


Abstract
Accessory subunits in heteromeric nicotinic receptors (AChRs) do not take part in forming ACh binding sites. α5 and β3 subunits can function only as accessory subunits. We show that both α5 and β3 efficiently assemble in human α4β2* AChRs expressed in permanently transfected human embryonic kidney (HEK) cell lines. Only (α4β2)2α5, not (α4β2)2β3 AChRs, have been detected in brain. The α4β2α5 line expressed 40% more AChRs than the parent α4β2 line and was equally sensitive to up-regulation by nicotine. The α4β2β3 line expressed 25-fold more AChRs than the parental line and could not be further up-regulated by nicotine. Relative sensitivity to activation by ACh depends on the accessory subunit, β2 conferring the greatest sensitivity, α5 less, and β3 and α4 much less. Accessory subunits form binding sites for positive allosteric modulators, as illustrated by the observation that α5 conferred high sensitivity to galanthamine. In the presence of α5 or β3, stable, partially degraded, dead end intermediates accumulated within the cells. These may have the form α5α4β2α5. The efficiency with which α5 and β3 assemble with α4 and β2 and the necessity of avoiding formation of potentially toxic intermediates may explain why α5 and β3 seem to be transcribed at low levels in brain. Autosomal dominant nocturnal frontal lobe epilepsy can be caused by the α4 mutation S247F. This mutant did not produce functional AChRs unless cells were cotransfected with α5, β3, or α6 to replace α4 as accessory subunit.
Heteromeric neuronal nicotinic acetylcholine receptors (AChRs) contain two ACh binding sites formed at the interfaces of α and β subunits in two αβ subunit pairs and a fifth accessory subunit, all arranged like barrel staves to form a central cation channel (Gotti et al., 2007). α5 and β3 subunits can function only as accessory subunits, forming AChRs with stoichiometries such as (α4β2)2α5 or (α4β2)2β3, whereas α2 to 4 and β2 or β4 can either form ACh binding sites or assemble in the accessory position to produce AChRs with (αβ)2α or (αβ)2β stoichiometries (Nelson et al., 2003; Kuryatov et al., 2005; Briggs et al., 2006; Drenan et al., 2008).
When expressed in permanently transfected human cell lines, most human α4β2 AChRs are in the (α4β2)2α4 stoichiometry, which has low sensitivity to ACh and rapid desensitization relative to the (α4β2)2β2 stoichiometry (Nelson et al., 2003). Nicotine binds to partially assembled AChRs, acting as a pharmacological chaperone to selectively increase assembly of the (α4β2)2β2 stoichiometry (Kuryatov et al., 2005; Sallette et al., 2005). This stoichiometry has high sensitivity to ACh and slow desensitization.
α4β2* AChRs are the major brain subtypes with high affinity for nicotine, and 11 to 37% of these, depending on brain region, are (α4β2)2α5 AChRs (Gerzanich et al., 1998; Brown et al., 2007; Gotti et al., 2007; Mao et al., 2008). Knockout of α5 AChRs in mice reduced activation of high-sensitivity brain AChRs without reducing the total number of AChRs (Brown et al., 2007) and caused resistance to nicotine-induced seizures and hypolocomotion (Salas et al., 2003; Kedmi et al., 2004).
Human (α4β2)2α5 AChRs have the high sensitivity to ACh of (α4β2)2β2 AChRs, but higher permeability to Ca2+ when expressed in Xenopus laevis oocytes using linked α4 and β2 subunits in combination with free α5 subunits to force formation of this stoichiometry (Tapia et al., 2007). In this system, (α4β2)2β3 and (α4β2)2α4 AChRs have low sensitivity to ACh but high permeability to Ca2+.
α5 subunits in human α3* AChRs expressed in X. laevis oocytes increased the Ca2+ permeability and desensitization rates of all α3 AChRs (Gerzanich et al., 1998). α5 increased the sensitivity of α3β2 but not α3β4 AChRs to activation by ACh. When human α3* AChRs were expressed in permanently transfected HEK cell lines, expression in the α3β2α5 line was 2.8-fold greater than the α3β2 line. Both α3β2 and α3β2α5 lines were up-regulated by nicotine, but α3β4 and α3β4α5 were not (Wang et al., 1998).
β3-containing AChRs are located in aminergic neurons in association with α6 subunits, and have been found as (α6β2)2β3, (α6β4)2β3, and (α4β2)(α6β2)β3 AChRs (Champtiaux et al., 2003; Gotti et al., 2007; Salminen et al., 2007). Because ventral tegmental area neurons (which are involved in addiction to nicotine) and substantia nigra neurons (which are involved in Parkinson's disease) express α4, β2, β3, and α6 subunits, (α4β2)2β3 AChRs should have the opportunity to be formed but have not been immunoisolated from brain (Gotti et al., 2007; Perry et al., 2007; Mao et al., 2008). Presynaptic (α4β2)(α6β2)β3 AChRs modulate the release of dopamine and neuroprotection by nicotine, are exceptionally sensitive to activation by nicotine, and are thought to be especially important in Parkinson's disease and its primate models (Quik et al., 2007; Salminen et al., 2007).
β3 subunits expressed in permanently transfected HEK cell lines promote assembly of (α6β2)2β3 and (α6β4)2β3 AChRs with increased sensitivity to up-regulation by nicotine (Tumkosit et al., 2006).
Autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE) can be caused by the α4 mutation S247F in which a small hydrophilic serine in the M2 sequence lining the channel is replaced by a bulky hydrophobic phenylalanine (Klaassen et al., 2006; Teper et al., 2007). When this mutant is expressed in X. laevis oocytes at subunit mRNA ratios resulting primarily in the (α4β2)2β2 stoichiometry, functional AChRs are formed that lack Ca2+ permeability, but coexpression with α5 restores Ca2+ permeability (Kuryatov et al., 1997). When expressed in a cell line in which the (α4β2)2α4 stoichiometry predominates, no function was observed, presumably because the presence of three phenylalanines blocks the channel but the mutant AChRs are expressed efficiently and nicotine increases their assembly, as with wild-type AChRs (Kuryatov et al., 2005).
Here we report the properties of (α4β2)2α5 and (α4β2)2β3 AChRs expressed in permanently transfected HEK cell lines, demonstrating effects of accessory subunits on ACh assembly, sensitivity to activation by agonists, and modulation by allosteric modulators. We also report that replacement of the α4 accessory subunit in the ADNLFE cell line with other AChR subunits permits ion channel function and alters sensitivity to activation.
Materials and Methods
cDNAs and Chemicals. Human α4 and β2 cDNAs were cloned in this lab as described previously (Kuryatov et al., 1997; Wang et al., 1998). The cDNA for human α5 was provided by Dr. F. Clementi (CNR University of Milan, Milan, Italy) and subcloned in pCEP4 vector (Invitrogen) (Wang et al., 1998). Human β3 was obtained from Christopher Grantham (Janssen Research Foundation, Beerse, Belgium) and subcloned into pCEP4/Hygromycin(+) for transfection using HindIII and XhoI restriction enzymes. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise noted.
Tissue Culture and Transfection. The HEK tsA201 parental cell line expressing human α4β2 AChRs was described previously (Nelson et al., 2003; Kuryatov et al., 2005). All cell lines were maintained in Dulbecco's modified Eagle's medium (high glucose; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM l-glutamine (Invitrogen) at 37°C, 5% CO2 at saturating humidity.
For transient transfection, 100-mm dishes of 25% confluent α4β2 cells were transfected with 6 μg of β3 or α5 cDNAs using the FuGENE 6 DNA transfection kit (Roche Diagnostics, Indianapolis, IN). After 48 h, cells were collected using ice-cold PBS, and AChRs were extracted.
