


Abstract
Glucocorticoids (corticosteroids) are highly effective in combating inflammation in the context of a variety of diseases. However, clinical utility can be compromised by the development of side effects, many of which are attributed to the ability of the glucocorticoid receptor (GR) to induce the transcription of, or transactivate, certain genes. By contrast, the anti-inflammatory effects of glucocorticoids are due largely to their ability to reduce the expression of pro-inflammatory genes. This effect has been predominantly attributed to the repression of key inflammatory transcription factors, including AP-1 and NF-κB, and is termed transrepression. The ability to functionally separate these transcriptional functions of GR has prompted a search for dissociated GR ligands that can differentially induce transrepression but not transactivation. In this review, we present evidence that post-transcriptional mechanisms of action are highly important to the anti-inflammatory actions of glucocorticoids. Furthermore, we present the case that mechanistically distinct forms of glucocorticoid-inducible gene expression are critical to the development of anti-inflammatory effects by repressing inflammatory signaling pathways and inflammatory gene expression at multiple levels. Considerable care is therefore required to avoid loss of anti-inflammatory effectiveness in the development of novel transactivation-defective ligands of GR.
For the past half a century, glucocorticoids (corticosteroids or glucocorticosteroids) have been among the most effective and widely used medications for the treatment of inflammatory diseases (Rhen and Cidlowski, 2005; Barnes, 2006). Despite this, however, a complete picture of how glucocorticoids operate as anti-inflammatory agents is still lacking. Typically, glucocorticoids, such as the synthetic dexamethasone, bind the ligand-binding domain of the glucocorticoid receptor (GR) to promote nuclear translocation (Pratt et al., 2004). In the nucleus, the DNA binding domain directs dimerization on imperfect DNA palindromes (consensus: 5′-GGT ACA NNN TGT TCT-3′) known as simple glucocorticoid response elements (GREs) (Fig. 1A) to transcriptionally activate (transactivate) genes, including tyrosine amino transferase (TAT) or tryptophan oxygenase (Danesch et al., 1987; Jantzen et al., 1987; Rhen and Cidlowski, 2005). It is noteworthy that roles for glucocorticoid-inducible genes, such as phosphoenol pyruvate carboxykinase in regulating gluconeogenesis or TAT in amino acid catabolism, suggested that transactivation by GR was key to the metabolic effects of glucocorticoids (for a comprehensive review, see Schäcke et al., 2002). By contrast, some genes [including lipocortin I (also known as Annexin I) and, p11/calpactin binding protein (S100A10), which inhibit phospholipase A2 activity; secretory leukocyte protease inhibitor, a protease inhibitor; or the type II interleukin (IL)-1 receptor, a molecular decoy] were also identified as glucocorticoid-inducible and may contribute toward the anti-inflammatory properties of glucocorticoids (Flower and Rothwell, 1994; Re et al., 1994; Abbinante-Nissen et al., 1995; Yao et al., 1999). However, despite such findings, the principal reason for the effectiveness of glucocorticoids as anti-inflammatory agents lies in their ability to reduce inflammatory gene expression, and this was not primarily attributed to transactivation by GR (Rhen and Cidlowski, 2005; Barnes, 2006).
Transrepression and Anti-Inflammatory Effects of Glucocorticoids
Transrepression at Negative GREs. Initial examples of transcriptional repression (transrepression) by GR included the pro-opiomelanocortin (POMC) and prolactin gene promoters where cis-acting simple negative GRE sites (nGREs) were proposed to mediate repression via direct binding of GR to DNA (Fig. 1A) (Sakai et al., 1988; Drouin et al., 1989a,b). Thus binding of GR to a simple nGRE that overlaps the TATA box in the osteocalcin promoter was presumed to block transcription (Strömstedt et al., 1991; Meyer et al., 1997). However, nGRE sites correspond poorly to the GRE consensus and, in the case of POMC, prolactin, and other neuroendocrine genes, repression is more recently attributed to tethering mechanisms in which positive transcriptional regulators that bind DNA are then targeted by GR (Subramaniam et al., 1998; Chandran et al., 1999; Martens et al., 2005) (Fig. 1A).

Schematics of the transcriptional responses elicited by the GR. A, GR is recruited to transcriptional promoter regions by binding DNA as a homodimer in the context of simple GRE sites (i, iv), or in conjunction with other transcription factors (X, Y) at composite sites (ii, v), or finally via interaction with another transcription factor without actually contacting the DNA itself (i.e., tethering) (iii, vi). In each scenario, there may be positive or negative effects on transcription, and the GRE site is described accordingly. The nature of the response elicited by GR is communicated to the basal transcriptional machinery, represented here as TATA binding protein (TBP) and RNA polymerase II (Pol II). B, communication between GR and the transcriptional machinery is achieved via the recruitment of coactivators and corepressors to the transcription factor complex. i, to activate simple GRE-dependent transcription the coactivator CREB-binding protein (CBP) and a p160 protein family member (p160), often SRC-1, is bound by GR and links with the basal machinery. The intrinsic and the associated histone acetylase (HAT) activities of CBP, and others, results in histone, and transcription factor, acetylation. Subsequent opening of the chromatin structure around the promoter region facilitates transcriptional activation. ii, at tethering nGREs, transcriptional activators, such as NF-κB or AP-1 (X and Y), bind DNA in the context of coactivators such as CBP. GR exerts a repressive effect by further recruiting p160 family members (not shown) and one or more histone deacetylases (HDAC), which deacetylate both the DNA in the promoter region leading to closing of the chromatin structure and GR to promote interaction with NF-κB. Finally, GR-dependent loss of the Pol II C-terminal domain kinase P-TEFb reduces phosphorylation of Pol II and reduces transcription of target genes.
