


Abstract
Among the nongenomic effects of steroids, control of vasomotion has received increasing attention. Lithocholate (LC) and other physiologically relevant cholane-derived steroids cause vasodilation, yet the molecular targets and mechanisms underlying this action remain largely unknown. We demonstrate that LC (45 μM) reversibly increases the diameter of pressurized resistance cerebral arteries by ∼10%, which would result in ∼30% increase in cerebral blood flow. LC action is independent of endothelial integrity, prevented by 55 nM iberiotoxin, and unmodified by 0.8 mM 4-aminopyridine, indicating that LC causes vasodilation via myocyte BK channels. Indeed, LC activates BK channels in isolated myocytes through a destabilization of channel long-closed states without modifying unitary conductance. LC channel activation occurs within a wide voltage range and at Ca2+ concentrations reached in the myocyte at rest and during contraction. Channel accessory β1 subunits, which are predominant in smooth muscle, are necessary for LC to modify channel activity. In contrast, β4 subunits, which are predominant in neuronal tissues, fail to evoke LC sensitivity. LC activation of cbv1+β1 and native BK channels display identical characteristics, including EC50 (46 μM) and Emax (≈300 μM) values, strongly suggesting that the cbv1+β1 complex is necessary and sufficient to evoke LC action. Finally, intact arteries from β1 subunit knockout mice fail to relax in response to LC, although they are able to respond to other vasodilators. This study pinpoints the BK β1 subunit as the molecule that senses LC, which results in myocyte BK channel activation and, thus, endothelial-independent relaxation of small, resistance-size arteries.
Acute, nongenomic effects of steroids are receiving increasing attention because, in some cases, they have led to the discovery of novel receptor proteins (Watson and Gametchu, 2003), with the possible development of steroidal analogs that have therapeutic use. Among these nongenomic effects, it is particularly noticeable that several physiologically relevant steroids have vasoactive properties. Although neuronal, endocrine, and endothelial factors regulate vascular reactivity and tone, vascular tone and contractility are ultimately determined by signaling molecules and ion channels that operate in the vascular myocyte itself. Some steroids, such as estradiol (Valverde et al., 1999), xenoestrogens (Dick and Sanders, 2001; Pérez, 2005), and glucocorticoids (Lovell et al., 2004), modify ion channel function via direct interactions with ion channel proteins. Others, such as aldosterone (Asher et al., 1996) and estradiol (White et al., 2002), target signaling systems that, in turn, modulate ion channels. Some others, such as cholesterol (Bolotina et al., 1989), alter the physicochemical properties of the lipid microenvironment in which channel proteins are embedded, with modification of channel conformation and function.
Bile acids (cholane-derived steroids) attenuate vascular reactivity in vitro and reduce systemic blood pressure in vivo (Pak and Lee, 1993; Bomzon and Ljubuncic, 1995). Furthermore, a spillover of bile acids from the portal to the systemic circulation is responsible for the systemic hypotension observed in patients with liver damage and/or significant portosystemic circulatory shunt. In some of these subjects, systemic circulating levels of bile acids may reach more than 100 μM (Ostrow, 1993). It has even been speculated that bile acids may serve as endogenous vasodilators (Bomzon and Ljubuncic, 1995), and bile acid relaxation of vascular smooth muscle seems to be caused by steroid actions on the smooth muscle itself (Ljubuncic et al., 2000). It is remarkable that the vasoactive properties of bile acids have not been tested in small, resistance-size arteries that develop myogenic tone; these arteries are critical to determining pressure and blood flow. Moreover, the molecular targets and mechanisms by which bile acids cause vasodilation remain largely unknown.
In a previous study, we found that lithocholate (LC), other naturally occurring bile acids, and LC synthetic analogs increase the activity of large-conductance, Ca2+-activated K+ (BK) channels in myocytes isolated from large arteries (Dopico et al., 2002). Because BK channel activation is a mechanism that opposes constriction and attenuates arterial tone (Jaggar et al., 2000), our previous finding raised the speculation that LC-mediated activation of BK channels in small, resistance-size artery smooth muscle is responsible for arterial tone modification by this steroid.
Here, we demonstrate that acute application of LC readily and reversibly increases the activity of native BK channels freshly isolated from small, resistance-size arteries. Vascular smooth muscle BK channels are made of channel-forming α (KCNMA1)- and regulatory β1 (KCNMB1)-subunits (Orio et al., 2002). In contrast, BK α+β4 (KCNMB4)-subunits are predominant in neuronal tissues (Brenner et al., 2000a; Meera et al., 2000). After cloning α-subunits (termed “cbv1”; AY330293) from myocytes freshly isolated from rat resistance-size cerebral arteries, we used recombinant BK channels to demonstrate that the channel β1 subunit acts as the LC sensor. In contrast, β4 subunits fail to render BK channels sensitive to LC. Moreover, pharmacological block of BK [but not other voltage-gated K+ (KV) channels that also control myocyte tone] or genetic ablation of BK β1 subunits prevents LC from dilating small, resistance arteries. Finally, endothelial removal does not modify LC-induced vasodilation, raising the possibility that LC-based compounds could be used as effective endothelium-independent vasodilators.
Materials and Methods
Artery Diameter Measurement. Middle cerebral arteries were isolated from adult male Sprague-Dawley rats (≈250 g) or 8- to 12-week-old β1 knockout and C57BL/6 control mice. Rats and mice were decapitated using a guillotine and sharp scissors, respectively. These procedures were approved by the Institutional Animal Care and Use Committee from The University of Tennessee Health Science Center, an Association for Assessment and Accreditation of Laboratory Animal Care-accredited institution. Pressurization of arteries was performed as described previously (Liu et al., 2004). Endothelium was removed by passing an air bubble into the vessel lumen for 90 s. Diameter changes were monitored through an inverted microscope (Nikon Eclipse TS100; Nikon Corporation, Tokyo, Japan), recorded on camera (Sanyo VCB-3512T; Sanyo Electric Corporation, Tokyo, Japan), and transferred to a computer. Diameter data were acquired and analyzed using IonWizard 4.4 software (Ion-Optics Corporation, Milton, MA).
Pressurized arteries were extraluminally perfused with physiological saline solution (PSS) (Liu et al., 2004) at a constant rate of 3.75 ml/min using a peristaltic pump Dynamax RP-1 (Rainin Instrument, Inc., Oakland, CA). At this rate, complete washout of the iberiotoxin (Ibtx) effect required >45 min. To keep basal tone under steady behavior, we shortened this period by increasing the flow rate approximately three times during washout of Ibtx, which sometimes evoked a flow-induced dilation (Fig. 1A). Equal volumes (25 ml) of vehicle-versus LC-containing solutions were applied at an equal, constant rate (see above) to the pressurized arterial segment in the chamber via a gravity system. Drugs were dissolved to make stock solutions (see Chemicals) and diluted in PSS to final concentration.