For permanent transfection, 35-mm dishes of 50% confluent α4β2 cells were transfected with β3 or α5 cDNAs using the FuGENE 6 DNA transfection kit (Roche Diagnostics). Hygromycin (Roche Diagnostics) was added at 0.1 mg/ml for α5 and β3 selection, 0.5 mg/ml Zeocin (Invitrogen) was added for α4 selection, and 0.6 mg/ml G418 (Invitrogen) was added for β2 selection. The transfected cells were passed onto 10-cm dishes before they were passed and plated on a 96-well plate for serial dilution. The 96-well plate was checked for the growth of single colonies; after the colony occupied approximately a quarter of the size of the well, it was plated onto a 24-well plate and then passed to three 35-mm dishes. Each promising clone was then tested to determine how much AChR was present. Screening for cells and extraction of stable clones continued as described previously by Tumkosit et al. (2006). Solid-phase assays for β2-containing AChRs were performed with mAb 295-coated wells, and assays for α5 and β3 containing AChRs were performed with mAb 210-coated wells.
Antiserum and mAbs. A rat antiserum to bacterially expressed α4 subunit sequences (excluding the transmembrane domains) was raised as described previously (Kuryatov et al., 2000). The rat mAb 210 binds to the main immunogenic region of human α1, α3, α5 (Lindstrom, 2000), and β3 (Tumkosit et al., 2006). The rat mAb 295 binds to the extracellular domain of native β2 subunits with high affinity only when they are associated with α3, α4, or α6 subunits (Lindstrom, 2000).
AChR extracts were incubated in mAb-coated microtiter wells for solid-phase radioimmunoassay, or with mAb-coupled to activated CH-Sepharose (GE Healthcare, Chalfont St. Giles, Buckinghamshire, UK) for purifying AChRs for use in immunoblot assays or loaded directly onto 5-ml sucrose gradients [5-20% sucrose (w/w)] for sedimentation analysis (Kuryatov et al., 2005).
For immunoprecipitation of AChRs with subunit-specific antibodies, the extract was incubated overnight with mAb or antiserum in the presence of [3H]epibatidine (2 nM). The AChR-antibody complexes were immunoprecipitated with sheep anti-rat IgG for rat antibodies. [3H] Epibatidine-labeled AChRs in the pellet were quantified using liquid scintillation counting. Nonspecific precipitation was measured using either normal mouse serum or normal rat serum.
AChR Extraction and Determining α5 and β3 Incorporation. Cells from which AChRs were to be extracted were collected in ice-cold PBS (100 mM NaCl and 10 mM sodium phosphate, pH 7.4) then centrifuged at 13,000g for 15 min in Eppendorf tubes with 1 ml of buffer A (50 mM NaPO4, pH 7.5, 50 mM NaCl, 5 mM EDTA, 5 mM EGTA, 5 mM benzamidine, 15 mM iodoacetamide, and 2 mM phenylmethylsulfonyl fluoride). The pellets were resuspended in buffer A plus 2% Triton X-100 and incubated for 1 h at room temperature to solubilize AChRs. Insoluble material was removed by centrifugation at 13,000g for 15 min. Total protein concentration of solubilized AChRs was determined using a BCA protein assay kit (Pierce Chemical, Rockford, IL).
The most stable α4β2β3 clones and α4β2α5 clones were selectively screened and tested for high expression of β3 and α5 based on liquid phase radioimmune assays with mAb 295 and mAb 210 as described previously (Kuryatov et al., 2005; Tumkosit et al., 2006). AChR-antibody complexes were immunoprecipitated with sheep anti-rat IgG. For immunoprecipitation of AChRs with subunit-specific antibodies, the extract was incubated overnight with mAb or antiserum in the presence of [3H]epibatidine (2 nM) (PerkinElmer Life And Analytical Sciences, Waltham, MA). [3H]Epibatidine-labeled AChRs in the pellet were quantified using liquid scintillation counting. Nonspecific precipitation was measured using normal rat serum.
Sucrose Gradients. Aliquots of 150 μl of cell extract in 2% Triton X-100 in buffer A were layered onto 11.3 ml of linear 5 to 20% sucrose gradients (w/v) in 0.5% Triton X-100 solution of PBS, 5 mM EDTA, 5 mM EGTA, and 1 mM NaN3 at pH 7.5. The gradient was centrifuged for 16 h at 40,000 rpm in a Beckman SW41 rotor. An aliquot (1 μl) of 2 mg/ml purified Torpedo californica electric organ AChR was added to the cell extract as an internal sedimentation standard. After centrifugation, 17-drop fractions were collected from the bottom. Immulon 96-well 4HBX plates (Thermo Fisher Scientific, Walthman, MA) were coated with mAb 295 to detect β2 subunits, mAb 210 to detect α5 or β3 subunits, or mAb 299 or mAb 371 to detect α4 subunits. Aliquots (20 μl) from each gradient fraction were added to appropriate wells to detect epibatidine binding or α-bungarotoxin binding.

Immunoisolation shows that nearly all of the AChRs expressed by the α4β2α5 and α4β2β3 cell lines contain the expected accessory subunit. mAbs 295 and 210 were coupled at 2 mg/ml to activated CH-agarose 4B. Triton X-100 extracts containing 15 and 30 pM AChR for α4β2α5 and 100 and 200 pM AChR for α4β2β3 cell lines were labeled with 2 nM [3H]epibatidine and assayed by immune precipitation with mAb 295. Aliquots of extract (20 μl) were adsorbed overnight on a shaker at 4°C with aliquots (20 μl) of mAb agarose, then the supernatants were re-assayed the next day.
Biotinylation. Cells from two 10-cm dishes of α4β2β3 were collected using ice-cold PBS and then washed in the same buffer. The cell suspension was labeled by EZ-link Sulfo-NHS-LC biotin (Pierce) at 1 mg/ml at 0°C for 1 h. The reaction was stopped by washing in PBS + 100 mM glycine. The pellet was solubilized in Triton X-100 as described above. Biotinylated AChRs from the cell surface were immunoisolated from sucrose gradients fractions on microwells coated with streptavidin.
Binding of [3H] Epibatidine. Surface expression in α4β2α5 cells was determined similarly to Kuryatov et al. (2005). Surface expression of α4β2β3 cells has to be done on collagen-coated 24-well plates (BD Discovery Labware, Bedford, MA) because of low adhesion of this cell line. When the α4β2β3 cells reached more than 50% confluence, 0.5 nM [3H]epibatidine was added to wells to label AChRs. Binding to AChRs in the cell surface was inhibited by 1 mM butyrylcholine chloride (Sigma-Aldrich), a membrane impermeable quaternary amine, to determine the internal pool of AChRs. Nonspecific binding was determined by addition of 100 μM nicotine. After incubation for 30 min on ice, the cells were washed three times with 0.5 ml of Dulbecco's modified Eagle's medium and dissolved in 200 μl of 0.1 N NaOH. The bound radioactivity was determined in Eppendorf tubes with 1 ml per tube of OptiPhase “Supermix” scintillation fluid using a 1450 Trilux Microbeta liquid scintillation counter (Perkin-Elmer Life and Analytical Sciences)
Up-Regulation of Epibatidine Binding Sites In Stably Transfected Cell Lines. Cells were plated in 100 μl of medium at a density of 70,000 to 100,000 cells per well on 96-well white clear-bottomed plates (Corning Incorporated, Corning, NY). The next day, nicotine was added. After incubation for 24 h, cells were fixed by adding 100 μl of 4% phosphate buffered formaldehyde (Fisher Scientific, Fair Lawn, NJ) per well for 1 h. Then AChRs were measured using [3H]epibatidine as described above.