Transrepression of Inflammatory Gene Transcription. In the context of inflammatory gene promoters, consensus cis-acting sequences by which GR binds DNA and directly exerts transrepression are not generally described. Instead, such genes show binding sites for transcription factors, including activator protein (AP)-1, the functionally related activating transcription factors (ATFs), CCAAT/enhancer binding proteins (C/EBPs) and, in particular, nuclear factor (NF)-κB. It is noteworthy that these sites, which are key to transcriptional activation, are also necessary for glucocorticoid-dependent inhibition of inflammatory transcription (Barnes, 2006). Thus glucocorticoid-dependent repression of IL-8, inducible nitric-oxide synthase, or rat cytokine-induced neutrophil chemoattractant expression correlated with transcriptional inhibition, principally via NF-κB sites (Mukaida et al., 1994; Kleinert et al., 1996; Ohtsuka et al., 1996). Although glucocorticoids repressed NF-κB DNA binding activity in these studies, the repression of intercellular adhesion molecule-1 and E-selectin expression also involved NF-κB but did not involve reduced NF-κB DNA binding (van de Stolpe et al., 1994; Brostjan et al., 1997). Such discrepancies are widely reported and are likely to depend on the repressive mechanism(s) prevailing in any given situation. Thus, in A549 pulmonary cells, dexamethasone shows little inhibition of IL-1β-induced NF-κB DNA binding induced for up to 2 h, whereas stimulation for 6 h, or after long (24 h) glucocorticoid pretreatments, significantly reduces NF-κB DNA binding (Newton et al., 1998a). Such effects may involve reduced expression of the NF-κB subunits p50 (NFκB1) (Newton et al., 1998a; Simpson and Morris, 1999) or possibly p65 (RelA) (Simpson and Morris, 1999; Kurokouchi et al., 2000) and/or induced expression of the NF-κB inhibitor IκBα (see below).
Given that protein synthesis inhibitors prevent the repression of NF-κB DNA binding by dexamethasone, new gene expression is implicated; indeed, glucocorticoids can increase IκBα transcription and expression to reduce NF-κB DNA binding (Auphan et al., 1995; Scheinman et al., 1995a). However, the induction of IκBα is not necessary for repression of NF-κB-dependent transcription in many cells (Heck et al., 1997; Wissink et al., 1998). For example, in pulmonary type II and endothelial cells stimulated with tumor necrosis factor α, IL-1β, or lipopolysaccharide, IκBα expression was not increased, and NF-κB DNA binding was unaltered over time frames in which glucocorticoid-dependent repression of gene expression occurred (Brostjan et al., 1996; Ray et al., 1997; Newton et al., 1998a).
Likewise, glucocorticoid-dependent repression of the collagenase 1 promoter was localized to an AP-1 site that is key to transcriptional activation and this alone conferred glucocorticoid sensitivity (Jonat et al., 1990; Schüle et al., 1990). Furthermore, although direct interaction between AP-1 and GR was proposed to account for repression, one study noted no effect on AP-1 DNA binding (Jonat et al., 1990), whereas another reported inhibition (Yang-Yen et al., 1990). However, subsequent analysis of AP-1 site occupancy revealed no effect of glucocorticoids and direct interference of transcription (i.e., a tethering nGRE) represents the current model of inhibition (Fig. 1A) (König et al., 1992).
Nuclear Events Mediate Transrepression. As with AP-1, a direct interaction between NF-κB and GR, without DNA binding by GR, is suggested to account for transrepression by glucocorticoids (Ray and Prefontaine, 1994; Caldenhoven et al., 1995; Scheinman et al., 1995b). It is noteworthy that this process is independent of IκBα expression, NF-κB DNA binding (De Bosscher et al., 1997; Heck et al., 1997), or NF-κB site occupancy (Nissen and Yamamoto, 2000) and is described as a tethering nGRE (Fig. 1A) (De Bosscher et al., 2003). An apparent consequence of direct interference was a mutual antagonism between GR and both AP-1 and NF-κB that was suggested to result from competition for coactivators, particularly cAMP response element-binding protein binding protein (CBP) (Kamei et al., 1996; McKay and Cidlowski, 1998; Sheppard et al., 1998; McKay and Cidlowski, 2000). However, other studies dispel this notion and instead suggest that interference with the basal transcriptional apparatus explains transrepression, possibly via recruitment of the p160 family member, GR interacting protein (GRIP) (De Bosscher et al., 2000, 2001, 2003; Rogatsky et al., 2001, 2002). Alternatively, phosphorylation of the C-terminal domain of RNA polymerase (Pol) II is prevented by glucocorticoids and this may mediate promoter-selective inhibition of NF-κB-dependent transcription via the GR-dependent loss of a regulatory kinase complex (Nissen and Yamamoto, 2000; Luecke and Yamamoto, 2005). Likewise, and in keeping with the fact that histone acetylation is required for activated transcription (Adcock et al., 2004), glucocorticoids may also decrease the acetylation at inflammatory gene promoters by reducing CBP-associated histone acetylase activity and recruiting histone deacetylase (HDAC) 2 to the NF-κB (p65)-CBP complex (Ito et al., 2000, 2001). This produces a transcriptionally less favorable promoter conformation and may occur via deacetylation of GR to promote interaction with NF-κB (Ito et al., 2006). It is noteworthy that the tethering of GR and recruitment of HDAC2 to deacetylate histone H4 also explains repression at the POMC promoter, and this suggests a general physiological relevance for such a mechanism (Fig. 1B) (Bilodeau et al., 2006).