Myocyte Isolation. Basilar and middle cerebral arteries were dissected out from each brain under a stereozoom microscope (Nikon C-PS) and placed into ice-cold dissociation medium (DM) containing 0.16 mM CaCl2, 0.49 mM EDTA, 10 mM HEPES, 5 mM KCl, 0.5 mM KH2PO4, 2 mM MgCl2, 110 mM NaCl, 0.5 mM NaH2PO4, 10 mM NaHCO3, 0.02 mM phenol red, 10 mM taurine, and 10 mM glucose. Each artery was cut into 1- to 2-mm long rings (∼30 rings/experiment). Rings were put in 3 ml of DM containing 0.03% papain, 0.05% bovine serum albumin, and 0.004% dithiothreitol for 15 min at 37°C in a polypropylene centrifuge tube and then incubated in a shaking water bath at 37°C and 60 oscillations/min for 15 min. The preparation was then centrifuged several times as described previously (Liu et al., 2004). After the final centrifugation, the supernatant was discarded, and the pellet was resuspended in 3 ml of DM containing 0.06% soybean trypsin inhibitor. Finally, the tissue was pipetted using a series of borosilicate Pasteur pipettes having fire-polished, diminishing internal diameter tips. The procedure rendered a cell suspension containing relaxed, individual myocytes (≥5 myocytes/field using a 40× objective) that could be easily identified under microscope (Olympus IX-70; Olympus America, Woodbury, NY). The cell suspension was stored in ice-cold DM containing 0.06% bovine serum albumin, and the cells were used for patch-clamping up to 4 h after isolation.
cRNA Preparation and Injection into Xenopus laevis Oocytes. Full-length cDNA coding for cbv1 subunits was cloned from rat cerebral artery myocytes by polymerase chain reaction and ligated to the PCR-XL-TOPO cloning vector (Invitrogen, Carlsbad, CA) (Jaggar et al., 2005). cDNA coding for cbv1 subunits was cleaved from the cloning vector by BamHI (Invitrogen) and XhoI (Promega, Madison, WI) and directly inserted into the pOX vector for expression in X. laevis oocytes. pOX-cbv1 was linearized with NotI (Promega) and transcribed in vitro using T3 polymerase. β1 Subunit cDNA inserted into the EcoRI/SalI sites of the pCI-neo expression vector was linearized with NotI and transcribed in vitro using T7 polymerase. β4 Subunit cDNA inserted into the pOx vector was linearized by NotI and transcribed using T3 polymerase. The mMessage-mMachine kit (Ambion Inc., Austin, TX) was used for transcription. The pOX vector and the cDNA coding for β1 subunits were generous gifts from Aguan Wei (Washington University, St. Louis, MO) and Maria Garcia (Merck Research Laboratories, Rahway, NJ).
Oocytes were removed from X. laevis and prepared as described previously (Dopico et al., 1998). cRNA was dissolved in diethyl poly-carbonate-treated water at 5 (cbv1) and 15 (β1 or β4) ng/μl; 1-μl aliquots were stored at -70°C. Cbv1 cRNA was injected alone (2.5 ng/μl) or coinjected with either β1 or β4 (7.5 ng/μl) cRNAs, giving molar ratios ≥6:1 (β:α). cRNA injection (23 nl/oocyte) was conducted using a modified micropipette (Drummond Scientific Co., Broomall, PA). The interval between injection and patch-clamp recordings was 48 to 72 h.
Electrophysiology. Oocytes were prepared for patch-clamp recordings as described previously (Dopico et al., 1998). Single-channel and macroscopic currents were recorded from inside-out (I/O) or outside-out (O/O) patches. For experiments with oocytes, both bath and electrode solutions contained 135 mM K+ gluconate, 5 mM EGTA, 1 mM MgCl2, 15 mM HEPES, and 10 mM glucose, pH 7.35. For experiments with myocytes, KCl substituted for K+ gluconate. In all experiments, free Ca2+ in solution was adjusted to the desired value by adding CaCl2. In most studies, free Ca2+ concentration in the electrode solution was 10 μM. In O/O studies with 17β-estradiol, however, free Ca2+ concentration in the electrode solution was 0.3 μM. Nominal free Ca2+ was calculated with MaxChelator Sliders (C. Patton, Stanford University, Stanford, CA) and validated experimentally using Ca2+-selective electrodes (Corning Incorporated Science Products Division, Corning, NY).

Lithocholate dilates pressurized arteries via activation of BK channels independently of an intact endothelium. A, rat cerebral arterial diameter trace showing that after artery development of myogenic tone, acute application of 45 μM LC causes sustained yet fully reversible dilation. LC action is practically abolished by 55 nM Ibtx, a selective BK channel blocker (the vasodilatory “rebound” after Ibtx wash is caused by increased flow rate; see Materials and Methods). B, diameter trace showing that LC-induced dilation is unaffected by 0.8 mM 4-AP, a blocker of KV channels other than BK. In A and B, vertical dotted lines indicate the times at which arterial diameter was determined. C, averaged diameter in response to LC (n = 17), Ibtx+LC (n = 4), and 4-AP+LC (n = 3). LC-specific action on diameter is highlighted by displaying data as the percentage of changes from values obtained in vehicle with (second and third column) or without (first column) K+ channel blockers. D, LC-induced dilation is similar in intact versus endothelium-denuded arteries (n = 5). The presence of a functional endothelium was assessed by responses to endothelium-dependent [10 μM acetylcholine (Ach); n = 4] and independent [10 μM sodium nitroprusside (SNP); n = 5] vasodilators. **, different from intact arteries (p < 0.01).
Patch-recording electrodes were made as described previously (Dopico et al., 1998). Immediately before recording, the tip of each electrode was fire-polished on a microforge WPI MF-200 (World Precision Instruments, Inc., Sarasota, FL) to give resistances of 5 to 9MΩ when filled with solution. An agar bridge with gluconate or Cl- as the main anion (for oocyte and myocyte experiments, respectively) was used as ground electrode. After excision from the cell, the membrane patch was exposed to a stream of bath solution containing each agent at final concentration. Solutions were applied onto the patches using a pressurized system DAD12 (ALA Scientific Instruments, New York, NY) via a micropipette tip with an internal diameter of 100 μm. Experiments were carried out at room temperature (21°C).
Currents were recorded using an EPC8 amplifier (HEKA Electronics, Lambrecht/Pfalz, Germany) at 1 kHz using a low-pass, eight-pole Bessel filter 902LPF (Frequency Devices, Haverhill, MA). Data were digitized at 5 kHz using a Digidata 1320A A/D converter and pCLAMP 8.0 (Molecular Devices, Sunnyvale, CA). For macropatch recordings, G/Gmax - V data were fitted to the Boltzmann function: G(V) = Gmax/(1 + e(-V + V0.5)/k).
Using the slope (k) of the G/Gmax versus V plots, the effective valence, z (i.e., 1/k), was calculated as 1/k = RT/F, where R is the gas constant, T is absolute temperature, and F is the Faraday constant. As an index of channel steady-state activity, we used the product of the number of channels in the patch (N) and the channel open probability (Po). NPo was obtained from all-points amplitude histograms from ≥30 s of continuous recording under each experimental condition.