FLEXstation Experiments. AChR function was determined in the cell lines using a FLEXstation II (Molecular Devices, Sunnyvale, CA) bench-top scanning fluorometer as described by Kuryatov et al. (2005). The day before the experiment the cells were plated at 100,000 cells/well on poly(d-lysine)-coated black-walled/clear-bottomed 96-well plates (BD Biosciences). Membrane potential and Ca2+ assay kits (Molecular Devices, Sunnyvale, CA) were used according to the manufacturer's protocol. Serial dilutions of drugs were prepared in V-shaped 96-well plates (Fisher Scientific Co., Pittsburgh, PA) and were added in separate wells at a rate of 20 μl/s during recording. Each point on the curves represents the average of three to four responses from different wells. The Hill equation was fitted to the concentration-response relationship using a nonlinear least-squares error curve-fit method (Kaleidagraph; Synergy Software, Reading, PA): I(x) = Imax [xn/(xn + EC50n)], where I(x) is the current measured at the agonist concentration x, Imax is the maximal concentration for the half-maximal response, and n is the Hill coefficient.
Results
Construction of Human α4β2α5 and α4β2β3 AChR-Expressing Cell Lines in tsA201 HEK Cells. As expected, transient transfection of tsA201 HEK cells with the α4β2 subunit combination resulted in functional AChRs, whereas α5 and β3 acted as obligate accessory subunits and did not form epibatidine binding sites when expressed in α4β3, α4α5, α5β2, or β3β2 combinations (data not shown).
Construction of permanent cell lines started with the α4β2 AChR cell-line described previously (Nelson et al., 2003; Kuryatov et al., 2005). This was transfected with Huα5/pCEP4 or Huβ3/pCEP4 to produce lines expressing α5 or β3 accessory subunits. Total AChR expression was measured by immunoisolation of [3H]epibatidine-labeled AChRs using mAb 295 to β2 subunits. Assays of incorporation of α5 and β3 used mAb 210, which was made to the main immunogenic region of α1 subunits through immunization of rats with bovine muscle AChR (Lindstrom, 2000) but also cross-reacts with human α1 (Lindstrom, 2000), α3 (Wang et al., 1998), α5 (Kuryatov et al., 1997), and β3 (Tumkosit et al., 2006). Adsorption of AChR extracts with mAb 210 coupled to Sepharose beads could adsorb virtually all of the AChRs from the cell lines, showing that virtually all of the AChRs incorporated either α5 or β3 subunits (Fig. 1). This also implies that α5 and β3 were expressed in amounts nearly equal to or greater than the amount of α4 and β2.
The α4β2α5 line expressed substantial amounts of [3H]epibatidine binding sites (1.75 ± 0.25 pmol/mg protein), approximately 40% more than the amount expressed by the parent α4β2 cell line (1.25 ± 0.35 pmol/mg protein). The α4β2β3 line expressed 25-fold more AChR (30 ± 10 pmol/mg protein). Thus, both α5 and β3 are efficiently incorporated into α4β2* AChRs. β3 seems to substantially promote assembly of AChRs with the α4β2 line, even more than it does in the cases of α6β2 and α6β4 AChR cell lines (2- to 6-fold) (Tumkosit et al., 2006). Transient transfections of the α4β2 cell line with either α5 or β3 (resulting in incorporation levels of 29 and 49%, respectively) did not significantly increase the total amount of AChR. However, in two other permanently transfected cell lines, (α4β2)2β3 AChRs were expressed in similarly remarkably high levels. One of these lines was the α6β2β3 line described in Tumkosit et al. (2006) transfected with α4, and the other was an α4α6β2 line transfected with β3. Thus, although there may be selection biases associated with cloning a particular line, after long-term selection, the presence of both α4 and β3 (expressed in excess) was associated with very high levels of (α4β2)2β3 AChR expression but not of greatly increased amounts of α6* AChRs when α6 was also present.

Activation of AChRs in the α4β2α5 and α4β2β3 cell lines assayed using a Ca2+-sensitive fluorescent indicator in a FLEXstation. A-C, dose/response curves for α4β2α5 cells. D-F, dose/response curves for α4β2β3 cells.
Functional Properties of (α4β2)2α5 and (α4β2)2β3 AChRs. Function was assayed using a fluorescent indicator for Ca2+ concentration in microwell cultures and a FLEXstation scanning fluorometer. The initial α4β2 line exhibited a high sensitivity (α4β2)2β2 AChR component (ACh EC50 = 0.23 μM, comprising 23% of the total response) and a low sensitivity (α4β2)2α4 AChR component (ACh EC50 = 57 μM, comprising 77% of the total response) when assayed using a Ca2+ sensitive indicator (Kuryatov et al., 2005). When a membrane potential-sensitive indicator was used, 50% of the response was from the more sensitive (α4β2)2β2 stoichiometry and 50% of the response from the less sensitive (α4β2)2α4 stoichiometry (Kuryatov et al., 2005). This is because electrophysiological studies in X. laevis oocytes showed that the sensitive (α4β2)2β2 stoichiometry had lower permeability to Ca2+ than the less sensitive (α4β2)2α4 stoichiometry (Tapia et al., 2007). Very similar two component dose/response curves are seen for α4β2* AChRs in synaptosomes from mouse thalamus assayed by agonist-induced Rb+ flux (Marks et al., 2007). Thus, it is likely that similar mixtures of α4β2 AChR stoichiometries exist in brain. Using linked α4 and β2 subunits expressed in oocytes, we found the (α4β2)2α5 subtype to be as sensitive to ACh as are (α4β2)2β2 AChRs but with higher permeability to Ca2+ (Tapia et al., 2007).