GR Mutants, Dissociated Steroids, and the Dim Mouse. Given that the metabolic side effects of glucocorticoids were largely ascribed to transcriptional activation, whereas repression of inflammatory gene transcription was attributed to transrepression, there is considerable interest in GR ligands that dissociate these two functions (Uings and Farrow, 2005). Clues to this possibility came from GR mutants that do not transactivate classic GRE-dependent transcription yet transrepress AP-1- and NF-κB-dependent transcription (Yang-Yen et al., 1990; Heck et al., 1994, 1997; Liden et al., 1997; Tao et al., 2001). For example, mutation of alanine 458 to threonine (A458T) within the dimerization, or D, loop of GR allows transrepression but prevents dimerization, DNA binding, and simple GRE-mediated transcription (Dahlman-Wright et al., 1991; Heck et al., 1994). Furthermore, replacement of wild-type GR with this (dim) mutant yields mice (GRdim/dim) that are defective in dexamethasone-induced GRE-dependent transcription and endogenous TAT gene expression yet are competent at repression of AP-1-dependent and classic inflammatory genes (Reichardt et al., 1998; Tuckermann et al., 1999; Reichardt et al., 2001). Likewise, GR mutants, which cannot bind coactivators or transactivate but can still transrepress NF-κB, further indicates that functional dissociation is achievable (Wu et al., 2004).
GR function may also be dissociated by ligands, including derivatives of RU486, that display limited simple GRE transactivation ability yet can still transrepress AP-1 reporters (Heck et al., 1994). Likewise, the steroidal compound RU24858 can be a poor transactivator but can efficiently repress AP-1-dependent responses (Vayssière et al., 1997). It is noteworthy that these compounds also repress NF-κB-dependent transcription and show anti-inflammatory properties in vivo (Vayssière et al., 1997; Vanden Berghe et al., 1999; Belvisi et al., 2001). However, despite these encouraging data, RU24848 also induces side effects, including loss of body weight and bone mass (Belvisi et al., 2001). Furthermore, in human eosinophils, RU24858 induced glucocorticoid-dependent genes, including lipocortin 1, to an extent similar to that of dexamethasone (Janka-Junttila et al., 2006). Conversely, in osteoblastic cells, RU24858 and related compounds were poor inducers, relative to prednisolone, of receptor activator of NF-κB ligand, a gene that promotes bone resorption (Humphrey et al., 2006). Thus the gene-specific and cell-type dependent transactivation ability of RU24858, even at simple GREs, suggests the existence of further regulatory determinants other than binding of ligand (Vayssière et al., 1997; Vanden Berghe et al., 1999; Tanigawa et al., 2002; Eberhardt et al., 2005; Chivers et al., 2006).
