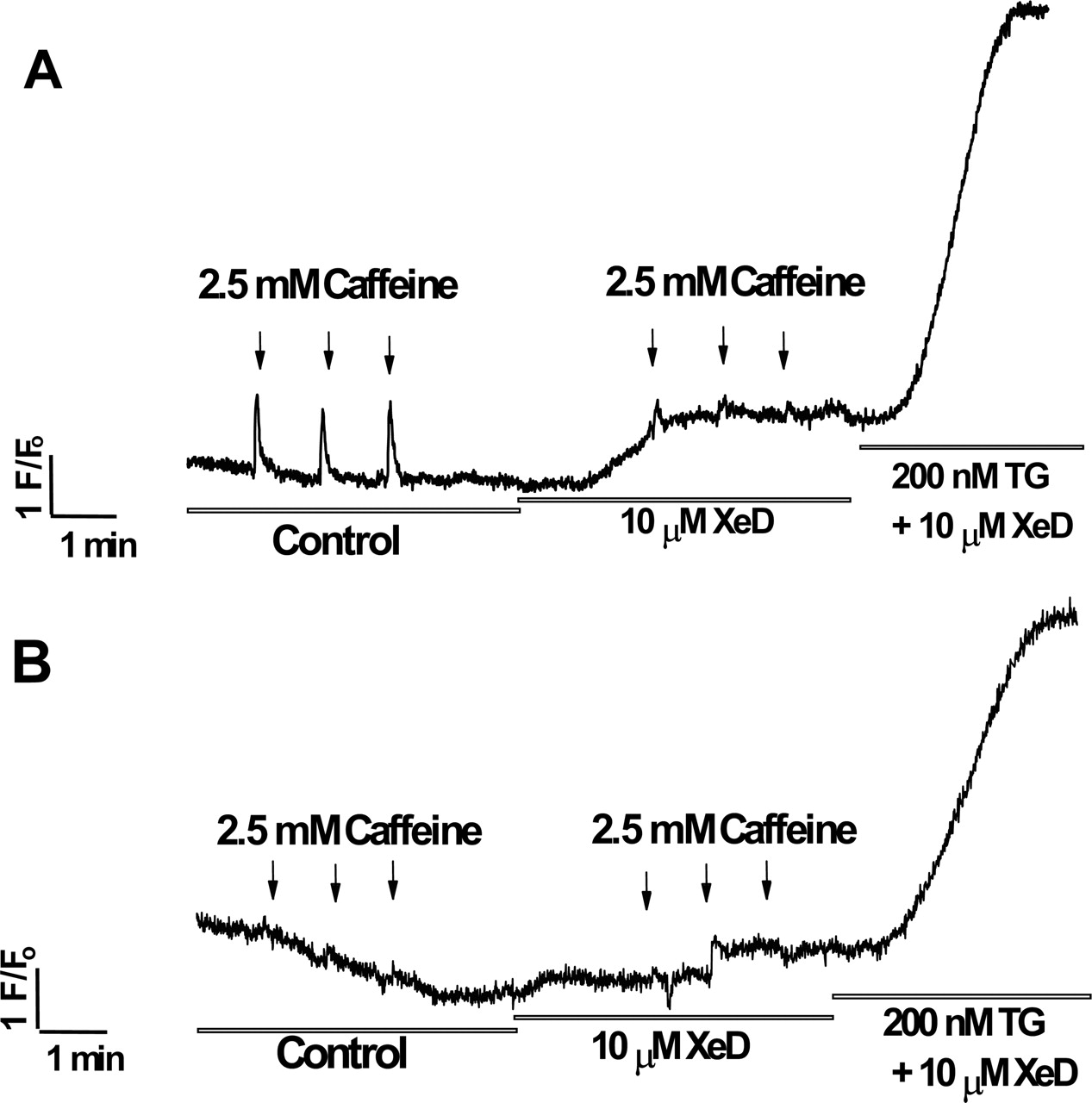
XeD Mobilizes Ca2+ from RyR-Sensitive Stores of Intact Myotubes. The specificity of XeD to activate RyR-mediated Ca2+ release was tested in intact murine myotubes using Ca2+ imaging of Fluo-4. Figure 6A shows that myotubes responded to repetitive application of 2.5 mM caffeine to the external medium, and the responses were of consistent magnitude and duration. Myotubes subsequently treated with 10 μM XeD in the medium responded with a transient increase in [Ca2+]o (Fig. 6A). During this phase, challenges with 2.5 mM caffeine produced Ca2+ transients of significantly reduced amplitude compared with those in the absence of XeD (Fig. 6A). High micromolar concentrations of ryanodine have been shown to persistently block RyR1 channels (Buck et al., 1992; Zimanyi et al., 1992). Myotubes pretreated with 500 μM ryanodine showed responses to neither 2.5 mM caffeine nor 10 μM XeD (Fig. 6B). To further test whether XeD inhibits SERCA in intact cells, 200 nM TG was added to the medium containing XeD in the presence or absence of ryanodine pretreatment. Regardless, TG caused a pronounced increase of intracellular Ca2+ resulting from both the release of calcium stores and store-operated Ca2+ entry.