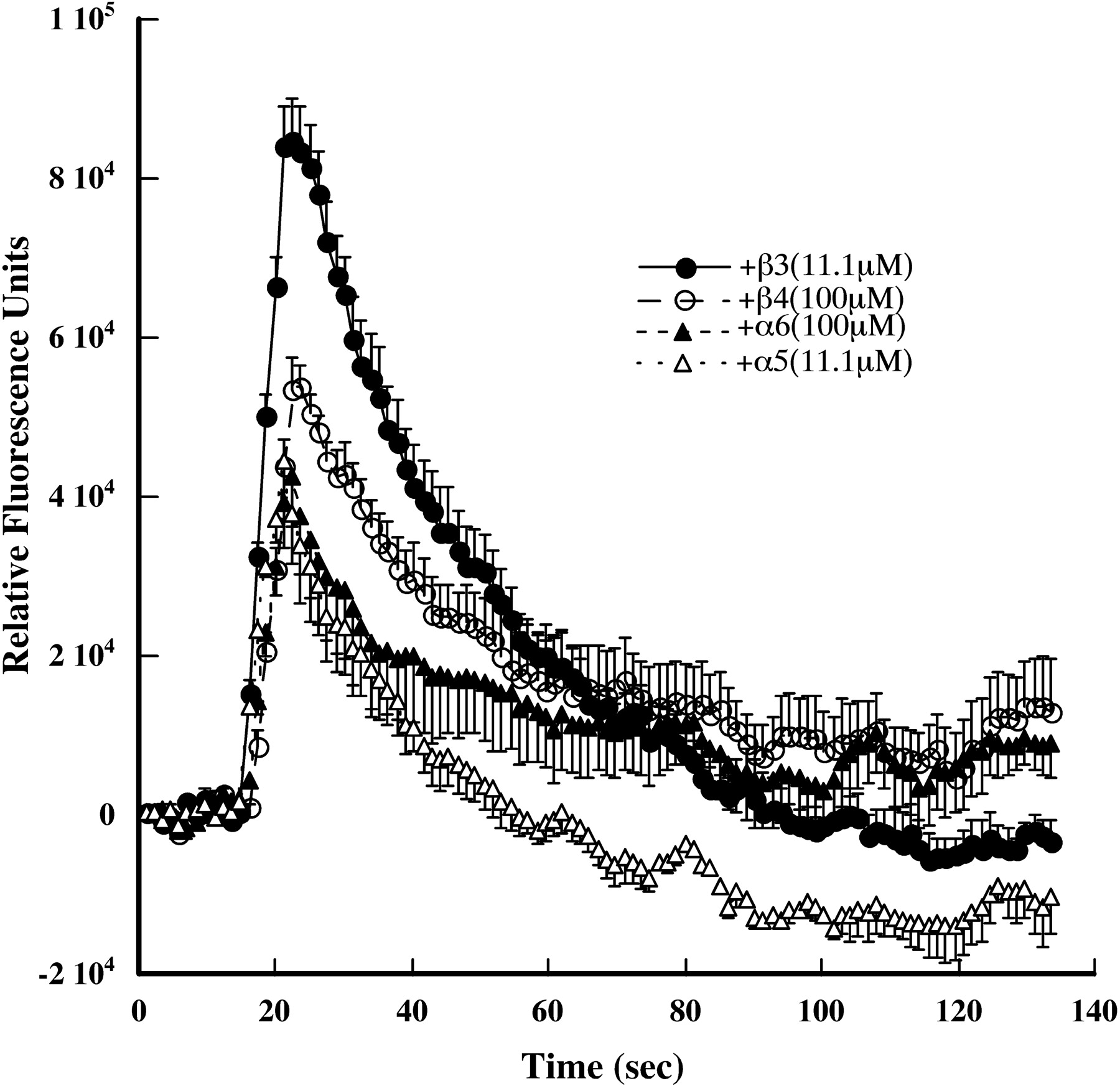
As expected for a cell line expressing on its surface exclusively (α4β2)2α5 AChRs, there were single component dose/response curves for activation by agonists (Fig. 2, A-C). (α4β2)2α5 AChRs in the cell line were 46-fold more sensitive to ACh than were (α4β2)2α4 AChRs but 5-fold less sensitive than were (α4β2)2β2 AChRs (Tables 1 and 3; Fig. 2. A-C). Sensitivity to activation by nicotine of (α4β2)2α5 AChRs was 3-fold less than the (α4β2)2β2 stoichiometry but 7-fold more than the (α4β2)2α4 AChRs. Cytisine behaved as a 25% agonist on (α4β2)2α5 AChRs in the cell line. Cytisine has been reported to not act as an agonist on the (α4β2)2β2 stoichiometry and to act as a 22% partial agonist on the (α4β2)2α4 stoichiometry expressed in X. laevis oocytes using high ratios of either β2 or α4 to produce one stoichiometry or the other (Moroni et al., 2006). In our hands, using linked α4 and β2 subunits plus free β2, α5, or α4 subunits to produce defined AChR stoichiometries expressed in X. laevis oocytes, cytisine shows 3.6% efficacy on (α4β2)2β2 AChRs, 8.3% efficacy on (α4β2)2α5 AChRs, and 22% efficacy on (α4β2)2α4 AChRs (data not shown). Thus, the accessory subunit in α4β2* AChRs has a large effect on the extent to which cytisine is a partial agonist.
α4β2* AChRs are more sensitive to block of function by desensitization caused by prolonged exposure to nicotine than to blockage by competitive or noncompetitive antagonists, or than to activation by most agonists
Comparison of properties of α4β2 AChR subtypes
In oocytes expressing linked α4 and β2 subunits, the (α4β2)2β3 subtype was found to be approximately as insensitive to ACh as the (α4β2)2α4 stoichiometry and similarly high in permeability to Ca2+ (Tapia et al., 2007). The α4β2β3 cell line exhibited a biphasic dose/response curve with approximately 18% exhibiting the same high sensitivity to ACh of the (α4β2)2β2 stoichiometry and the remainder exhibiting low sensitivity (Table 1, Fig. 2, D-F). These results suggest that 18% of the surface AChRs in this cell line have the (α4β2)2β2 stoichiometry, whereas more than 80% have the (α4β2)2β3 stoichiometry. Consistent with this, Fig. 1 showed that 13% of the AChRs did not incorporate a β3 subunit. (α4β2)2β3 AChRs were 5-fold less sensitive to nicotine than were (α4β2)2α5 AChRs. Cytisine was a 19% agonist on (α4β2)2β3 AChRs expressed in the cell line. (α4β2)2β3 AChRs exhibited 12-fold more nicotine sensitivity than (α4β2)2α4 AChRs and 7.8-fold lower sensitivity than (α4β2)2α5 AChRs.
Agonists both activate and desensitize AChRs. AChRs in cell lines can be assayed for acute desensitization by agonists and for desensitization over long periods that reflect the time period that they would be exposed to drugs in vivo. α4β2* AChRs are more sensitive to block of function by desensitization than they are to most competitive or noncompetitive antagonists or than they are to activation by most agonists (Tables 1 and 3). The parent α4β2 line is 14-fold more sensitive to long-term desensitization by nicotine than it is to acute competitive block by DHβE and 126-fold more sensitive to long-term desensitization by nicotine than it is to acute channel block by mecamylamine. Although acute desensitization of the (α4β2)2α4 stoichiometry is more rapid (Nelson et al., 2003), both stoichiometries seem to desensitize to the same final state after exposure to nicotine. There is a monotonic long-term desensitization curve to nicotine, indicating that both stoichiometries are equally sensitive to long term desensitization.
(α4β2)2α5 AChRs are similarly sensitive to long-term desensitization by nicotine as are AChRs in the parent line (Table 1). (α4β2)2β3 AChRs are 4.5-fold less sensitive to long-term desensitization by nicotine. During exposure to nicotine over 2 min, (α4β2)2α5 AChRs desensitize more rapidly to a lower plateau level (15 ± 1%) than do α4β2 (68 ± 9%) AChRs (Fig. 3). These results, assayed using a Ca2+-sensitive fluorescent indicator and the FLEXstation, are consistent with results obtained electrophysiologically with (α4β2)2α5 AChRs expressed in X. laevis oocytes (Ramirez-Latorre et al., 1996; Kuryatov et al., 1997). (α4β2)2β3 AChRs also acutely desensitize more rapidly than do α4β2 AChRs but less rapidly than (α4β2)2α5 AChRs. At the 0.1 to 0.2 μM concentrations of nicotine sustained in the sera of smokers (Benowitz, 1996), the IC50 = 0.009 μM (α4β2)2α5 AChRs or the IC50 = 0.027 μM (α4β2)2β3 would result in most of these AChRs being desensitized. Most (α4β2)2α5 AChRs would also be desensitized by the 0.0054 μM plasma concentration of nicotine produced by 1 to 2 puffs of a cigarette (Brody et al., 2006).
(α4β2)2α5 AChRs are 3-fold less sensitive to acute blockage by DHβE or mecamylamine than are the predominantly (α4β2)2α4 AChRs of the parent line (Tables 1 and 3). Thus, accessory subunits can have substantial effects on activation, desensitization, and antagonist effects, even though accessory subunits do not contribute to formation of ACh binding sites. This is consistent with the effects of α5 observed on α3β2 and α3β4 AChRs (Gerzanich et al., 1998).