Nonclassical GR-Dependent Transactivation
Combinatorial Transactivation. In considering transcriptional activation by GR, it is important to note transactivation from nonsimple GREs. Thus, GR and AP-1 may synergize at composite and tethering promoter sites (Fig. 1A) (Pearce and Yamamoto, 1993; Teurich and Angel, 1995; Pearce et al., 1998). Likewise, glucocorticoids can positively regulate NF-κB-dependent responses, and synergy between NF-κB and GR occurs at appropriately spaced sites in artificial constructs and real genes (Wang et al., 1997; Hofmann and Schmitz, 2002; Webster et al., 2002). Furthermore, even at simple GREs, GR has long been known to synergize with other transcription factors, whereas positive combinatorial responses between GR and a variety of factors including signal transducer and activator of transcription (STAT) 1, STAT3, STAT5, C/EBP, Ets, Egr-1, and AP-2 are widely described (Schüle et al., 1988; Strähle et al., 1988; Aittomäki et al., 2000; Mullick et al., 2001; Tai et al., 2002). Thus IL-6 and dexamethasone combination, as could occur in the resolution or treatment of inflammation, synergistically induces the rat serine protease inhibitor-3 and the α2-macroglobulin genes via STAT3 and C/EBP α. (Kordula and Travis, 1996; Takeda et al., 1998; Lerner et al., 2003). Given that the latter did not involve GR DNA binding, a positive-tethering GRE (Fig. 1A) is indicated in the context of a multiprotein complex that also contains c-Jun (Lerner et al., 2003). Similar synergy between STAT5 and GR may variously be described as a positive composite or a tethering GRE depending on the requirement for GR DNA binding (Stöcklin et al., 1996; Cella et al., 1998). Furthermore, because C/EBPβ/STAT5 cooperativity requires GR (Wyszomierski and Rosen, 2001), and C/EBP proteins are glucocorticoid-inducible and can synergize with the GR, there is obvious potential for dramatic modulation of the transcriptional response by glucocorticoids (Strähle et al., 1988; Kordula and Travis, 1996; Gotoh et al., 1997). Likewise, cooperative interaction between GR, STAT5, and isoforms of nuclear factor (NF) 1 also highlights the importance of multiprotein complexes (Mukhopadhyay et al., 2001). Thus interaction between transcription factors, coactivators, including CBP and the steroid receptor cofactor (SRC) p160 family of proteins, allows differential and combinatorial responses at different promoters (Kamei et al., 1996; Freedman, 1999; Wyszomierski and Rosen, 2001; Lerner et al., 2003; Kabotyanski et al., 2006).
Dimerization-Independent Transcription. Depending on conditions and exclusion criteria, global gene expression analyses reveal that 1 to 20% of sequences are regulated by glucocorticoids and, of these, 45 to 70% are induced (Galon et al., 2002; Planey et al., 2003; Wang et al., 2003; Donn et al., 2007). Rogatsky et al. (2003) identified approximately 90 directly dexamethasone-inducible genes, 10 of which were analyzed in cell lines expressing either wild-type or mutant GRs. Although all genes showed glucocorticoid-inducibility by wild-type GR, mutants of the two GR activation functions (AF1 and AF2) or the dim mutant resulted in expression of different gene subsets. For example, with dim GR, 4 of 10 genes were induced to greater than 50% of their normal response, and only 3 of the 10 genes showed less than 20% of the wild-type response. This suggests a requirement for different aspects of GR function by different glucocorticoid-inducible genes. Consequently, while preventing classic GRE-dependent transcription and TAT induction, GRdim/dim mice may allow induction of other glucocorticoid-inducible genes to exert either anti-inflammatory or side effects. This is illustrated by phenylethanolamine N-methyltransferase (Tai et al., 2002), which is required for epinephrine synthesis, and the anti-inflammatory mitogen-activated protein kinase (MAPK) phosphatase (MKP-1) (Kassel et al., 2001), because these are both glucocorticoid-inducible by the GR dim mutant (Adams et al., 2003; Abraham et al., 2006). Likewise, survival of STAT5/GR interaction and resultant gene induction in GRdim/dim animals further questions the effect of dissociated GR ligands (Tronche et al., 2004).
Transactivation Plays an Important Anti-Inflammatory Role
A Partial Role for Transcriptional Repression. Despite the discussion above, it is clear that the transcriptional repression does not fully explain the repression of inflammatory gene expression by glucocorticoids (Table 1). Thus, in A549 pulmonary cells, repression of NF-κB-dependent transcription by dexamethasone was no more than 40 to 50% and this correlated with the transcription rate of NF-κB-dependent genes (Newton et al., 1998a,b; Chivers et al., 2006). Likewise, repression of IL-8 expression in primary airway epithelial cells, or transforming growth factor β-induced IL-11 in A549 cells, by dexamethasone was not primarily via reduced transcription (Wang et al., 1999; Chang et al., 2001). Likewise, in primary human airway smooth muscle cells, dexamethasone again reduced NF-κB-dependent transcription by no more than 50%, yet many NF-κB-dependent genes and the two bradykinin receptor genes in which post-transcriptional processes predominated were strongly repressed (Haddad et al., 2000; Catley et al., 2006). Moving over to inflammatory cells, analysis of the granulocyte/macrophage colony-stimulating factor and IL-5 promoters and both NF-κB- and AP-1-dependent transcription in primary human T cells and T cell lines also failed to document substantial a role for transrepression under conditions in which endogenous gene expression was highly repressed by dexamethasone (Staples et al., 2003; Bergmann et al., 2004). Finally, in primary human monocytes, lipopolysaccharide induced IL-1β transcription, yet the repression by dexamethasone was primarily post-transcriptional (Kern et al., 1988). Thus, using endogenous GR in multiple cells, including primary human cells, only a partial role for transcriptional repression in the repression of inflammatory gene expression by glucocorticoids is documented.
Examples of genes showing post-transcriptional regulation by glucocorticoids Selected examples of genes that are inhibited by glucocorticoids. Details of the mode of repression, cell type, and inducing stimulus are provided. The ability of either transcriptional or translational inhibitors, when added after the glucocorticoid, to block the repressive effects of glucocorticoids (Y), is given in the column “Sensitivity to Transcriptional or Translational Blockade.”