Chemicals. All chemicals were purchased from Sigma (St. Louis, MO), with the exception of 5β-cholanic acid 3α-ol (LC) (Steraloids, Inc., Newport, RI) and Ibtx (Alomone Labs Ltd., Jerusalem, Israel). On the day of the experiment, an LC stock solution (333 mM) in dimethyl sulfoxide (DMSO) was freshly prepared by sonication for 5 min. For arterial tone experiments, the LC stock was diluted 1/10 in DMSO and further diluted in PSS to render final LC concentration. Solution containing vehicle [0.1% DMSO (v/v)] was used as control perfusion. For electrophysiological recordings, the LC stock solution was diluted 1/10 in 95% ethanol and further diluted with bath solution to render final LC concentration (3-1000 μM). The DMSO/ethanol vehicle (≤0.1/≤0.86% final concentrations) in bath solution was used as control.
Data Analysis. Artery diameter response to each compound is shown as a percentage of the diameter obtained before compound application. Arterial diameter and electrophysiological data were analyzed with IonWizard 4.4 (IonOptics) and pCLAMP 8.0 (Molecular Devices). Further analysis, plotting, and fitting were conducted using Origin 7.0 (OriginLab Corp, Northampton, MA) and InStat 3.0 (GraphPad Software Inc., San Diego, CA).
Statistical analysis was conducted using either one-way analysis of variance and Bonferroni's multiple comparison test or paired Student's t test; significance was set at p < 0.05. Data are expressed as mean± S.E.M.; n = number of patches/arteries.
Results
Lithocholate Dilates Small, Resistance-Size Arteries via BK Channels. After myogenic tone development at 60 mm Hg, intact arteries reached a diameter of 154.4 ± 5.2 μm (n = 17). Maximal contraction and dilation were checked by perfusing the vessel with 60 mM KCl at the beginning and with Ca2+-free solution at the end of each experiment (Fig. 1, A and B). In all cases (n = 17), application of 45 μM LC, that is, a concentration well below LC's critical micellar concentration (CMC) (≥1 mM under our recording conditions) (Roda et al., 1995), caused a significant increase in peak arterial diameter: +6% on top of a transient increase in diameter caused by vehicle-containing solution (Fig. 1, A-C). LC-induced dilation was not only larger than that caused by vehicle but also more sustained (≈2.8 times longer); for example, by the time the vehicle effect had totally vanished, LC-induced dilation still represented 109.9 ± 1.7% of the initial arterial diameter determined before any compound application (n = 17; p < 0.01). The differential vasodilation caused by LC versus vehicle is most evident from the area under the curve values (integrals) corresponding to the change in diameter as a function of time: 18,369 ± 3964 versus 10,262 ± 2574 (p < 0.01) (Table 1). It is noteworthy that the net increase in cerebral artery diameter caused by LC over pre-LC values (+9.9%) is expected to cause a marked increase (∼30%) in cerebral blood flow (CBF), because changes in artery diameter are related to changes in CBF by a factor of ∼3 (Gourley and Heistad, 1984).
Characteristics of lithocholic acid- versus vehicle-induced vasodilation
Rat cerebral artery diameter is critically controlled by myocyte BK channel activity (Jaggar et al., 2000). Because these channels are selectively blocked by nanomolar concentrations of Ibtx (Liu et al., 2004), we used this peptide to determine any possible contribution of BK channels to LC dilation. As expected, bath application of 55 nM Ibtx caused a robust decrease in the diameter of intact arteries (-11.8 ± 3.4%) (n = 4) (Fig. 1A). It is remarkable that LC dilation was completely lost when the steroid was applied on top of Ibtx (Fig. 1, A and C). In the presence of Ibtx, LC caused some reduction in diameter (-3.4%), which could be related to the well-known increase in cytosolic Ca2+ caused by bile acids (Thibault and Ballet, 1993). In brief, our data indicate that LC fails to dilate small, resistance arteries when BK channels are specifically blocked.
Cerebrovascular smooth muscle tone is also controlled by KV channels other than BK (Faraci and Sobey, 1998). To determine the selectivity of BK channel involvement in LC dilation, we evaluated LC action in the presence of 4-aminopyridine (4-AP), which, at submillimolar to low millimolar concentrations, blocks most KV but not BK channels in rat cerebral arteries (Liu et al., 2004). Applying 0.8 mM 4-AP caused an immediate decrease in diameter (-9.7 ± 2.3%, n = 4) (Fig. 1B). In contrast to the Ibtx results, the change in peak diameter caused by LC in the presence of 4-AP was identical to that determined in the absence of KV channel blocker (+6% over pre-LC values; Fig. 1C). Although we cannot rule out some contribution of KV channels other than BK to LC dilation, our results indicate that KV channels other than BK do not play a major role in LC dilation of pressurized small, resistance-size cerebral arteries. Furthermore, 45 μM LC on top of 4-AP almost totally reverted the vasoconstriction caused by the KV channel blocker (Fig. 1B), underscoring the effectiveness of BK channel-targeting by LC in reversing cerebrovascular constriction driven by voltage-dependent mechanisms. In contrast to LC dilation, the small and transient increase in diameter caused by vehicle was unmodified by Ibtx (Fig. 1A) but was somewhat decreased by 4-AP (Fig. 1B). The mechanism(s) involved in the transient dilation evoked by vehicle is out of the scope of this study. Nevertheless, the differences in time course and magnitude (Table 1), together with their differential modulation by selective channel blockers, clearly indicate that LC and vehicle dilation of cerebral arteries are mediated by different ionic mechanisms, the former via BK channels.
Finally, to rule out that endothelial factor(s) could be mediating or, at the least, modulating LC-induced dilation, we studied LC action in de-endothelialized arteries and compared it with that in intact vessels. LC-induced dilation is similar in intact versus endothelium-denuded arteries (n = 5). The presence of a functional endothelium was assessed by vascular responses to endothelium-dependent (acetylcholine; 10 μM) and -independent (sodium nitroprusside; 10 μM) vasodilators. Indeed, although vasodilation in response to acetylcholine was lost (n = 4), sodium nitroprusside-induced dilation was fully preserved in de-endothelialized arteries (n = 5) (Fig. 1D). It is noteworthy that LC increase in diameter of de-endothelialized arteries was not significantly different from that of intact arteries (Fig. 1D). Thus, LC-induced dilation of small, resistance-size cerebral arteries is independent of a functional endothelium. Collectively, data shown in Fig. 1 suggest that LC targeting of myocyte BK channels causes LC dilation of small cerebral arteries.