Galanthamine has been reported to act as a positive allosteric modulator (PAM) of human α4β2 AChRs expressed in permanently transfected HEK 293 cells (Samochocki et al., 2003). We observed that very low concentrations of galanthamine (EC50 = 0.25 nM) increased the response of (α4β2)2α5 AChRs to 1 μM ACh by up to 220% (Fig. 4). Only small potentiation (20%) of either α4β2 or (α4β2)2β3 AChRs was detected using FLEXstation assays. Galanthamine at concentrations of 1 μM and above inhibited all three AChR subtypes, consistent with the results of Samochocki et al. (2007). These experiments illustrate the principle that a particular AChR accessory subunit can confer high sensitivity to a PAM.
Nicotine-Induced Up-Regulation. Nicotine acts as a molecular chaperone to selectively increase assembly of the (α4β2)2β2 stoichiometry in the parent α4β2 cell line with an EC50 = 0.035 μM and an extent of 4.9-fold (Kuryatov et al., 2005).
Nicotine up-regulated the amount of (α4β2)2α5 AChRs with an EC50 = 0.0353 ± 0.0078 μM and an extent of 4.8-fold (Tables 2 and 3). Thus, α5 does not alter the sensitivity or extent of nicotine-induced up-regulation of α4β2* AChRs. This might suggest that nicotine acts to promote assembly of α4β2 subunit dimer or α4β2α4β2 subunit tetramer assembly intermediates before assembly with α5, because α5 decreased sensitivity to activation by nicotine 3-fold and greatly increased sensitivity to rapid desensitization. However, the IC50 for desensitization by nicotine after exposure for hours is approximately the same for the α4β2 and α4β2α5 lines, and this may be the most relevant parameter for nicotine-induced up-regulation if a desensitized conformation is what promotes assembly. The up-regulated AChRs continued to incorporate α5 efficiently, as shown by the ability of mAb 210 (to α5) to immune precipitate all of the AChRs, which could be immune-precipitated by mAb 295 (to β2 subunits) (Fig. 5).
Upregulation of α4β2* AChRs in cell lines

Acute desensitization of responses to 30 μM ACh assayed using a Ca2+ fluorescent indicator in a FLEXstation. Both (α4β2)2α5 and (α4β2)2β3 AChRs desensitize much more rapidly than does the mixture of (α4β2)2β2 and (α4β2)2α4 AChRs in the α4β2 cell line. The responses shown are the averages of four microwell cultures from each line.

The PAM galanthamine selectively potentiates (α4β2)2α5 AChRs. ACh and galanthamine were added simultaneously, and activity was assayed using a Ca2+-sensitive indicator in the FLEXstation. ACh was used as agonist at 1 μM for both the α4β2 line and α4β2α5 line and at 30 μM for the α4β2β3 line in the experiments shown. ACh was also tested on the α4β2 line at 0.1 μM ACh so as to be below the EC50 for the (α4β2)2β2 stoichiometry. At this concentration, as at 1 μM, only a small positive allosteric effect was observed. Concentrations of galanthamine below 10 nM (EC50 = 0.25 nM) increase the response of the α4β2α5 line to an EC50 concentration of ACh by a maximum of 2.2-fold. The responses to near EC50 ACh concentrations of the α4β2 or α4β2β3 cell lines were increased by <20%.
Sensitivity to up-regulation of the α4β2α5 line by cytisine (EC50 = 0.0058 ± 0.0014 μM) (Table 2) was similar to that of the parent α4β2 cell line (EC50 = 0.0075 ± 0.0027 μM) (Kuryatov et al., 2005). Note that the sensitivities to upregulation (Tables 2 and 3) and desensitization (Table 1) are much greater than the sensitivities to activation (e.g., 3- to 19-fold in the case of nicotine). Note also that agonists are more potent at up-regulation than the competitive antagonist. These results suggest that a desensitized conformation of α4β2 intermediates assembles more efficiently than a resting or active conformation.
By contrast with (α4β2)2α5 AChRs, (α4β2)2β3 AChRs were not up-regulated at all by nicotine (Tables 2 and 3). This may be relevant to the observation that the α4β2β3 line expressed AChRs at 25 times the level of the parent α4β2 line, approximately the level obtained when the parental line was maximally up-regulated by nicotine and all of the α4 and β2 subunit pools were incorporated into mature AChRs (Kuryatov et al., 2005), leaving no possibility for a further effect of nicotine. In the case of α6 AChRs, β3 increased the level of expression in cell lines selected from α6β2 or α6β4 parental lines 1.5- to 3.6-fold but also increased the sensitivity to nicotine-induced up-regulation by 6.6- to 11-fold (Tumkosit et al., 2006). In these lines, α6β2 AChRs and α6β4 AChRs were expressed at levels 5% that of the α4β2 line. The presence of β3 resulted in both more mature α6 AChRs and more α6 detected in Western blots, suggesting that α6 was unstable unless incorporated into mature AChRs. By contrast, similar α3β2 and α4β2 cell lines (Wang et al., 1998; Kuryatov et al., 2005) have large stable pools of partially assembled subunits, which can be quickly assembled into many more mature AChRs in the presence of nicotine without requiring the synthesis of new subunits.
Surface Membrane Expression. (α4β2)2β3 AChRs were very efficiently (67 ± 15%) expressed on the cell surface. For comparison, in the α4β2 line, 81% of AChRs are on the surface normally and 60% after up-regulation by nicotine (Kuryatov et al., 2005). Thus, not only are (α4β2)2β3 AChRs efficiently assembled but they are also expressed on the cell surface with high efficiency comparable with the α4β2 cell line. By contrast, (α4β2)2α5 AChRs were much less efficiently (20 ± 6%) expressed on the cell surface, and efficiency of expression on the surface was not further increased by up-regulation with nicotine. Specific association of α5 with a postsynaptic scaffold protein may be required for optimal expression on the cell surface (Conroy et al., 2003). These assays measured the fraction of epibatidine binding sites that were expressed on the cell surface. The percentage of mature pentameric AChRs expressed on the cell surface is actually higher than this would indicate because, as will be described later, within the cell nearly half of the epibatidine binding sites are on partially assembled AChRs.
Effects of Chaperones on Incorporation of α5 and β3. A possible explanation for why (α4β2)2β3 AChRs have not been observed in immunoisolation studies from brain (Gotti et al., 2007; Perry et al., 2007; Mao et al., 2008), despite the evidence presented here that β3 can assemble very efficiently with α4β2, is that a specific chaperone present in neurons inhibits the incorporation of β3 on the minus side of α4 subunits. We investigated transient transfection of the α4β2α5 and α4β2β3 cell lines with Lynx1 (Ibañez-Tallon et al., 2002) and Ric-3 (Lansdell et al., 2005) as candidate chaperones. Neither decreased incorporation of β3 into AChRs (data not shown). Transfection with Lynx1 caused up to a 40% decrease in incorporation of α5 and caused an equal amount of a second low affinity (EC50 = 85.7 μM) component in the α4β2α5 dose/response curve [probably (α4β2)2α4 AChRs]. Transfection with Ric-3 greatly increased expression of mature α7 AChRs in an α7 cell line (data not shown). Thus, Lynx1 and Ric-3 were functional when expressed in tsA201 HEK cell lines, so lack of effect on incorporation of β3 was not due to lack of function of Lynx1 and Ric-3.