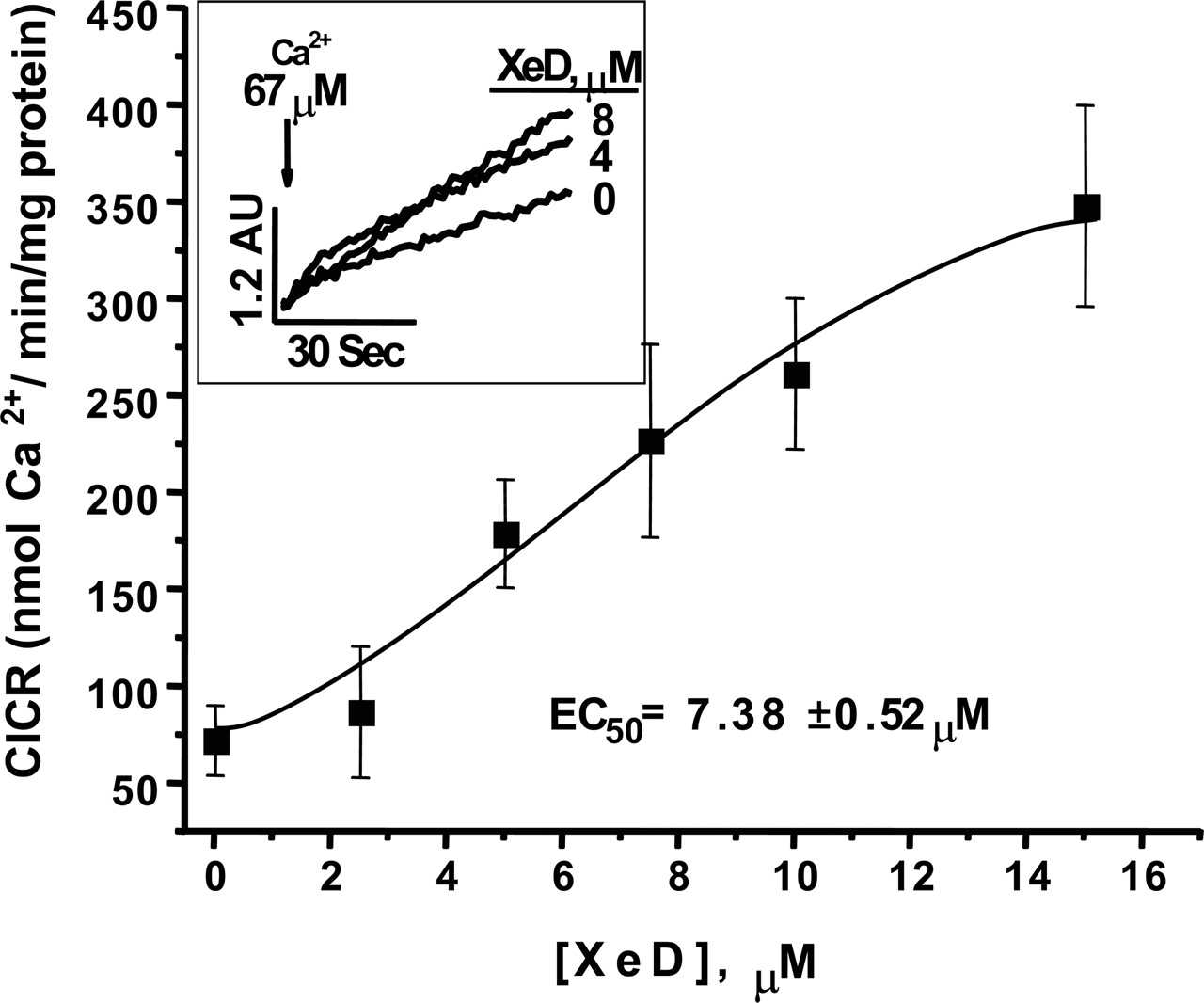
XeD enhances the CICR release rate from SR vesicles. Measurements were performed at 37°C with constant stirring. SR vesicles were actively loaded with four sequential additions of 45 nmol CaCl2 that constituted approximately 80% of their total (100%) capacity (2.605 ± 0.19 mmol Ca2+/mg). Inset, the original recordings of CICR upon bolus addition of 100 nmol CaCl2. The rate of Ca2+ flux across SR vesicles (nmol Ca2+/min/mg protein) was measured by obtaining the liner slope of extravesicular changes in free Ca2+. In the presence of XeD, the CICR rate increase in a dose-dependent manner, with an EC50 value of 7.4 ± 0.5 μM (n = 4).
Discussion
The present study examines the effect of XeC and its hydroxylated derivatives to address the disagreement as to whether these compounds block SERCA and to identify an explanation for their apparent ability to mobilize Ca2+ from ER stores in some preparations. Because more than 200 published studies have used XeC as a pharmacological tool to study calcium mobilization in various systems, understanding the molecular and cellular activities of this group of compounds is essential. In the present study, XeC blocked IP3-induced Ca2+ release from cerebellar microsomes with a potency consistent with that given in our previous report (Gafni et al., 1997). We also investigated the potency of three naturally occurring hydroxylated derivatives (XeD, 7-OH-XeA, and ArC) and found them to have 10- to 15-fold lower potency for the block of IP3-induced Ca2+ release using microsomes isolated from the cerebellum.
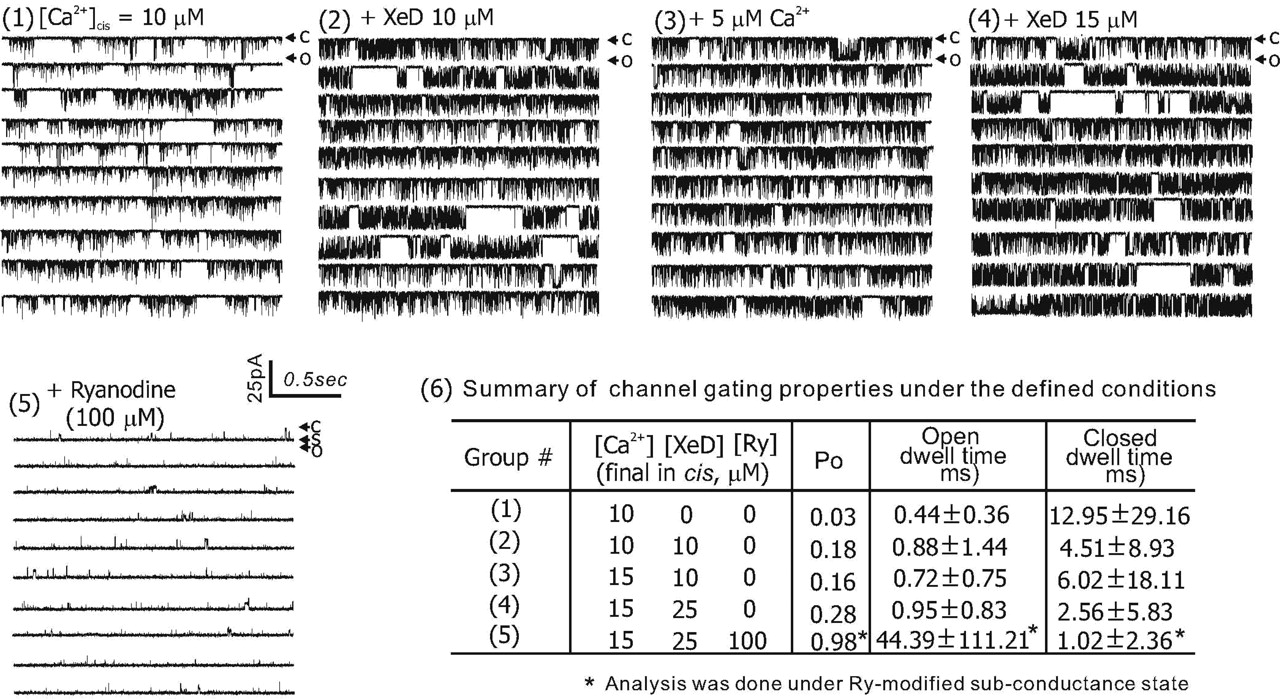