Post-Transcriptional Repression and Glucocorticoid-Dependent Gene Expression. Because 1) total inhibition of transcription (for example, by actinomycin D) when added after an inducing stimulus can be insufficient to repress inflammatory gene expression and 2) glucocorticoids only partially inhibit transcription, it is not surprising that post-transcriptional events, particularly mRNA destabilization, are also central to glucocorticoid-dependent repression (Table 1) (Newton, 2000; Stellato, 2004). It is noteworthy that such effects are frequently blocked by inhibitors of transcription or translation and a requirement for glucocorticoid-dependent gene expression is therefore indicated (Table 1). Indeed AU-rich elements (AREs), in the 3′ untranslated regions of unstable mRNAs can mediate both signal-induced mRNA stabilization and destablization by glucocorticoids (Peppel et al., 1991; Lasa et al., 2001; Fan et al., 2005). Furthermore, the ARE binding protein tristetraprolin, which promotes mRNA deadenylation and destabilization (Lai et al., 1999), may be induced by dexamethasone to post-transcriptionally repress inflammatory gene expression (Smoak and Cidlowski, 2006) (but see Jalonen et al., 2005).
MKP-1, an Anti-Inflammatory Glucocorticoid-Inducible Gene. The involvement of p38 MAPK in ARE-mediated stabilization of inflammatory genes provides a further mechanism for glucocorticoid-mediated destabilization via a process that requires glucocorticoid-induced gene expression (Winzen et al., 1999; Lasa et al., 2000, 2001; Clark, 2003). In this case, MKP-1, or dual specificity phosphatase 1, is strongly induced by glucocorticoids and dephosphorylates the active, phosphorylated form, of p38 MAPK to reduce both p38 activity and the stability of inflammatory gene mRNAs (Kassel et al., 2001; Chen et al., 2002; Lasa et al., 2002; Zhao et al., 2005) (Fig. 2). In addition, the p38 MAPK pathway may activate, or induce the expression of, inflammatory transcription factors including ATF-1, ATF-2, and AP-1 (see Wesselborg et al., 1997; Newton and Holden, 2003). Therefore induction of MKP-1 by glucocorticoids may also lead to transcriptional repression of inflammatory genes, such as E-selectin, that are regulated by AP-1 and/or ATF factors (Fürst et al., 2007). Likewise, p38 MAPK is an established positive regulator of NF-κB-dependent transcription (Wesselborg et al., 1997; Newton and Holden, 2003), and in such cases, glucocorticoid-induced MKP-1 expression is predicted to reduce expression of NF-κB-dependent genes.
However, MKP-1 specificity may not be limited to p38 MAPK, because both the extracellular signal-regulated kinases (ERKs) and c-Jun N-terminal kinases (JNKs) may be substrates for inhibition by this phosphatase (Chu et al., 1996; Franklin and Kraft, 1997; Kassel et al., 2001; Slack et al., 2001). Thus, in human airway smooth muscle cells, the induction of MKP-1 by dexamethasone represses JNK phosphorylation and is partly responsible for the repression of GROα expression (Issa et al., 2007). In addition, inhibition of JNK by MKP-1 directly represses AP-1-dependent transcription (Liu et al., 1995) and prevents both Elk-1 and serum response element activation (Wu et al., 2005), again indicating a role in the repression of transcription. Furthermore, the JNKs, like the p38 MAPK (Lee et al., 1994), are also implicated in the translational control of cytokine biosynthesis and indeed this process is also targeted by glucocorticoids (Swantek et al., 1997). Thus, bone marrow-derived macrophage from MKP-1-/- mice reveal impaired dexamethasone-dependent repression of many inflammatory genes, and this illustrates the considerable anti-inflammatory potential of glucocorticoid-induced MKP-1 (Abraham et al., 2006). Furthermore, the robust induction of MKP-1 by dexamethasone in GRdim/dim macrophage, indicates that GR dimerization is not necessary and suggests that GR ligands, which do not induce simple GRE responses, may still induce MKP-1 (Abraham et al., 2006). Indeed, despite poor activation of a simple GRE reporter, the induction of MKP-1 by RU24858 may account for the sensitivity of RU24858-dependent repression of cyclooxygenase-2, IL-8, and inducible nitric-oxide synthase expression to inhibitors of transcription and translation (Korhonen et al., 2002; Chivers et al., 2006).