Assembly of AChRs Analyzed Using Sucrose Gradient Sedimentation. Partially assembled α4β2 AChRs are disrupted by Triton X-100, and [3H]epibatidine binding is detected to only mature pentameric AChRs on sucrose gradients unless assembly intermediates are stabilized using a cross-linking reagent (Kuryatov et al., 2005). The presence of either α5 or β3 subunits resulted in the formation of partially assembled AChRs, which accounted for approximately half of the total [3H]epibatidine binding sites on the gradients (Fig. 6). These partially assembled AChRs must contain both α4 and β2 to form epibatidine binding sites and must contain accessory subunits to prevent dissociation by Triton X-100. Immunoisolation showed that mature and partially assembled AChRs contained α5 or β3, but the limited affinity of mAb 210 did not allow isolation of all complexes containing α5 or β3 when mAb 210 was used on coated microwells. α4 was immunologically detectable virtually only in mature pentamers, even though α4 had to be present in partially assembled AChRs to permit formation of epibatidine binding sites. This suggests that the α4 in the partially assembled AChRs, especially the large cytoplasmic domain where epitopes recognized by antiserum to α4 and mAb371 are located, was partially proteolytically degraded.
Unassembled or partially assembled α4 and β2 subunits in the parent cell line are stable in large pools, and essentially all of the subunits in the pools can be assembled into mature pentameric AChRs as a result of adding nicotine, which binds to assembly intermediates, thereby promoting assembly of mature AChRs (Kuryatov et al., 2005).
Up-regulation of α4β2α5 cells using nicotine resulted in increased amounts of both mature and partially assembled AChRs (Fig. 7). Thus, the molecular chaperone effect of nicotine, which promotes assembly, probably acts on an intermediate containing at least the one α4 and one β2 subunit needed to form an ACh binding site (e.g., α4β2, α5α4β2, α4β2α5, α4β2α4β2) but before formation of dead-end partially assembled AChRs (e.g., α5α4β2α5).
The accumulation of partially assembled AChRs in the α4β2α5 line can be prevented by transient cotransfection with more α4 (Fig. 8). This suggests that depletion of the pool of α4 subunits is the step that limits the extent of AChR assembly and nicotine-induced up-regulation.
The partially assembled AChRs may be productive intermediates such as α4β2 subunit pairs or α4β2α5 trimers, which can form mature α4β2α4β2α5 AChRs with the assembly of additional subunits, or they may be dead-end tetramers like α5α4β2α5, which cannot further assemble productively with the addition of another subunit. The loss of α4 epitopes in these partially assembled AChRs suggests that most were dead-end complexes that could not form mature pentamers and advance from the endoplasmic reticulum to the Golgi apparatus and so remained in the endoplasmic reticulum, where they were partially degraded. Their large size is consistent with an α5α4β2α5 subunit composition. This arrangement contains only known subunit interfaces but does not permit assembly of mature pentamers of expected stoichiometries because the presence of two α5 subunits prevents the assembly of an additional α4β2 pair. The capping by two α5 subunits may prevent disruption or dissociation by Triton X-100.

AChRs up-regulated by nicotine still efficiently incorporate α5 subunits. After overnight exposure to the indicated concentrations of nicotine, AChRs in Triton X-100 extracts were immune precipitated by either mAb 295 to measure total AChRs or mAb 210 to measure those that incorporated α5 subunits.

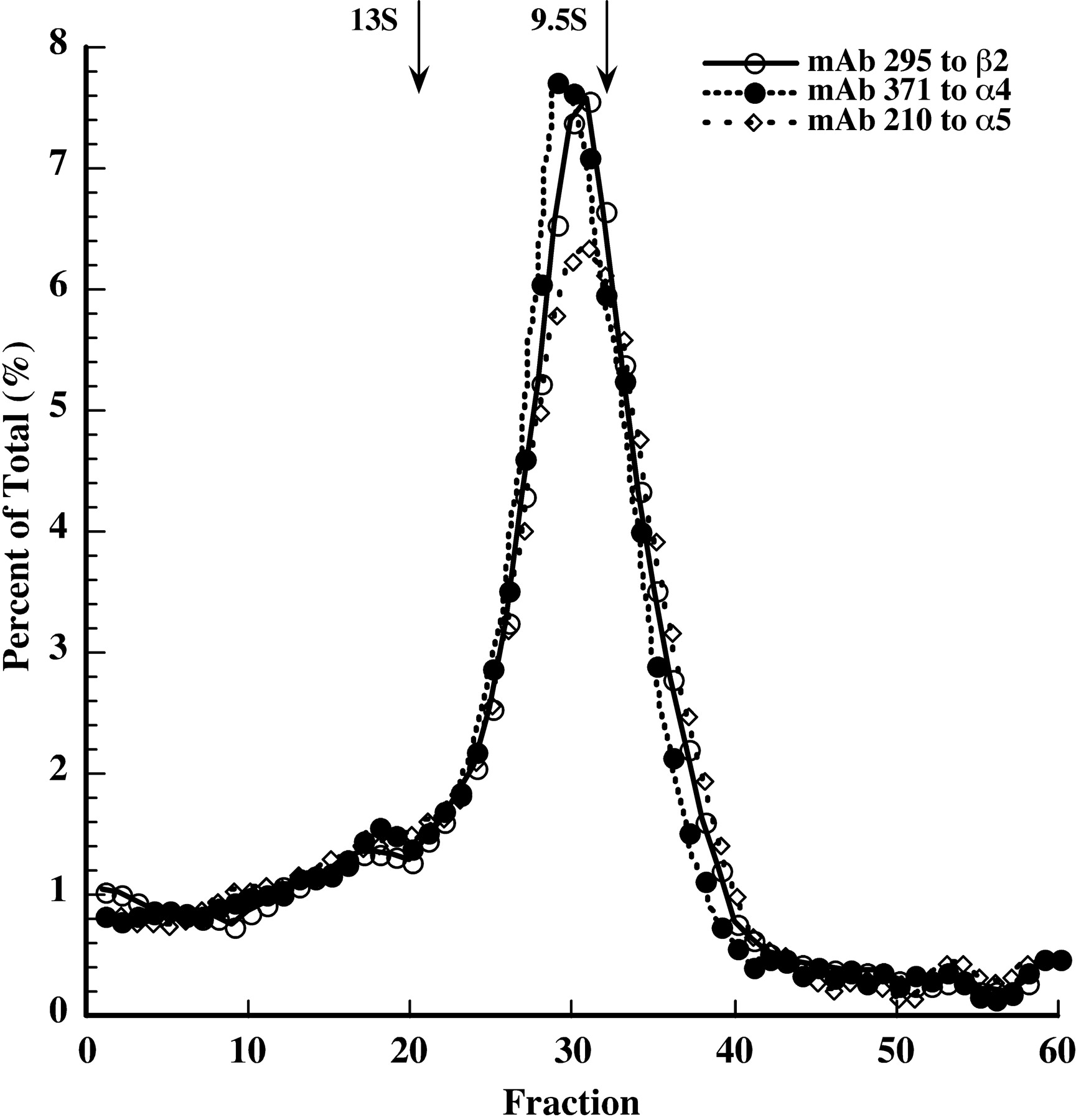
Sucrose gradient sedimentation reveals large amounts of partially assembled AChRs. Cosedimentation on the gradients of Torpedo AChR 9.5S monomers and 13S dimers immunoisolated on mAb210 coated wells and labeled with 125I-α-bungarotoxin (indicated by arrows) provided internal standards for identifying the 10 S mature pentameric AChRs peaking around fraction 30 and the 8.5 S partially assembled AChRs peaking near fraction 40 after having sedimented from the top of the gradient collected in fraction 60. [3H]epibatidine-labeled AChRs containing β2 subunits were isolated using mAb295-coated microwells. Those containing immunologically recognizable α4 subunits were immunoprecipitated using antiserum to bacterially expressed α4. AChR containing α5 and β3 subunits were isolated on mAb 210 coated microwells.