XeD dose-dependently activates RyR1 single channel reconstituted in planar lipid bilayer. Purified RyR1 protein preparation was introduced into the cis bilayer chamber, which consisted of 800 μl of 500 mM CsCl, 10 μM Ca2+, and 20 mM HEPES, pH 7.4. The cytosolic sidedness was verified with 100 μM ryanodine by the end of each experiment. The channel was recorded under each defined condition (denoted in the figure) for at least 10 min; C, O, and S indicate the current amplitude when channel was at zero, maximal, and subconductance states, respectively. Similar results were observed from a total of n = 3 independent BLM experiments that two were on reconstituted channel from purified RyR1 protein and one on incorporated RyR1 channel fused from skeletal JSR vesicle. The channel Po was calculated using pClamp 9.0.
Logit analysis of the IP3-inhibition curves revealed that the slopes for XeC, XeD, and 7-OH-XeA were not significantly different from each other. In contrast, ArC exhibited a shallower inhibition curve with a logit slope significantly less that unity (–0.54). Of the compounds tested, the molecular structure of ArC differs in two significant ways. First, it possesses two hydroxyl substitutions. Furthermore, the molecular structures of XeD, XeC, and 7-OH-XeA possess a trans ring junction in at least one oxaquinolizidine ring (Fig. 1A). In contrast, ArC possesses two cis ring junctions. The trans ring linkage is a more stable configuration than the cis configuration (Gafni et al., 1997). Thus, the cis configuration of ArC may provide more structural flexibility when occupying one or more sites on IP3Rs. These structural differences may explain, at least in part, the more shallow inhibition curve seen with ArC (logit slope =–0.54) compared with the other compounds that were tested and whose slopes were near unity.