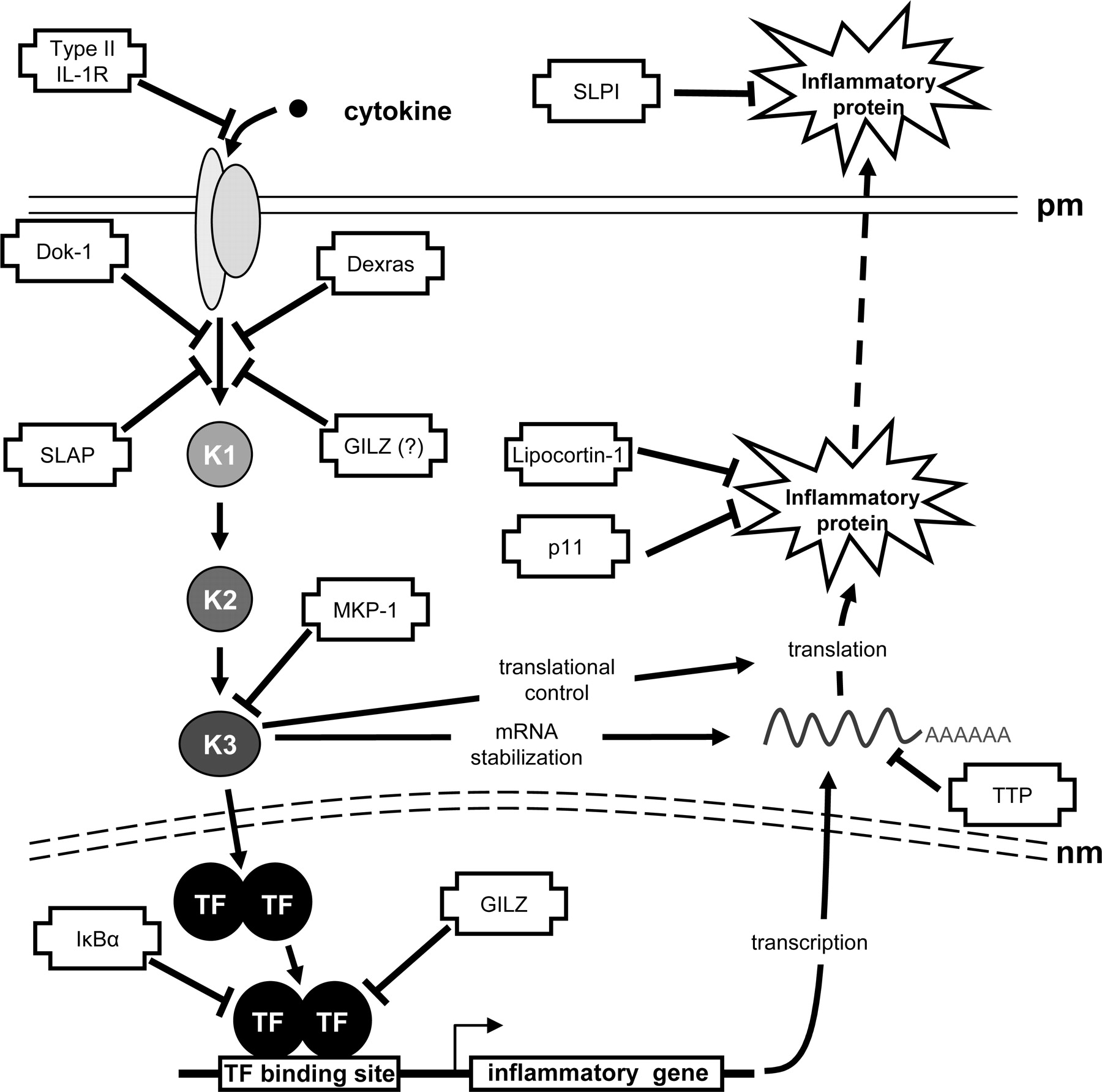
Global impact of glucocorticoid-inducible genes on inflammatory gene expression. A schematic representation of the cascades leading to inflammatory gene expression is depicted with possible targets and sites of action for putative anti-inflammatory glucocorticoid-inducible genes. Activation of a pro-inflammatory cascade, after binding of cytokine to its cognate receptor in the plasma membrane (pm), is shown occurring via a number of kinases (K1–3). The signal crossed the nuclear membrane (nm) and leads to transcription factor (TF) activation and the production of inflammatory gene mRNA. Under the influence of further kinase cascades (here K1–3), the mRNA is stabilized and translated into protein. Finally, many proteins are exported into the extracellular space for function. Sites of action of glucocorticoid-inducible genes are indicated. The glucocorticoid-inducible type II IL-1 receptor (IL-1R) acts as a decoy receptor to prevent activation of the cell by IL-1. Dexras, Src-like adaptor protein (SLAP) and downstream of tyrosine kinase (Dok)-1 all inhibit, through a variety of mechanisms, the activation of signal transduction cascades. A similar role for glucocorticoid-induced leucine zipper (GILZ) protein is suggested. Mitogen-activated protein kinase phosphatase (MKP)-1 is an inhibitor of the MAP kinase family and therefore affects numerous cellular mechanisms, including activation of transcription, mRNA stability, and translation. Inhibitor of κB (IκB) α and GILZ inhibit key inflammatory transcription factors (NF-κB and AP-1). Tristetraprolin (TTP) promotes deadenylation and degradation of mRNA. Lipocortin-1 and p11/calpactin binding protein inhibit phospholipase A2. Finally, secretory leukocyte protease inhibitor (SLPI) is a potent inhibitor of serine proteases.