The α4β2β3 line also exhibits nearly equal amounts of mature and partially assembled AChRs. In addition, similar to the α4β2α5 line, all of the epitopes for mAb 371 to the α4 cytoplasmic surface are destroyed (as are epitopes for anti-serum to α4 and mAb 299, which has an extracellular epitope, data not shown) (Fig. 9). Biotinylation of the cell surface with a membrane-impermeable reagent before solubilization permitted identification of AChRs that were on the cell surface by isolation on streptavidin-coated wells. As expected, only mature AChRs were expressed on the cell surface (Fig. 9). Thus, in both the α4β2α5 and α4β2β3 lines, there are large amounts of partially assembled AChRs stabilized by their accessory subunits. Probably in each case these are formed as a result of large amounts of accessory subunits and limiting amounts of α4 (Fig. 8). In the presence of large amounts of β3 relative to α4, a α4β2β3 assembly intermediate trimer might assemble with β3 to form a stable dead end β3α4β2β3 complex before it could assemble with α4 then β2 or with an α4β2 pair to form a mature (α4β2)2β3 AChR. The partially assembled AChRs detected on the sucrose gradients are probably partially degraded dead-end complexes of the form β3α4β2β3. Only mature AChRs get to the cell surface where their function can be assayed.

Nicotine up-regulates both partially and fully assembled α4β2α5 AChRs. The positions of the 9.5 S monomer and 13 S dimer of T. californica AChRs on the gradients are shown by arrows.
The presence of nearly half of the epibatidine binding sites as intracellular dead end intermediates means that higher proportions of mature AChRs are on the cell surface than was calculated by measurements of the proportion of epibatidine binding sites on the cell surface. Correcting for the amount of binding sites present on intracellular dead end intermediates, virtually all mature (α4β2)2β3 AChRs are expressed on the cell surface as are 34% of mature (α4β2)2α5 AChRs.
Obligate Accessory Subunits (α5 or β3) and Other Subunits (β4 and α6) Can Rescue Function of ADNFLE α4S247Fβ2 Mutant AChRs. These mutant AChRs do not form functional AChRs in the transfected cell line, presumably because HEK cells preferentially produce (α4β2)2α4 AChRs resulting in three phenylalanine groups in the lumen of the channel (Kuryatov et al., 2005).
Displacing the α4 in the accessory position of α4S247Fβ2 mutant AChRs by cotransfection with α5 or β3 results in functional AChRs (Fig. 10). This is consistent with the observation that, when this mutant is expressed in oocytes with subunit ratios that promote assembly of the (α4β2)2β2 stoichiometry, functional AChRs are produced (Kuryatov et al., 1997). β3 was most potent in rescuing function, consistent with its exceptional efficiency in assembling with wild-type AChRs in the α4β2β3 line. β4 subunits could also displace the α4 accessory subunit to form functional AChRs (Fig. 10). They may also have displaced some β2 subunits in forming ACh binding sites, but only displacing the accessory α4 subunits in forming ACh binding sites would reduce the number of phenylalanines blocking the channel. α6 subunits could also rescue function (Fig. 10). These could displace either α4 acting as an accessory subunit or α4 forming part of an ACh binding site. (α4β2)(α6β2)β3 AChRs have been found in brain (Gotti et al., 2006). In HEK cell lines, it has been difficult, requiring special conditions, to get α4 and α6 to coassemble into AChRs where both α4 and α6 subunits participate in forming ACh binding sites (A.K. and J.L., unpublished observations).
Mutant (α4β2)2α5 and (α4β2)2β3 AChRs were activated by a variety of agonists (Fig. 10, Table 4). α5 in the mutant resulted in 7-fold more sensitivity to ACh than wild-type (α4β2)2α5 AChRs (Tables 1 and 4). β3 had remarkable effects, increasing ACh sensitivity 1334-fold, nicotine sensitivity 94-fold, and cytisine sensitivity 850-fold compared with wild-type (α4β2)2β3 AChRs (Tables 1 and 4). Cytisine was a remarkably potent (EC50 = 0.0056 μM) full agonist on mutant (α4β2)2β3 AChRs. Thus, in the presence of these obligate accessory subunits, the presence of phenylalanines just on the two binding site α4 subunits not only does not block the channel but also greatly increases sensitivity to its opening. There is precedent for mutations in M2 greatly increasing sensitivity to activation by agonists (Labarca et al., 2001).
Activation of S247Fα4β2* AChRs in a cell line
Discussion
The obligate accessory subunits α5 and β3 are efficiently incorporated with human α4 and β2 AChR subunits expressed in permanently transfected HEK cell lines, thereby revealing properties of (α4β2)2α5 and (α4β2)2β3 AChRs that are likely to be relevant to the expression of these subtypes in neurons. Expression of excess accessory subunits resulted in their incorporation in nearly all of the AChRs in these cell lines.
(α4β2)2α5 AChRs are known to be expressed in brain and elsewhere (Gotti et al., 2007). We have shown that α5 increased Ca2+ permeability compared with (α4β2)2β2 AChRs and sensitivity to activation compared with (α4β2)2α4 AChRs (Tapia et al., 2007), and that α5 restored Ca2+ permeability to (S247Fα4β2)2β2 AChRs (Kuryatov et al., 1997). The α4β2α5 line expressed 40% more AChRs than the parental α4β2 line. This permitted further up-regulation of expression by nicotine and other agonists with the same sensitivity as the α4β2 line. α5 increased sensitivity to activation by agonists compared with (α4β2)2α4 AChRs, increased the rate of acute desensitization by nicotine, but did not change sensitivity to long-term desensitization.

Transient transfection with additional α4 causes all AChRs to be assembled as mature (α4β2)2α5 AChRs. Sedimentation on sucrose gradients resolved only mature 10 S AChRs. Immunoisolation of [3H]epibatidine labeled AChRs on mAb coated microwells showed that α4, β2, and α5 subunits were present in these AChRs.

Sucrose gradient sedimentation analysis of the α4β2β3 line. Microwells coated with mAbs were used to immunoprecipitate [3H]epibatidine-labeled AChRs from gradient fractions revealing that, as with the α4β2α5 line, there are nearly equal amounts of mature and partially assembled AChRs and that α4 epitopes are detectable in the mature AChRs and not the partially assembled AChRs. The α4β2β3 cells were surface labeled with biotin before solubilization, then streptavidin coated wells were used to isolate AChRs that derived from the cell surface. Only mature AChRs and no partially assembled AChRs were found on the cell surface.

Function of the S247Fα4β2 cell line AChRs is rescued by transient transfection with α5, β3, β4, or α6 subunits. After transfection overnight with the indicated subunits, function was assayed using the FLEXstation with a Ca2+-sensitive indicator. The strongest responses to application of ACh are shown.
(α4β2)2β3 AChRs are not usually considered among brain ACh subtypes, but because α4, β2, β3, and α6 subunits are all assembled in the endoplasmic reticulum of dopaminergic neurons like those of the ventral tegmental area or the substantia nigra, which are known to assemble (α6β2)(α4β2)β3 AChRs (Gotti et al., 2007), there is ample opportunity for their synthesis. In the transfected line, (α4β2)2β3 AChRs assemble with exceptional efficiency so that very high levels of AChRs are expressed on the cell surface. No further upregulation by nicotine was observed, probably because α4 and β2 subunit pools were depleted.