Lithocholate Directly Activates Myocyte BK Channels via the Channel β1 Subunit. To determine whether LC directly targets BK channels in cerebral artery myocytes, we studied drug action on channel activity by using I/O patches with the membrane potential and free [Ca2+]i set at values (-40 to -30 mV and 3 μM) similar to those obtained in cerebrovascular myocytes during contraction (Knot and Nelson, 1998; Pérez et al., 2001). After excision, the patch was exposed to vehicle-containing solution, and BK NPo was recorded for no less than 1 min. Then, applying LC-containing (1-1000 μM) solution reversibly increased NPo in a concentration-dependent fashion: EC50 = 46 ± 6 μM, Emax ∼300 μM (Fig. 2, A and B). At Emax, NPo reached 350% of control, and this ceiling remaining steady up to 1 mM LC. Concentrations greater than 1 mM (i.e., close to the CMC for LC under our recording conditions) (Roda et al., 1995) systematically resulted in loss of gigaseals, probably caused by a micelle-mediated detergent effect. Thus, LC maximally increases BK channel activity at aqueous concentrations in which LC monomers predominate, as opposed to a detergent action on the membrane caused by micelle formation in solution. LC increase in NPo was observed in membrane patches that were excised from the myocyte >5 min before applying LC under continuous bath perfusion in the absence of nucleotides. Therefore, LC action does not require cell integrity or the continuous presence of cytosolic messengers. Rather, it is caused by a direct interaction between the steroid and the BK channel complex itself and/or its immediate proteolipid environment.

Lithocholate at submillimolar concentrations activates native BK channels in freshly isolated rat cerebral artery myocytes. A, single-channel recordings from an I/O patch excised from an arterial myocyte before, during, and after 45 μM LC. Vehicle-containing solution was applied before (Vhcl) and after (Washout) LC-containing solution. Openings are shown as downward deflections; arrows indicate the baseline; Vm =-40 mV, free [Ca2+]i ≈3 μM. B, LC action is concentration-dependent: EC50 = 46 ± 6 μM; Emax ≈300 μM, at which NPo reaches ∼350% of control (n ≥ 3).
To determine which subunit of the channel complex is involved in sensing LC with an eventual increase in NPo, we performed electrophysiological recordings in I/O patches from X. laevis oocytes expressing either homomeric cbv1 or heteromeric cbv1+β1 channels under identical conditions. To evoke measurable levels of Po within a second-minute time frame in the absence of β1 subunits, these studies were conducted at Ca2+i of 10 μM at either positive or negative Vm (+20 or -20 mV); considering that the LC effect on BK channel NPo is voltage-independent (Dopico et al., 2002; and below), data obtained at both voltages were pooled. Cbv1 subunits expressed in oocytes rendered macro- and microscopic currents that showed all major biophysical and pharmacological features of BK currents (Jaggar et al., 2005). The presence of functional β1 subunits was confirmed by macroscopic current characteristics (Brenner et al., 2000a) (slower activation kinetics, increased apparent Ca2+ sensitivity with a shift in V0.5 of ∼20 mV toward negative potentials), and channel activation by bath application of 10 μM 17β-estradiol to the extracellular surface of O/O patches (Valverde et al., 1999) (Supplemental Figs. S1, A and B, and S2B).
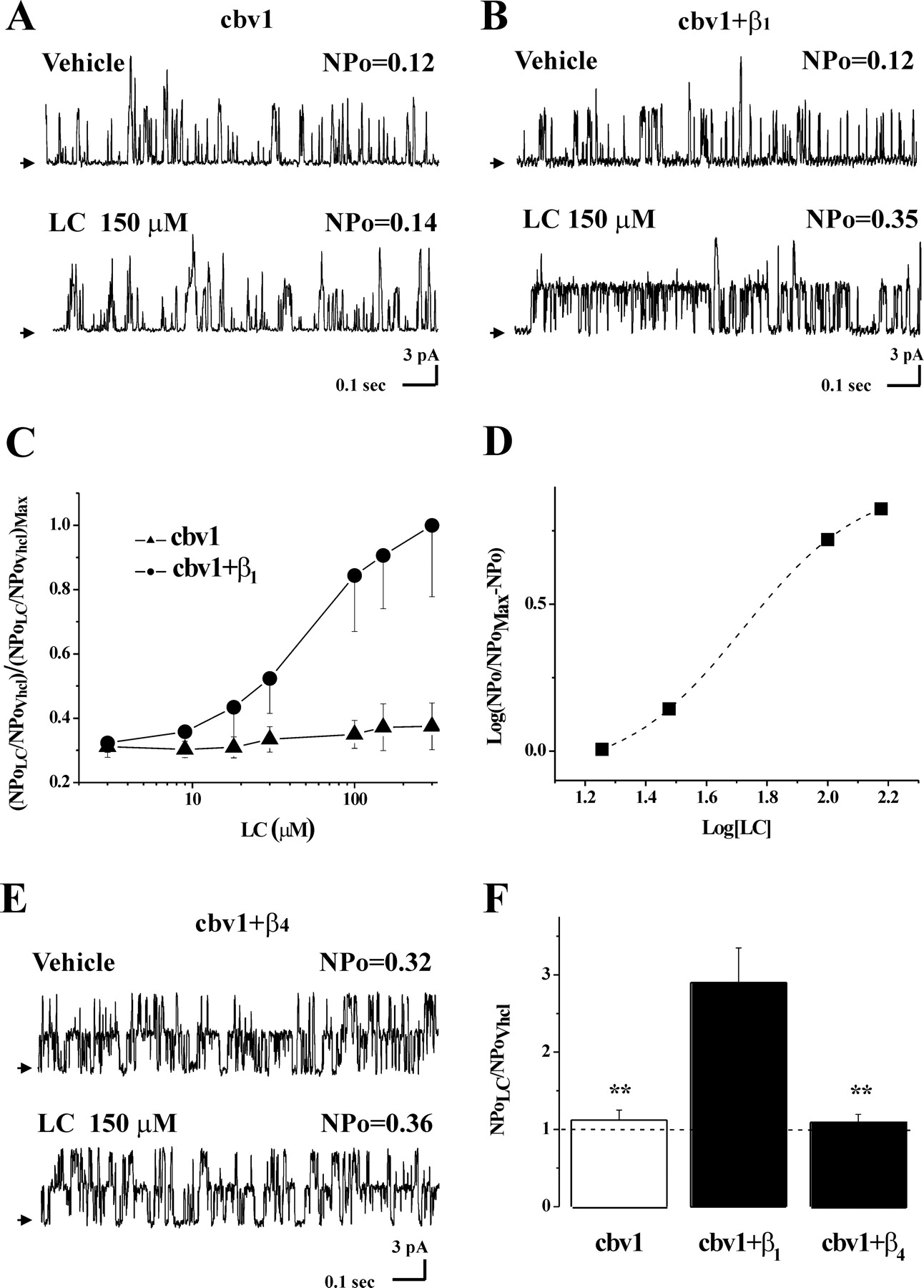
In contrast to the results obtained with native BK channels in cerebrovascular myocytes, application of LC concentrations as high as 150 μM (on top of vehicle) to the internal side of I/O patches failed to activate homomeric cbv1 channels, with average NPo values reaching 112 ± 13% of control (p > 0.05; n = 4) (Fig. 3, A and C). Thus, LC activation of cerebrovascular BK channels requires the presence of β1 subunits and/or some other component of the myocyte membrane that is missing in the heterologous expression system. As found with native BK channels, however, LC (3-300 μM) caused a reversible and concentration-dependent increase in NPo of heteromeric cbv1+β1 channels (Fig. 3, B and C), with EC50 = 43.5 ± 4.7 μM and Emax ∼300 μM, at which NPo reached 290 ± 45% of control. These values are practically identical to those of native BK channels (see above), indicating that differences in composition/organization between rat cerebrovascular myocyte and X. laevis oocyte membranes are not critical in LC action on BK channels. The identical LC responses of native cerebrovascular BK and cbv1+β1 channels seem to indicate the involvement of a common target(s) mediating LC action in these two systems, possibly the β1 subunit itself.