Other Glucocorticoid-Inducible Genes with Anti-Inflammatory Potential. Partial glucocorticoid-dependent repression of inflammatory gene expression in MKP-1-/- animals suggests the existence of further repressive mechanisms (Abraham et al., 2006). Indeed, although a number of glucocorticoid-inducible genes with anti-inflammatory effects have been mentioned, numerous others also exist (Fig. 2). For example, glucocorticoids induce the expression of Clara cell secretory 10-kDa protein and thymosin β4 sulfoxide, which are protective in the context of pulmonary allergic inflammation and neutrophilic responses, respectively (Young et al., 1999; Chen et al., 2001). Likewise, the repression of inflammatory gene expression may be targeted by glucocorticoid-inducible leucine zipper (GILZ), which is highly glucocorticoid-inducible in T cells, macrophages, mast cells, and epithelial cells and represses both AP-1- and NF-κB-dependent transcription (Mittelstadt and Ashwell, 2001; Berrebi et al., 2003; Godot et al., 2006; Eddleston et al., 2007). Likewise, Dexras1, a glucocorticoid-inducible Ras-like protein that prevents activation of ERK, may target inflammatory signal transduction (Kemppainen and Behrend, 1998; Graham et al., 2002). Furthermore, interaction with, and inhibition of, protein kinase C δ by Dexras1 (Nguyen and Watts, 2006), or up-regulation of the adaptor proteins “downstream of tyrosine kinase” (Dok)-1 and Src-like adaptor protein (SLAP) by dexamethasone may also reduce inflammatory signaling, antigen-induced extracellular signal-regulated kinase activation, Ca2+ transients or activation of Syk kinase Syk (Hiragun et al., 2005, 2006).
A further mechanism by which glucocorticoids exert anti-inflammatory and immunosuppressive effects is by preventing proliferative responses and promoting apoptosis of certain immune cells (Newton, 2000). These effects are due in part to the repression of critical proliferative or antiapoptotic factors, for example IL-2, IL-3, granulocyte macrophage–colony-stimulating factor, and IL-5 with respect to T-cells and eosinophils. However, glucocorticoid-dependent arrest of lymphoid cells is also due to post-transcriptional mRNA destabilization of the G1 progression factor, cyclin D3 (Reisman and Thompson, 1995). As with the glucocorticoid-dependent repression of inflammatory genes, this effect is blocked by transcriptional inhibition, suggesting once more an involvement of glucocorticoid-inducible genes. Likewise, the proapoptotic effect of glucocorticoids on thymocytes requires GR transactivation, which in this case is not sustained by the dim GR mutant (Reichardt et al., 1998). Thus, glucocorticoid-induced proapoptotic genes in thymocytes and T cells may also contribute to reduced inflammation (Wang et al., 2003; Wang et al., 2006b).
GR Ligands and Function
Dissociated GR Ligands and Differential GR Function. Along with RU24858, numerous steroidal and nonsteroidal ligands of GR show various degrees of dissociated function based on standard reporter assays (Elmore et al., 2001, 2004; Shah and Scanlan, 2004; De Bosscher et al., 2005; Barker et al., 2006; Wang et al., 2006a). Although a number of these compounds show anti-inflammatory activity, detailed descriptions with respect to inducible genes or side effects are not reported. However, Elmore et al. (2004) characterized GR ligands in standard GRE transactivation and E-selectin promoter (transrepression) assays as well as on inflammatory and side effect genes. Thus certain high-affinity ligands revealed considerable differences in potency with respect to transrepression of the E-selectin promoter compared with repression of either IL-6 or collagenase expression. This suggests the existence of different mechanisms of inhibition with respect to each outcome and could reflect the distinct forms of transrepression for AP-1 and NF-κB that have been functionally established using both GR ligands and mutants (Shah and Scanlan, 2004; Bladh et al., 2005; Martens et al., 2005). However, an alternative explanation is that at least some of the observed “transrepression” is attributed to the glucocorticoid-dependent induction of genes (e.g., GILZ, MPK-1), which may mimic true transrepression. With respect to the compounds used in the study by Elmore et al. (2004), this concept is supported because classic GRE-dependent transactivation did not correlate with TAT or aromatase expression (Elmore et al., 2004). Thus, despite poor simple GRE-dependent transactivation, these ligands, as occurs for RU24858 (Belvisi et al., 2001; Chivers et al., 2006), may induce genes that relate to side effects or anti-inflammatory properties.
Explaining the Differential Effects of Ligands on GR Function. Binding of ligand to GR elicits conformational change in the receptor (Xu et al., 1999; Bledsoe et al., 2002; Kauppi et al., 2003). This may influence the precise interactions of GR within the nucleus, such that in the presence of antagonist, GR associates with the nuclear receptor corepressor (NCoR), whereas binding of agonist recruits the coactivator SRC-1 (Kauppi et al., 2003; Garside et al., 2004). Complexity is introduced to the system as the concentration-function relationship between a ligand and a response may be modulated by the local concentration of GR, coactivators, or their splice variants, as well as the nature of the interaction with DNA (Chen et al., 2000; Cho et al., 2005; Meijer et al., 2005). Indeed, mutually antagonistic effects of corepressors and coactivators with respect to the dose-response relationship to GR agonists means that the relative intracellular concentrations of each will determine final responsiveness, and such findings may explain the observed variability (potency and efficacy) of GR-dependent responses (Wang et al., 2004). Furthermore, interaction of nuclear hormone receptors with multiple chromatin remodelling enzymes, including CBP, p300, SRC-1, p300/CBP associated factor, as well as methylases and basal transcription factors (Freedman, 1999; McKenna and O'Malley, 2002; Uings and Farrow, 2005), means even for the simplest promoters that there are numerous interactions, that could be differentially modulated by the alternate conformations of GR bound by ligand. If the scope of transcriptional responses by GR, for example at simple GREs, composite GREs, tethering GREs (Fig. 1A), and the intermediates that lie between these conceptual extremes is considered, then the complex differential effects of GR ligands in modulating transcription becomes difficult to predict.