In 1999, De Smet and coworkers (1999) reported that XeC (>10 μM) inhibited active 45Ca2+ uptake in saponin-permeabilized A7r5 smooth-muscle cells. The finding was interpreted as proof of a second activity of XeC, that of a SERCA pump inhibitor. In fact, no measurements of SERCA pump activity were presented in their study (De Smet et al., 1999). In the present study, SERCA1 (isolated from rabbit skeletal muscle) and SERCA2a (isolated from porcine cardiac muscle) were directly measured using a coupled enzyme assay and were defined as the TG-sensitive component of Ca2+-(Mg2+)ATPase activity. Contrary to the results of De Smet et al. (1999), we found no direct evidence that XeC or its hydroxylated derivatives (up to 50 μM) inhibited either SERCA isoform tested. Although the effect of XeC and its hydroxylated derivatives on nonmuscle SERCA2b was not measured, considering their highly conserved structure and catalytic function, it is unlikely that SERCA2b is a selective target. Because XeC was found to inhibit rather than promote activation of RyR1, the activity of XeC reported by De Smet et al. could be the result of hydroxylated derivatives of XeC present in their preparation.

XeD mobilizes Ca2+ from ryanodine-sensitive stores of intact myotubes. A, calcium mobilization in cultured primary myotubes was measured in response to 2.5 mM caffeine stimulations in the absence (control) and presence of 10 μM XeD. XeD enhanced transient calcium through the RyRs in skeletal myotubes (n = 6). B, RyRs were blocked by preincubating myotubes with 500 μM ryanodine for 10 min before the recording. Ryanodine blocked the effect of XeD on the ryanodine-sensitive stores; hence, no responses to 2.5 mM caffeine or 10 μM XeD were observed (n = 8). Under both experiment conditions, the addition of 200 nM TG in the presence of 10 μM XeD effectively inhibited SERCA, causing the increase of intracellular Ca2+ caused by the release of calcium from stores and store-operated Ca2+ entry.