A Hill-like plot for LC activation of cbv1+β1 channels renders a slope (apparent Hill coefficient) of 1.3 (Fig. 3D), which suggests the involvement of at least two “sites” in the cbv1+β1 complex for LC to increase NPo. An increase in the number of channels (N) might contribute to the overall increase in NPo caused by LC. Data from patches in which n = 1 (Fig. 4A), however, show an increase in Po that is similar to the increase in NPo in patches containing an unknown N. Thus, LC action on BK steady-state activity seems to be solely determined by an increase in Po. Given the apparent Hill coefficient of 1.3, the increase in Po seems to require the interaction of at least two LC molecules with the β1 subunits of the channel complex.
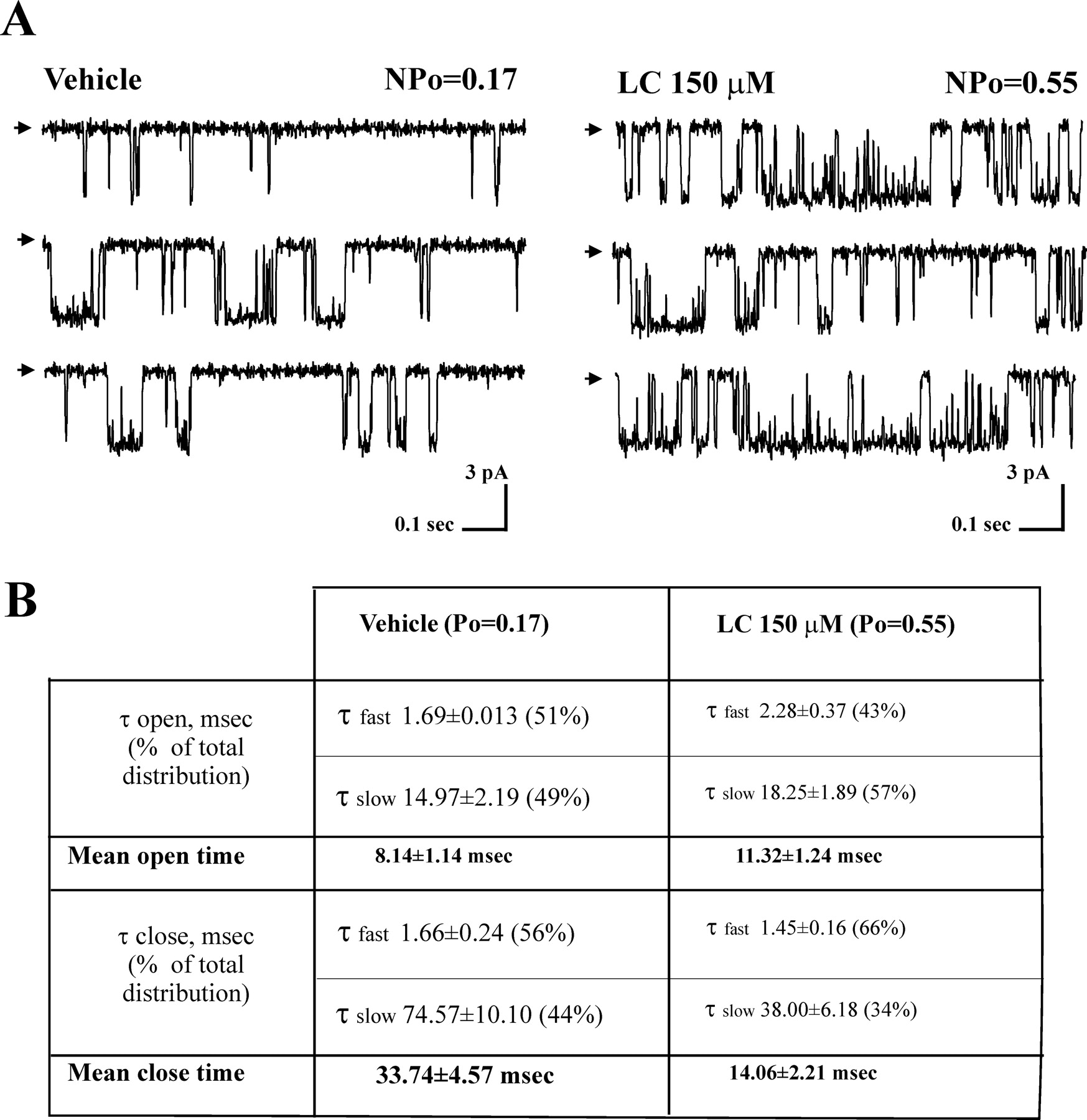
From the channel dwell-time distributions in patches (n = 2) in which N = 1 (Fig. 4A), we calculated both open and mean closed times (Dopico et al., 1998). Both distributions could be well-fitted with double exponential functions, indicating the existence of at least two open and two closed states. Lithocholate increased the channel mean open time, which reached 137% of control. This enhancement resulted from an LC-induced increase in the average duration of both short and long open channel events, with an accompanying mild shift in the open-channel distribution toward longer openings; the long open state(s) accounted for 49 and 57% of total open events in vehicle and LC, respectively (Fig. 4, A and B). In addition, LC drastically decreased the channel mean closed time, which reached 41% of control. This reduction was primarily caused by a robust reduction in the average duration of channel long closed events and a shift toward briefer closures; the long close state(s) accounted for 44 and 34% of total close events in vehicle and LC, respectively (Fig. 4, A and B). In brief, the steroid-induced increase in channel Po results primarily from LC-induced destabilization of channel long closed states, eventually reducing by more than half the channel mean closed time. These changes in channel kinetics with consequent increase in Po occurred in the absence of significant change in unitary conductance: 228.6 ± 3.7 versus 234.0 ± 4.6 pS in symmetric 135 mM K+ (n = 4; not significant). Thus, LC modification of BK channel function is limited to the modification of channel gating (see Discussion).