The stoichiometry and subunit composition of AChRs expressed in neurons may depend critically on transcriptional or translational regulation of subunit synthesis determining the pools of subunits available for assembly. In X. laevis oocytes, injecting an excess of α4 mRNA results in the (α4β2)2α4 stoichiometry, whereas injecting an excess of β2 mRNA results in the (α4β2)2β2 stoichiometry (Moroni et al., 2006).
Immunoisolation studies from rat brains indicate that the amount of α4 and β2 subunits always greatly exceeds the amount of α5 subunits (Gotti et al., 2007; Perry et al., 2007; Mao et al., 2008). This accounts for the observation that (α4β2)2α5 AChRs are only 11 to 37% of the total α4β2* AChRs, depending on the brain region (Mao et al., 2008). Long-term exposure to nicotine increases the amount of brain α4β2* AChRs but not the amount of (α4β2)2α5 AChRs (Mao et al., 2008). This might result from the synthesizing of α5 in limiting amount, all of which is assembled, leaving only pools of α4 and β2 to be assembled in response to the pharmacological chaperone effects of nicotine. We show here that when α5 is present in excess, it assembles efficiently with α4β2 and the amount of α4β2α5 AChRs is increased in the presence of nicotine. In rat brain, less β3 is expressed than α5, and the amounts of α4, β2, α5, and α6 exceed the amount of β3 (Gotti et al., 2007; Perry et al., 2007; Mao et al., 2008). Furthermore, α5 is always found in association with α4, and β3 is always in association with α6, never with α4 alone. As with α5, after up-regulation by nicotine, the amount of β3 remains constant, indicating that all of the limited amount of β3 is already incorporated in AChRs. Here we show that β3 can assemble efficiently with α4β2. The absence of (α4β2)2β3 AChRs in brain may result from a combination of the limiting amount of β3 and perhaps also greater affinity of β3 for assembling with α6 than α4 and greater affinity of α5 for assembling with α4 than α6.
We show that when α5 and β3 are expressed in amounts equal to or greater than α4 and β2, many dead-end, partially assembled AChRs are formed. Thus, the observation that only small amounts of α5 and β3 are usually expressed may reflect a biological necessity to avoid forming nonproductive assemblies. Both β3α4β2 and α4β2β3 subunit trimers have allowable subunit interfaces and can form mature AChRs by assembly with α4β2 dimers. With excess β3, β3α4β2β3 tetramers are likely to form, which have allowable interfaces but cannot form a mature pentamer with addition of another α4 or β2. The stability of putative β3α4β2β3 tetramers to dissociation by Triton X-100 and the proteolytic decay of the α4 within them indicate that they are not easily eliminated by conventional editing mechanisms and are thus potentially toxic.
Demonstration in cell lines that various AChR subunits can assemble efficiently and be up-regulated by nicotine, in combination with the concept that in neurons some subunits are synthesized in limiting amounts, can explain several conundrums. After treatment of rats with nicotine for 2 weeks, in the striatum α4β2* AChRs are increased (as measured by ligand binding), α6β2* AChRs are decreased, and the total amount of β3-containing AChRs remains constant (Perry et al., 2007). These results might be explained if α4β2 AChRs in GABAergic neurons were up-regulated and the total amount of α4 subunit in dopaminergic neurons remained constant (Nashmi et al., 2007), but in the dopaminergic neurons, α6 was displaced by α4 from (α6β2)(α4β2)β3 AChRs to form (α4β2)2β3 AChRs. This would be expected if nicotine acted on α4β2 and α6β2 subunit pairs to promote assembly because nicotine is much more potent at promoting the assembly of α4β2 than α6β2 AChRs (Kuryatov et al., 2005; Tumkosit et al., 2006). We show here that β3 avidly assembles with α4β2 and in Tumkosit et al. (2006) that it avidly assembles with α6β2; thus, all the β3 present will assemble. In rat superior colliculus, nicotine did not reduce the numbers of α6* AChRs (Perry et al., 2007). This would be expected if the retinal ganglia neurons that terminate in the superior colliculus expressed only (α6β2)2β3 AChRs and did not also express α4 to compete for assembly, as do ventral tegmental area dopaminergic neurons.
Both obligate accessory subunits (α5 and β3) and other subunits (β4 and α6) can restore function to the ADNFLE mutant cell line S247Fα4β2. Displacing the α4 in the accessory position to form, for example, (S247Fα4β2)2β3 AChRs leaves only two phenylalanines in the cation channel. Remarkably, this not only unblocks the channel but also greatly increases sensitivity to activation through some interaction between the phenylalanine groups and the β3 subunit. There is precedent for mutations in the M2 region of α4 and other subunits greatly increasing sensitivity to activation (Labarca et al., 2001).
As shown here and elsewhere, α5 and β3 subunits can have substantial effects on the efficiency of assembly of the AChR subtypes that contain them and on the pharmacological, conductance, and desensitization properties of these AChRs. In addition, α5 and β3 subunits may have important roles in targeting AChRs to particular locations (Gotti et al., 2007). In the ventral tegmental area, dopaminergic neurons, α6* AChRs containing β3 subunits, are selectively located at presynaptic endings (Champtiaux et al., 2003; Quik et al., 2007). In transfected N2a cells, α4β2 AChRs are localized to filopodia, but (α4β2)2β3 AChRs are not (Drenan et al., 2008). When expressed in HEK cells, α5 and α3 (but not α4 and β2) specifically associate with PSD-93a, PSD-95, and SAP102, proteins that are associated with postsynaptic densities (Conroy et al., 2003). Disrupting interactions with postsynaptic density proteins in neurons expressing (α3β4)2α5 AChRs impairs excitatory postsynaptic currents without altering the number of AChRs by disrupting alignment of pre- and postsynaptic elements. It seems likely that these accessory subunits could target (α4β2)2α5 and (α4β2)2β3 AChRs to particular post- and presynaptic locations.
GABAA receptors are homologous in structure to nicotinic AChRs. In these receptors, the presence of the accessory subunit γ is necessary to permit formation of a binding site for benzodiazepines at the interface between γ and α subunits (Cromer et al., 2002). PAMs such as benzodiazepines have great theoretical significance as drugs because they can promote the effectiveness of neurotransmission without altering the pattern of signaling and can sustain their effects, unlike competitive agonists, which may act as time-averaged antagonists through desensitizing the receptors. Potent PAMs have been reported for α7 AChRs (Hurst et al., 2005; Ng et al., 2007). Galanthamine binds at a subunit interface similar to that at which benzodiazepines bind (Hansen and Taylor, 2007) and acts as a PAM on AChRs (Samochocki et al., 2003). We showed that (α4β2)2α5, unlike α4β2 or (α4β2)2β3 AChRs, is uniquely sensitive to galanthamine. Unique interfaces, like those between α5 and α4 or between β3 and α6, could provide very specific drug targets for PAMs more easily discriminated than ACh binding sites for selectively targeting AChR subtypes. Cell lines like those described here could be critically important in screening for such positive allosteric modulators.
Acknowledgments
We thank Barbara Campling for comments on the manuscript.
Footnotes
-
This work was supported by grant NS11323 from the National Institutes of Health (to J.L.).
-
ABBREVIATIONS: AChR, acetylcholine receptor; ACh, acetylcholine; HEK, human embryonic kidney; ADNFLE, autosomal-dominant nocturnal frontal lobe epilepsy; PBS, phosphate-buffered saline; mAb, monoclonal antibody; PAM, positive allosteric modulator; DHβE, dihydro-β-erythroidine.
- Received February 29, 2008.
- Accepted March 31, 2008.
- The American Society for Pharmacology and Experimental Therapeutics





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}