The Prospect of Therapeutically Improved Dissociated Glucocorticoids. The above discussion reveals multiple processes by which glucocorticoids acting via GR can affect gene expression. For example, GR, plus glucocorticoid, elicits positive transcriptional responses not only from simple GRE elements but also in combination with other transcription factors and via promoter elements that do not necessarily involve GR dimerization or contact with DNA. Indeed, a recent study of 548 known or potentially glucocorticoid-inducible genes revealed that most contain GRE sites that do not conform to the simple GRE palindrome and are more typically represented as composite elements (So et al., 2007). Thus, ligands that are silent at simple GREs may still induce transcriptional responses from these other promoters. Therefore, in the design and characterization of novel “dissociated” GR ligands, it is important to consider which types of transcriptional responses relate to which functional outcomes. Indeed, referring back to the original paradigm for dissociation, if a ligand was silent with respect to all forms of transactivation, it is questionable whether such a ligand would show any anti-inflammatory potential at all. There are two reasons for this: first, if a GR ligand is silent with respect to positive transcriptional responses via tethering GREs, there could also be issues as to the effectiveness of such a ligand in transrepression from tethering sites; second, the existence of glucocorticoid-inducible genes that repress AP-1 and NF-κB raises a question as to the nature of “transrepression.” Thus, induction of MKP-1 or GILZ, to switch of AP-1 or NF-κB may appear as transrepression.
In terms of separating transactivation from transrepression to generate novel anti-inflammatory agents with improved safety profiles, it is clear that many, if not most, transactivation mechanisms are not represented in current screening protocols. Consequently, the dissociation that is actually achieved by existing compounds is unclear. Furthermore, accumulating evidence supports the concept that there are many glucocorticoid-inducible genes and that a number of these exert effects that are consistent with anti-inflammatory benefit (Fig. 2). Thus the collective induction of such genes offers the potential for multiple anti-inflammatory effects that are integrated at different regulatory levels (Fig. 2). Therefore, loss of any transactivational properties may reduce the expression of some anti-inflammatory genes, and this could attenuate aspects of anti-inflammatory effectiveness (Fig. 2). Although detailed accounts of anti-inflammatory efficacy versus expression of side effects genes remain to be reported, novel compounds that show reduced up-regulation of steroid-inducible genes may in fact show lessened side effect issues (Coghlan et al., 2003; Schäcke et al., 2004; De Bosscher et al., 2005). However, such benefits will need to be very carefully balanced against the possibility that anti-inflammatory functions will also be lost. Therefore final clinical efficacy will depend on the relative balance of both “desirable” and “undesirable” transactivation and transrepression events, which need to be carefully defined to optimize the design of future GR ligands. Consequently, rather than searching for compounds that discriminate between transactivation and transrepression, we propose the need to functionally screen to identify “differential” compounds that display the most favorable functional profiles. Finally, steroid hormones also show nongenomic modes of action and these are not considered in current strategies to design improved GR ligands (Losel et al., 2003).
Footnotes
-
R.N. is a Canadian Institutes of Health Research (CIHR) New Investigator and Alberta Heritage Foundation for Medical Research (AHFMR) Scholar. Work in the laboratory of R.N. is supported by CIHR and AHFMR as well as by research awards from AstraZenenca, Altana, and GlaxoSmithKline.
-
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
-
doi:10.1124/mol.107.038794.
-
ABBREVIATIONS: GR, glucocorticoid receptor; GRE, glucocorticoid response element; TAT, tyrosine amino transferase; IL, interleukin; POMC, pro-opiomelanocortin; nGRE, negative GRE site; AP-1, activator protein-1; ATF, activating transcription factor; C/EBP, CCAAT/enhancer binding protein; NF-κB, nuclear factor-κB; IκB, inhibitor of κB; CBP, cAMP response element-binding protein binding protein; RU24858; STAT, signal transducer and activator of transcription; MAPK, mitogen-activated protein kinase; MKP-1, mitogen-activated protein kinase phosphatase; ARE, AU-rich element; JNK, c-Jun N-terminal kinase; GILZ, glucocorticoid-inducible leucine zipper; Dok, downstream of tyrosine kinase; Pol, RNA polymerase; SRC, steroid receptor coactivator.
- Received June 5, 2007.
- Accepted July 10, 2007.
- The American Society for Pharmacology and Experimental Therapeutics
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵





{kind=link}
{kind=link}