β1 But not β4 subunits confer lithocholate sensitivity to BK channels. Records from I/O patches showing that 150 μM LC fails to increase homomeric cbv1 (i.e., rslo1) (A) but enhances heteromeric cbv1+β1 NPo (B) under identical conditions (Vm = -20 mV, free [Ca2+]i ≈10 μM). C, whereas LC fails to potentiate cbv1 channels even at 300 μM, LC activates cbv1+β1 channels in a concentration-dependent manner: Emax ≈300 μM; EC50 = 43.5 ± 4.7 μM. These values are almost identical with those obtained with native channels in rat cerebral artery myocytes (Fig. 2). D, a logit-log plot of LC action on cbv1+β1 shows data fitted to a sigmoidal function, which renders a slope = 1.32 (see Results). To construct this plot, Emax was calculated as the mean of NPo values obtained at 150 and 300 μM LC. E, LC at concentrations that maximally activate native BK and cbv1+β1 channels fails to activate cbv1+β4 channels. F, averaged ratios of NPo in the presence (NPoLC) and absence (NPo Vhcl) of 150 μM LC for cbv1 (n = 6), cbv1+β1 (n = 6), and cbv1+β4 (n = 9) channels expressed in X. laevis oocytes. **, different from cbv1+β1 (p < 0.01).
To determine whether LC increase in BK Po is selectively mediated by the β subunit type (β1) that is predominant in smooth muscle or could be mediated by other channel accessory subunits, we tested LC action on cbv1+β4 channels. When coexpressed with α subunits, β4 subunits introduce a hyperpolarizing shift in V0.5 similar to that caused by α+β1 coexpression. In addition, β4 subunits render the BK complex relatively resistant to Ibtx (Meera et al., 2000). This phenotype was confirmed in our study (Supplemental Fig. S2, A and B). Under conditions identical to those used with cbv1 and cbv1+β1 channels, cbv1+β4 channels were consistently refractory to LC action (8/8 patches), even when tested at concentrations (150 μM) that were close to Emax values in both cbv1+β1 and native BK channels (Fig. 3, E and F); cbv1+β4 NPo reached 109 ± 11% of control (not significant, also not significantly different from data with LC and cbv1 homomeric channels). Therefore, β1 but not β4 subunits confer LC sensitivity to cerebrovascular BK channels.

Lithocholate increases BK unitary currents by increasing Po caused by a mild increase in mean open time and a marked decrease in mean closed time. A, current records from an I/O patch containing a single cbv1+β1 channel expressed in X. laevis oocytes in the absence (left) and presence (right) of 150 μM LC. LC increase in Po (≈320%) is similar to LC increase in NPo (≈290 ± 45%; Figs. 3C and 4B, Results), strongly suggesting that LC action occurs without increase in the number of channels (N); Vm set to +20 mV; free [Ca2+]i ≈10 μM. Arrows on the left of the top traces of each indicate the baseline, and channel openings are shown as downward reflections. B, cbv1+β1 channel dwell times in the absence and presence of 150 μM LC. Both open and closed time distributions could be well-fitted with a double-exponential function, indicating the existence of at least two open (fast and slow) and two closed (fast and slow) states. The table shows both the average duration of each component (τ) and its contribution to the total time spent in open (closed) states (as percentage in parentheses). LC increase in Po (∼320%) is caused by a mild increase in the average duration of both short and long open events and a sharp decrease (-60%) in mean close time, the latter basically caused by LC-induced destabilization of channel long closures.
Lithocholate Effectively Activates BK Channels within Physiological Ranges of [Ca2+]i and Membrane Voltage. β1 Subunits modulate both Ca2+-dependent and -independent channel gating, resulting in an increase in the apparent Ca2+ sensitivity of the channel. This effect is more pronounced at [Ca2+]i that effectively increases Po (Meera et al., 1996; Nimigean and Magleby, 2000). On the other hand, the lateral chain of LC contains a carboxyl that is ionized at physiological pH, raising the possibility that LC action could be modified by transmembrane voltage. Thus, we explored the Ca2+- and voltage-dependence of LC action on cbv1+β1 channel Po by using a wide voltage range (±150 mV) and [Ca2+]i levels that expanded those in the myocyte under physiological conditions (0.15-0.3 μM at rest; up to 10-30 μM in the vicinity of BK channels during contraction) (Pérez et al., 2001; Liu et al., 2004). Even at nonphysiological, very positive voltages (+80 mV), LC potentiation of BK NPo was unnoticeable when recorded in solutions having zero Ca2+ added plus 10 mM EGTA to chelate trace amounts of the divalent (n = 3) (data not shown). This is consistent with LC modulating channel gating via a β1-mediated mechanism, because at “zero” or subactivatory [Ca2+]i, β1 subunit modification of gating does not translate into an evident change in overall Po (Nimigean and Magleby, 2000). Furthermore, LC activation (as a percentage of NPo in vehicle) increased with [Ca2+]i: from 139.9 ± 32.9 (n = 4; p < 0.05) at 0.1 μM Ca2+ to a maximal effect of 244.1 ± 58.9% (n = 3; p < 0.01) at 1 μM Ca2+. This maximum remained steady within the 1 to 10 μM [Ca2+]i range (n = 16), to decrease with higher [Ca2+]i (e.g., at 30 μM, NPo in LC reached 172.5 ± 9.5% of control; p < 0.05, n = 3) (Fig. 5A). These data demonstrate that LC activates BK channels within a Ca2+ range that spans from resting levels to those reached during myocyte contraction. It is remarkable that LC activation of BK channels is most effective at Ca2+ levels reached near the BK channel during cerebral artery myocyte contraction (Pérez et al., 2001).
Next, we evaluated LC action on cbv1+β1-mediated currents as a function of applied voltage, exposing I/O macropatches to [Ca2+]i at which LC activation of BK channels is robust: 0.3, 3, and 10 μM [Ca2+]i. From G/Gmax versus Vm plots fitted to a Boltzmann relationship, we obtained V0.5 = 101.9 ± 1.2 (n = 3), 74 ± 10 (n = 3), and 32.3 ± 11.2 mV (n = 5), respectively. At every [Ca2+]i tested, LC (150 μM) shifted the V0.5 value by ≈-17.7 mV (Fig. 5B) without changing the slope of the plot. Thus, at any [Ca2+]i, z (i.e., an index of the minimum number of elementary charges that cross the electric field to gate the channel) was similar in the absence or presence of LC (e.g., at 10 μM free [Ca2+]i: z = 1.24 ± 0.29 versus 1.26 ± 0.2). These data suggest that LC does not interfere with the voltage-sensing process of channel gating. The lack of LC effect on z is also consistent with a β1-mediated action on channel gating (Brenner et al., 2000a). Together, our data show that LC is an effective activator of BK channels via their β1 subunits at physiologically relevant [Ca2+]i and voltages.