Moreover, XeC did not interfere with the ability of TG to inhibit SERCA activity, as suggested by Castonguay and Robitaille (2002), because the addition of 200 nM TG subsequent to XeC was sufficient to fully block SERCA activity. The present results show that XeC and its derivatives do not compete with TG for a common binding site on SERCA.
A new finding in the present study that may help to explain this apparent paradox was that the hydroxylated analog of XeC (XeD) activated RyR channels in a similar concentration range required to inhibit IP3R. Thus, hydroxylation of one or both of the oxaquinolizidine rings was sufficient to maintain potency for blocking IP3R and imparting RyR activating activity rather that RyR inhibition.
RyR channel activation is also a likely mechanism to explain the recently reported actions of XeB (a hydroxylated xestospongin) in intact myotubes (Jaimovich et al., 2005). In this study, XeB was shown to suppress bradykinin-induced Ca2+ signals in neuroblastoma (NG108-15) cells and selectively blocked the slow intracellular Ca2+ signal mediated by IP3R without interfering with capacitative Ca2+ entry or SERCA-dependent SR stores. Nevertheless, rat myotubes pretreated with XeB exhibited larger Ca2+ transients elicited by the addition of external K+ to depolarize the cells (see Fig. 4, C and D, in Jaimovich et al., 2005).
In a recent study, Sawisky and Chang (2005) examined the important role of intracellular Ca2+ stores in regulating the actions of pituitary adenylate cyclase-activating polypeptide necessary for the release of growth hormone (GH) and maturational gonadotrophin secretion from goldfish pituitary cells. Although both XeC and XeD were found to effectively inhibit IP3R-dependent Ca2+ signaling, only XeD (1 μM) was able to enhance GH and maturational gonadotrophin responses, processes that require RyR-dependent elevation of intracellular Ca2+ concentration. In this study, only XeD (not XeC) enhanced activation of RyR-mediated CICR and, in turn, enhanced responses to GH response to pituitary adenylate cyclase-activating polypeptide, a finding which the authors called “surprising” (Sawisky and Chang, 2005). We propose that our new finding of the unique bifunctional activity of XeD toward blocking IP3R and activating RyR1 can largely account for the results of Sawisky and Chang.
In conclusion, XeC lacks SERCA inhibition activity, is a potent blocker of IP3-induced Ca2+ release, but is a much weaker blocker of RyRs. Hydroxylated derivatives also lack inhibitory activity toward SERCA but can enhance RyR activity in the same dose range in which they effectively block IP3R.
Acknowledgments
We thank Dr. Paul D. Allen for his generous gift in providing purified RyR1 for single-channel study.
Footnotes
-
This work was supported by National Institute of General Medical Sciences grant R01-GM57560, and National Institute of Environmental Health Sciences grants P01-ES11269, P42-ES04699, P42-ES05707, and T32-ES07059 (to T.A.T.).
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.105.019125.
-
ABBREVIATIONS: IP3, inositol-1,4,5-trisphosphate; IP3R, inositol-1,4,5-trisphosphate receptors; RyR, ryanodine receptor; XeC, xestospongin C; CICR, Ca2+-induced Ca2+ release; 7-OH-XeA, 7-hydroxy xestospongin A; ER/SR, endoplasmic/sarcoplasmic reticulum; SERCA, sarcoplasmic/endoplasmic reticulum Ca2+-ATPase; ArC, araguspongin C; TG, tharpsigargin; APIII, Antipyrylazo III; MOPS, 4-morpholinepropanesulfonic acid; A23187, calcimycin; [3H]Ry, [3H]ryanodine; BLM, bilayer lipid membrane; HPLC, high-performance liquid chromatography; f1–9, fractions 1–9; GH, growth hormone; Po, open probability.
- Received September 19, 2005.
- Accepted October 21, 2005.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}