Lithocholate activates BK channels within the physiological ranges of [Ca2+]i and membrane potential. A, NPo during exposure of intracellular side of I/O patches to 150 μM LC (NPo LC) versus NPo in vehicle-containing solution (NPo Vhcl) plotted as a function of free [Ca2+]i. Channel NPo was obtained after coexpression of cbv1 and β1-subunits in X. laevis oocytes. The membrane voltage was set within the range ±20 mV, and the bath solution contained 0.1 (n = 4), 0.3 (n = 3), 1 (n = 3), 3 (n = 7), 10 (n = 6), or 30 μM (n = 4) free [Ca2+]i. Each column represents the mean ± S.E.M. B, voltage needed for half-maximal increase in BK channel NPo (V0.5) as a function of [Ca2+]i in the vehicle-containing solution (Vhcl) and in the presence of 150 μM LC. V0.5 values were obtained from G/Gmax curves for I/O macropatches at 0.3, 3, and 10 μM [Ca2+]i. Voltage steps of 200-ms duration were applied from -150 to +150 mV with 10-mV increments; Vholding = 0 mV. Each data point represents the mean value ± S.E.M. from ≥4 patches (oocytes). At every [Ca2+]i, LC causes a similar leftward shift in V0.5 of ∼17.7 mV.

Lithocholate fails to dilate pressurized arteries from β1 subunit knockout mice. A, arterial diameter traces show that acute 45 μM LC and 55 nM Ibtx cause sustained diameter increase and decrease, respectively, in arteries from wt mice (gray trace) but not in arteries from BK β1 knockout mice (black trace). The small and transient dilation caused by vehicle (Vhcl) is similar in both mice. B, averaged diameter data in response to LC (left) and Ibtx (right) in wt (hollow) (n = 7) and β1 knockout (▪) (n = 5) mice. *, different from wt mice (p < 0.05); **, different from wt mice (p < 0.01).
Lithocholate Fails to Induce Cerebrovascular Dilation in β1 Knockout Mice. To determine the impact of LC targeting of BK β1 subunits on organ function, we evaluated LC action on the arterial diameter of pressurized cerebral arteries from β1 knockout versus wt C57BL/6 mice (controls). To verify the presence of functional β1 subunit-containing BK channels in controls, we tested artery diameter sensitivity to block by 55 nM Ibtx, as done with rat arteries (Fig. 1A). In control mice, Ibtx caused a significant vasoconstriction within 15 min of application (up to -8.7 ± 4.2% decrease from initial diameter; p < 0.01; n = 4) (Fig. 6, A and B). As reported (Brenner et al., 2000b), Ibtx decrease in diameter was largely attenuated in arteries from β1 knockout mice (-2.25 ± 0.44%; different from vasoconstriction in wt mice, p < 0.05; n = 4) (Fig. 6, A and B).
The mild and transient vehicle dilation found in rat arteries was also observed in mouse arteries. Consistent with results obtained in rat arteries showing the lack of Ibtx modulation of vehicle dilation (Fig. 1A), genetic ablation of β1 subunits failed to modify vehicle action (Fig. 6A), buttressing the idea that this mild and transient dilation does not involve BK channels.
More important, as found with rat cerebral arteries, 45 μM LC caused a sustained yet fully reversible increase in diameter of wt mouse cerebral arteries (+4.4 ± 0.9% from initial diameter; p < 0.01; n = 7). In sharp contrast, LC consistently failed to dilate arteries from β1 knockout mice (n = 5) (Fig. 6, A and B), indicating that in intact cerebral arteries, the presence of BK β1 subunits is crucial for LC dilation.
Discussion
We have demonstrated for the first time that LC is an effective dilator of pressurized, resistance-size arteries, and this relaxation is endothelium-independent. LC-induced vasodilation is caused by selective targeting of myocyte BK channel function. Whereas a variety of ion channels other than BK contribute to regulate cerebrovascular tone (Faraci and Sobey, 1998; Dietrich et al., 2005), making them putative targets of LC effect on vasomotion, the fact that genetic ablation of KCNMB1 or selective pharmacological block of BK (but not other KV) channels suppresses LC-mediated cerebrovascular dilation clearly indicates that the BK channel β1 subunit is the molecular effector of LC-induced cerebrovascular dilation. Lithocholate targeting of BK channels results in a reversible increase in Po due to several LC actions on channel dwell times, with destabilization of the channel long closed states being predominant. Lithocholate action on Po is evident within wide voltage and [Ca2+]i ranges, which include values measured in cerebrovascular myocytes both under resting conditions and during contraction (Knot and Nelson, 1998; Pérez et al., 2001).
Both vasodilation and full channel activation occur at LC concentrations well below this steroid CMC, which indicates that these actions are caused by the presence of LC monomers in the aqueous phase and not by nonspecific detergent effects on the membrane caused by LC micelles in solution. Lithocholate selectivity on β1 over β4 channel subunits also argues for a specific LC target interaction. Finally, some bile acid analogs that are effective “detergents” (positive curvature-forming lipids) fail to activate myocyte BK channels (Dopico et al., 2002). Collectively, these results strongly support the idea that LC activates BK channels via a selective interaction with a steroid target caused by the presence of LC monomers in solution.
Lithocholate monomers activate the channel independently of cell integrity, cytosolic mediators, or steroid metabolism. Instead, channel activation results from the interaction of LC with the channel protein complex and its immediate lipid microenvironment, the channel β1 subunit behaving as the “functional target” of LC. Whether channel activation requires LC binding to specific sites in the β1 subunit itself or the subunit sensing of LC located some-where else in the subunit proteolipid vicinity remains to be determined. However, it is clear from our data that the BK β1 subunit behaves as the specific LC sensor. Furthermore, this subunit is necessary for LC to dilate the intact artery. The fact that genetic ablation of β1 subunits prevents LC from dilating small, resistance-size arteries seems to indicate that the animal fails to develop compensatory mechanisms that could render the myocyte BK channel and the artery sensitive to LC dilation in the absence of β1 subunits. In brief, we have identified the BK β1 subunit as the functional target that mediates endothelium-independent LC dilation of intact and pressurized resistance arteries.
In particular, LC-induced dilation of small cerebral arteries (∼10% increase in diameter) will result in a robust increase in CBF of ∼30%, raising the speculation that LC-related analogs could be developed and, eventually, used clinically as cerebrovascular dilators. This possibility acquires particular relevance considering that 1) stroke remains the third leading cause of death and first cause of long-term disability in the United States (see http://www.americanheart.org); 2) >88% of strokes are ischemic (Williams et al., 2003), in which impaired vasomotion may be found; 3) biomedical research has largely failed to provide effective and safe cerebrovascular dilators (Legos et al., 2002); 4) endothelial-mediated vasodilation is impaired in several processes that affect cerebral vessels, such as atherosclerosis and vasospasm; it is noteworthy that LC dilation does not require a functional endothelium (Fig. 1); 5) whereas several other steroids [17β-estradiol (Valverde et al., 1999), xenoestrogens (Dick and Sanders, 2001; Pérez, 2005), androgens (Deenadayalu et al., 2001) and glucocorticoids (King et al., 2006)] activate BK channels, the effects of these agents on cerebrovascular myocyte BK channels and/or tone have not been demonstrated; and 6) these steroids and some of their analogs have widespread hormonal actions, which may preclude/limit their clinical use as vasodilators. Thus, pinpointing the LC myocyte BK β1 subunit interaction as a mechanism leading to cerebrovascular dilation may be a first step for designing newer and safer steroid-based agents to help in the pharmacological treatment of cerebrovascular ischemic disease.
Lithocholate actions differ in several critical aspects from those of other steroids reported to modulate BK channels. At micromolar (1-30 μM) concentrations, 17β-estradiol increases BK (hslo) channel activity by interacting with the channel β1 subunit (Valverde et al., 1999). In contrast to LC, 17β-estradiol was also found to be a potent activator of BK channels containing either β2 or β4 subunits (King et al., 2006). Furthermore, it has been reported that 17β-estradiol at submicromolar concentrations (0.01-1 μM) can modulate BK activity through an interaction between the steroid and the channel α subunit (Korovkina et al., 2004). Finally, it has been suggested that 17β-estradiol dilation of coronary arteries via BK channels is not the result of a direct action on the channel but mediated through NO/cGMP-mediated pathways (White et al., 2002).
Tamoxifen (a xenoestrogen) and analogs have complex actions on BK activity: increase and decrease in Po have both been reported, with this dual modulation being related to basal Po before drug application (Dick and Sanders, 2001; Duncan, 2005; Pérez, 2005). In contrast, LC increases Po at all voltages, [Ca2+]i, and levels of basal Po tested. Furthermore, under some conditions (Duncan, 2005; Pérez, 2005), the β1 subunit is not necessary for tamoxifen to evoke its complex actions on BK channels, with the α subunit being sufficient. Finally, tamoxifen and analogs decrease unitary current amplitude at concentrations as low as 1 to 10 μM (Duncan, 2005). This action might counterbalance the drug-induced increase in Po, with consequent reduction in drug potentiation of total BK current and, thus, vasodilation. Instead, the requirement for β1 subunits to increase Po and the lack of effects on unitary conductance are observed at all LC concentrations. Thus, in contrast to tamoxifen and analogs, LC modification of BK channel function is limited to that of a gating modifier.
Cholesterol at concentrations found in cell membranes reduces BK channel Po; not only is the final effect opposite to that of LC, but α subunits are sufficient for cholesterol action (Bolotina et al., 1989; Crowley et al., 2003). Finally, a recent article describes that corticosterone activates β4-containing BK channels more effectively than β2-containing BK channels, with the opposite being true for dehydroepiandrosterone. Testosterone, in turn, seems not to discriminate among channels containing these two β subunits (King et al., 2006). In contrast, LC concentrations that are maximally effective in activating cbv1+β1 channels completely fail to modulate cbv1+β4 channels.
Dehydrosoyasaponin-1 (DHS-1), a complex molecule that contains a steroidal nucleus, was reported to modulate BK channels through an interaction with the β1 subunit (Giangiacomo et al., 1998). DHS-1 is effective only when accessing the channel from the cytosolic side of the membrane, limiting its application to tissue/organ studies. In contrast, LC and structural analogs are similarly effective when applied to the external or internal membrane surface (Dopico et al., 2002). DHS-1 action is also strongly voltage-dependent, whereas LC is not. Finally, it is currently unknown whether other β subunits (other than β1) may render BK channels sensitive to nanomolar concentrations of DHS-1. It is noteworthy that the fact that LC is not sensed by α+β4 channel complexes allows us to speculate that LC and its analogs might be used to selectively target tissues/organs that contain high amounts of β1 subunits (i.e., smooth muscle) as opposed to others rich in α+β4 complexes [i.e., central nervous system, in which BK channel activation would affect neuronal excitability (Meredith et al., 2006) and/or neurotransmitter release (Brenner et al., 2005)]. In brief, based on these comparisons with other steroids that modulate BK channels, LC and probably its synthetic analogs (Dopico et al., 2002) may represent a unique tool to probe the presence of functional β1 subunits and/or modulate smooth muscle BK channel activity.
The exact locus of LC action remains speculation. The lateral chain of bile acids contains a carboxylate that is largely ionized at physiological pH (7.35-7.4) at which our experiments were conducted. The fact that LC action on cbv1+β1 channel Po is voltage-independent (suggesting that the ionized carboxylate is not sensed across the voltage field) is consistent with the charged lateral chain residing in or nearby the aqueous solution. The overall hydrophobicity of the steroid nucleus is very likely to place it within the lipid bilayer. It is remarkable that in contrast to other steroids, LC and analogs are planar amphiphiles. They present a bean-shaped molecule with two clearcut “planes” or “hemispheres”: a concave polar and a convex hydrophobic hemisphere. It is noteworthy that the planar polarity of the bile acid ring structure is critical for these steroids to increase BK channel Po (Dopico et al., 2002). Data using chimeric β1 and β4 subunits in which transmembrane-cytosolic end and the extracellular loop have been swapped indicate that the former region determines LC sensitivity (A. N. Bukiya, J. Liu, L. Toro, and A. M. Dopico, unpublished data). The β1 subunit transmembrane regions may bring ideal interfaces for LC membrane intercalation, with the hydrophobic hemisphere of the planar amphiphile facing the bilayer lipids and the hemisphere containing the polar hydroxyl facing the β1 subunit. In this putative model of LC location, however, the presence of polar groups on one side of the bile acid requires some polar surface to diminish the energetic cost of inserting the steroid polar groups within the hydrophobic environment of the bilayer core. It is interesting that the β1 subunit contains an unusually high number of threonine residues in its transmembrane segments. Furthermore, β4 subunits, which fail to sense LC, largely lack these polar residues in their transmembrane segments. Systematic mutagenesis combined with molecular modeling will determine which (if any) of the polar residues present in β1 but absent in β4 subunits are critical for interacting with bile acids. Although the exact locus of LC action on BK channels remains to be determined, it is clear from our study that the channel β1 subunit behaves as the bile acid sensor.
Acknowledgments
We deeply thank Robert Brenner (University of Texas at San Antonio) and Richard Aldrich (University of Texas at Austin) for their generous gift of β1 knockout mice, P. Liu for initially probing LC on cerebrovascular BK channels, Jonathan Jaggar for helpful comments, David Armbruster for critical reading of the manuscript, and M. Asuncion-Chin for technical assistance.
Footnotes
- Received January 19, 2007.
- Accepted April 27, 2007.
Supported by National Institutes of Health grants HL77424 and AA11560 (to A.M.D.) and HL54970 (to L.T.). J. L. is an American Heart Association Postdoctoral Fellow.
ABBREVIATIONS: LC, lithocholate; BK, large-conductance Ca2+-activated K+; PSS, physiological saline solution; Ibtx, iberiotoxin; wt, wild type; DM, dissociation medium; I/O, inside-out; O/O, outside-out; N, number of functional channels in the patch; DMSO, dimethyl sulfoxide; CMC, critical micellar concentration; CBF, cerebral blood flow; KV, voltage-gated K+; 4-AP, 4-aminopyridine; z, effective valence; DHS-1, dehydrosoyasaponin-1; NPo, product of the number of channels in the patch and the channel open probability.
↵
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.

- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}